
Mitochondrial Peroxiredoxin 3 Is Rapidly Oxidized and Hyperoxidized by Fatty Acid Hydroperoxides
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Hydroperoxide Concentration Measurements
2.3. Enzyme Expression and Purification
2.4. Reduction of HsPrx3
2.5. Catalytic Activity of HsPrx3
2.6. Kinetics of HsPrx3 Oxidation and Hyperoxidation by fFA-OOHs
2.7. SDS PAGE and Western Blot Analysis of HsPrx3 Hyperoxidation
2.8. Mass Spectrometry Analysis of HsPrx3 Modifications by FA-OOHs
2.9. Fatty Acids Binding to HsPrx3
2.10. Computer-Assisted Molecular Modeling of FA Binding to HsPrx3
2.11. Simulations of HsPrx3 Hyperoxidation by Fluxes of Mitochondrial Hydroperoxides in the Presence of Peroxidases
3. Results
3.1. HsPrx3 Is Rapidly Inactivated by fFA-OOHs
3.2. Kinetics of HsPrx3 Oxidation by fFA-OOH
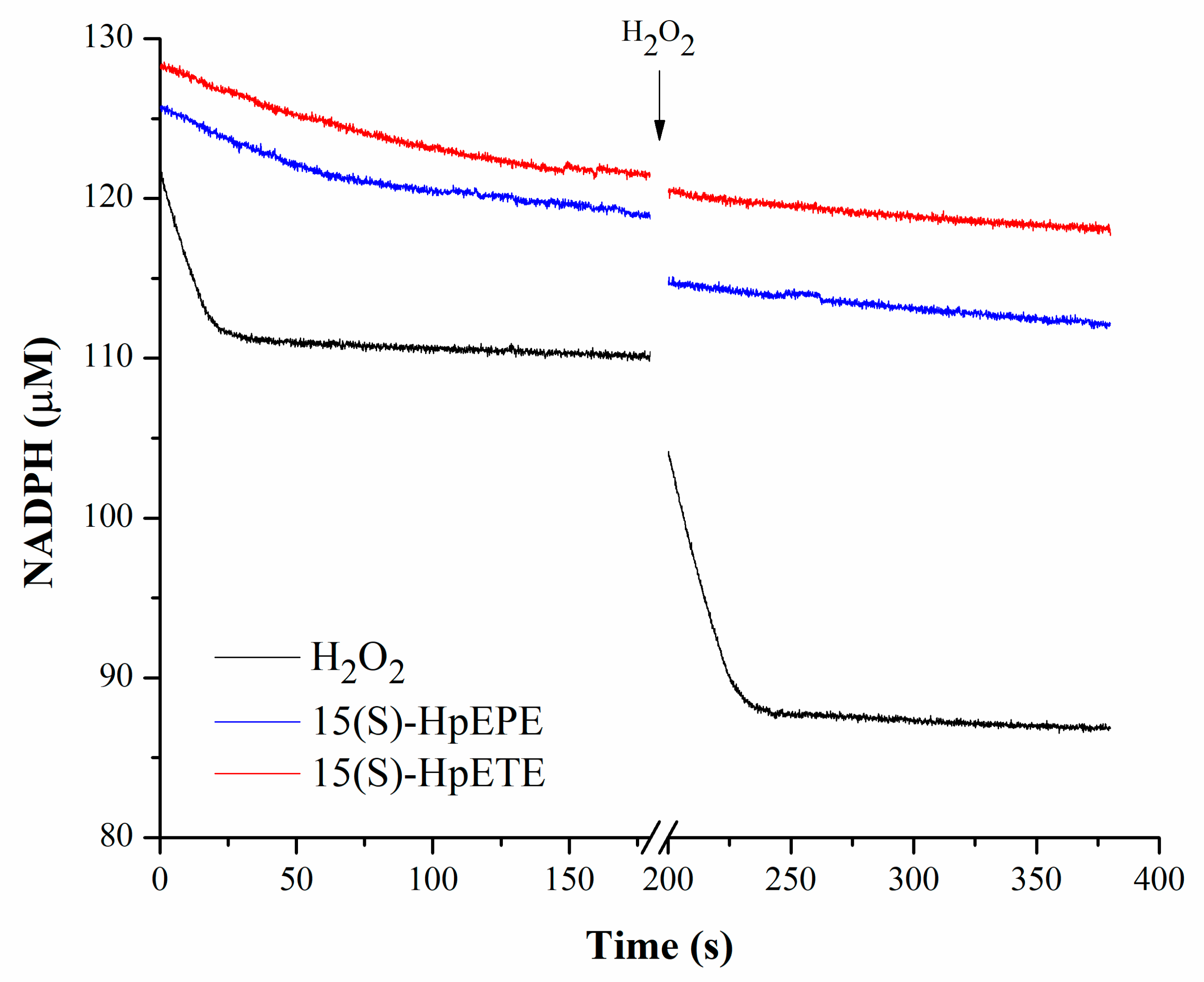
3.3. Consumption of Hydroperoxides by HsPrx3 under Non-Catalytic Conditions
3.4. HsPrx3 Is Hyperoxidized at Low Concentrations of fFA-OOHs
3.4.1. SDS PAGE and Western Blot Analysis
3.4.2. Mass Spectrometry Analysis
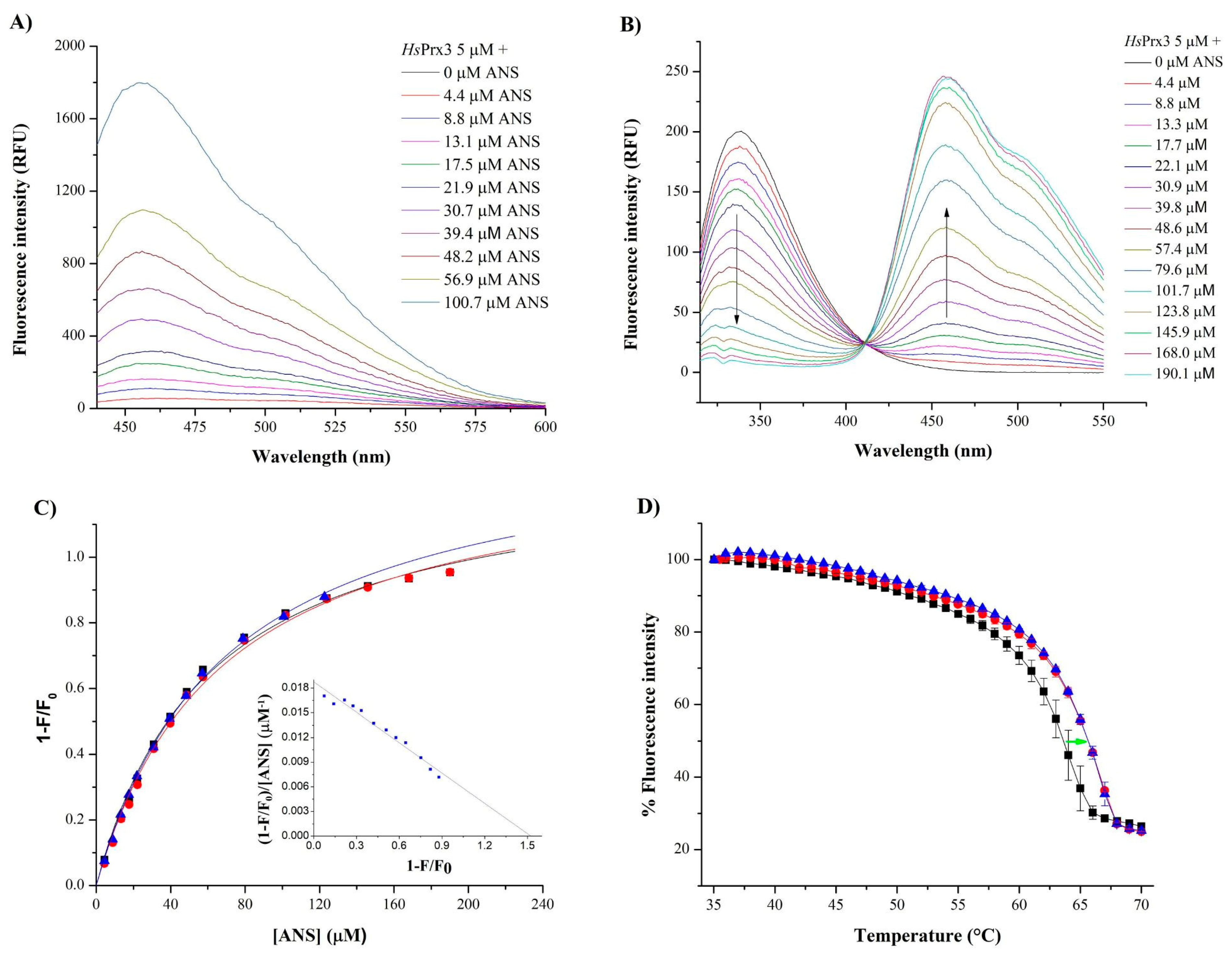
3.5. Biophysical Experiments Suggest the Binding of Hydrophobic Compounds to HsPrx3
3.5.1. Binding of ANS and AA to HsPrx3
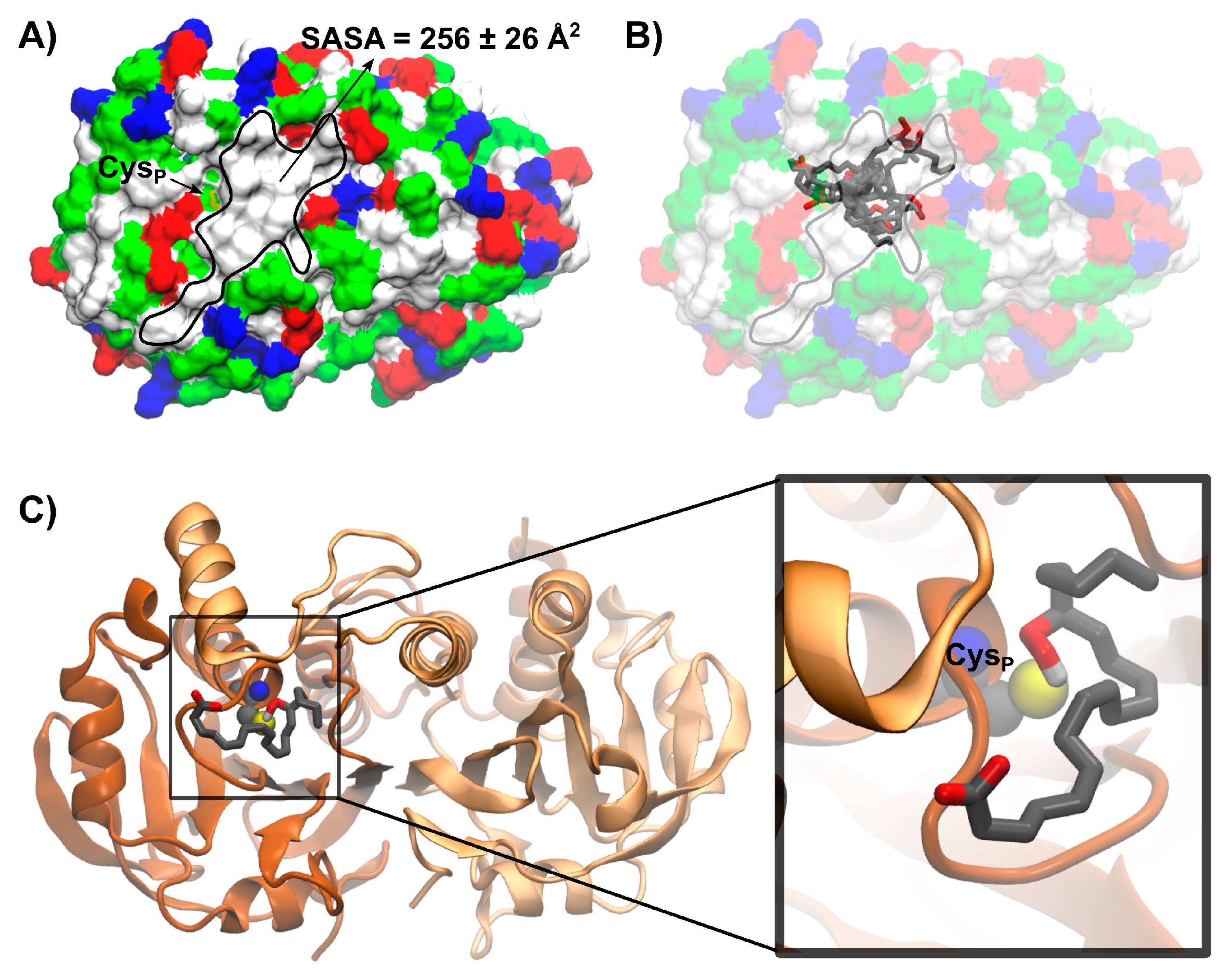
3.5.2. Modeling 15(S)-HpETE Binding to HsPrx3
4. Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | k (M−1s−1 or s−1) | Reference |
|---|---|---|
| (1) redPrx3 + H2O2 → Prx3SOH + H2O | 2 × 107 | [12] |
| (2) Prx3SOH → Prx3SS | 2 | [13] |
| (3) Prx3SOH + H2O2 → Prx3SO2H + H2O | 1.1 × 103 | [13] |
| (4) redPrx3 + fFA-OOH → Prx3SOH + fFA-OH | 3.5 × 107 | This work |
| (5) Prx3SOH + fFA-OOH → Prx3SO2H + fFA-OH | 1 × 107 | This work |
| (6) Prx3SS + redTrx2/Grx2 → redPrx3 + oxTrx2/Grx2 | 4 × 104 a | [54] |
| (7) redPrx5 + H2O2 → Prx5SOH + H2O | 3 × 105 | [80] |
| (8) redPrx5 + fFA-OOH → Prx5SOH + fFA-OH | 2 × 107 b | [80] |
| (9) Prx5SOH → Prx5SS | 15 | [80] |
| (10) Prx5SS + redTrx2 → redPrx5 + redTrx2 | 2 × 106 | [80] |
| (11) redGPx1 c + H2O2 → oxGPx1 + H2O | 4 × 107 | [28] |
| (12) redGPx1 + fFA-OOH → oxGPx1 + fFA-OH | 4 ×107 | [28] |
| (13) oxGPx1 + 2GSH d→ redGPx1+ GSSG | 2 × 105 | [28] |
| % of Prx3-SO2− with Respect to Initial Reduced Prx3 | ||||
|---|---|---|---|---|
| [Reduced Prx3] 20 µM a | ||||
| fFA-OOH (µM/s) | H2O2 (µM/s) | 0.4 | 1 | 4 |
| 0 | 2.8 × 10−3 | 2 × 10−2 | 0.37 | |
| 0.001 | 3.3 × 10−2 | 0.1 | 0.75 | |
| 0.01 | 0.30 | 0.8 | 4 | |
| 0.1 | 3.7 | 8.5 | 39 | |
| [Reduced Prx3] 100 µM a | ||||
| fFA-OOH (µM/s) | H2O2 (µM/s) | 0.4 | 1 | 4 |
| 0 | 1.5 × 10−4 | 9.3 × 10−4 | 1.6 × 10−2 | |
| 0.001 | 2.0 × 10−3 | 5.6 × 10−3 | 3.6 × 10−2 | |
| 0.01 | 1.9 × 10−2 | 4.8 × 10−2 | 0.22 | |
| 0.1 | 0.23 | 0.51 | 2.1 | |
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Pan, C.; Feng, C.; Yan, C.; Yu, Y.; Chen, Z.; Guo, C.; Wang, X. Role of mitochondrial reactive oxygen species in homeostasis regulation. Redox Rep. 2022, 27, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Piacenza, L.; Zeida, A.; Trujillo, M.; Radi, R. The superoxide radical switch in the biology of nitric oxide and peroxynitrite. Physiol. Rev. 2022, 102, 1881–1906. [Google Scholar] [CrossRef]
- Mauerhofer, C.; Philippova, M.; Oskolkova, O.V.; Bochkov, V.N. Hormetic and anti-inflammatory properties of oxidized phospholipids. Mol. Asp. Med. 2016, 49, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Muller, F.L.; Liu, Y.; Sabia, M.; Liang, H.; Song, W.; Jang, Y.C.; Ran, Q.; Van Remmen, H. Denervation induces cytosolic phospholipase A2-mediated fatty acid hydroperoxide generation by muscle mitochondria. J. Biol. Chem. 2009, 284, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Ademowo, O.S.; Dias, H.; Burton, D.G.; Griffiths, H.R. Lipid (per) oxidation in mitochondria: An emerging target in the ageing process? Biogerontology 2017, 18, 859–879. [Google Scholar] [CrossRef] [PubMed]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem. 2010, 345, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Fisher-Wellman, K.H.; Neufer, P.D. Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol. Metab. 2012, 23, 142–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quijano, C.; Trujillo, M.; Castro, L.; Trostchansky, A. Interplay between oxidant species and energy metabolism. Redox Biol. 2016, 8, 28–42. [Google Scholar] [CrossRef]
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 2010, 425, 313–325. [Google Scholar] [CrossRef] [PubMed]
- De Armas, M.I.; Esteves, R.; Viera, N.; Reyes, A.M.; Mastrogiovanni, M.; Alegria, T.G.P.; Netto, L.E.S.; Tortora, V.; Radi, R.; Trujillo, M. Rapid peroxynitrite reduction by human peroxiredoxin 3: Implications for the fate of oxidants in mitochondria. Free. Radic. Biol. Med. 2019, 130, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.G.; Pearson, A.G.; Pullar, J.M.; Jönsson, T.J.; Lowther, W.T.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin 3 is more resilient to hyperoxidation than cytoplasmic peroxiredoxins. Biochem. J. 2009, 421, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Peskin, A.V.; Pace, P.E.; Winterbourn, C.C. Enhanced hyperoxidation of peroxiredoxin 2 and peroxiredoxin 3 in the presence of bicarbonate/CO2. Free. Radic. Biol. Med. 2019, 145, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Esteras, N.; Abramov, A.Y. Mitochondria and lipid peroxidation in the mechanism of neurodegeneration: Finding ways for prevention. Med. Res. Rev. 2021, 41, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef]
- Colbeau, A.; Nachbaur, J.; Vignais, P. Enzymac characterization and lipid composition of rat liver subcellular membranes. Biochim. Et Biophys. Acta (BBA)-Biomembr. 1971, 249, 462–492. [Google Scholar] [CrossRef]
- Belikova, N.A.; Vladimirov, Y.A.; Osipov, A.N.; Kapralov, A.A.; Tyurin, V.A.; Potapovich, M.V.; Basova, L.V.; Peterson, J.; Kurnikov, I.V.; Kagan, V.E. Peroxidase activity and structural transitions of cytochrome c bound to cardiolipin-containing membranes. Biochemistry 2006, 45, 4998–5009. [Google Scholar] [CrossRef]
- Ježek, J.; Dlasková, A.; Zelenka, J.; Jabůrek, M.; Ježek, P. H2O2-activated mitochondrial phospholipase iPLA2γ prevents lipotoxic oxidative stress in synergy with UCP2, amplifies signaling via G-protein–coupled receptor GPR40, and regulates insulin secretion in pancreatic β-cells. Antioxid. Redox Signal. 2015, 23, 958–972. [Google Scholar] [CrossRef] [Green Version]
- Průchová, P.; Gotvaldová, K.; Smolková, K.; Alán, L.; Holendová, B.; Tauber, J.; Galkin, A.; Ježek, P.; Jabůrek, M. Antioxidant Role and Cardiolipin Remodeling by Redox-Activated Mitochondrial Ca2+-Independent Phospholipase A2γ in the Brain. Antioxidants 2022, 11, 198. [Google Scholar] [CrossRef] [PubMed]
- Ježek, J.; Jabůrek, M.; Zelenka, J.; Ježek, P. Mitochondrial phospholipase A2 activated by reactive oxygen species in heart mitochondria induces mild uncoupling. Physiol. Res. 2010, 59, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Pallast, S.; Arai, K.; Wang, X.; Lo, E.H.; Van Leyen, K. 12/15-Lipoxygenase targets neuronal mitochondria under oxidative stress. J. Neurochem. 2009, 111, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Lane, D.J.; Ayton, S.; Bush, A.I. Iron and Alzheimer’s disease: An update on emerging mechanisms. J. Alzheimer’s Dis. 2018, 64, S379–S395. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, Y. In Search of the Holy Grail: Toward a Unified Hypothesis on Mitochondrial Dysfunction in Age-Related Diseases. Cells 2022, 11, 1906. [Google Scholar] [CrossRef]
- Schuckelt, R.; Brigelius-Flohe, R.; Maiorino, M.; Roveri, A.; Reumkens, J.; Strabburger, W.; Ursini, F.; Wolf, B.; Flohe, L. Phospholipid hydroperoxide glutathione peroxidase is a seleno-enzyme distinct from the classical glutathione peroxidase as evident from cDNA and amino acid sequencing. Free. Radic. Res. Commun. 1991, 14, 343–361. [Google Scholar] [CrossRef]
- Toppo, S.; Flohé, L.; Ursini, F.; Vanin, S.; Maiorino, M. Catalytic mechanisms and specificities of glutathione peroxidases: Variations of a basic scheme. Biochim. Et Biophys. Acta (BBA)-Gen. Subj. 2009, 1790, 1486–1500. [Google Scholar] [CrossRef]
- Yant, L.J.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, T.A. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free. Radic. Biol. Med. 2003, 34, 496–502. [Google Scholar] [CrossRef]
- Schneider, M.; Forster, H.; Boersma, A.; Seiler, A.; Wehnes, H.; Sinowatz, F.; Neumuller, C.; Deutsch, M.J.; Walch, A.; Hrabe de Angelis, M.; et al. Mitochondrial glutathione peroxidase 4 disruption causes male infertility. FASEB J. 2009, 23, 3233–3242. [Google Scholar] [CrossRef] [Green Version]
- Maiorino, M.; Roveri, A.; Benazzi, L.; Bosello, V.; Mauri, P.; Toppo, S.; Tosatto, S.C.; Ursini, F. Functional interaction of phospholipid hydroperoxide glutathione peroxidase with sperm mitochondrion-associated cysteine-rich protein discloses the adjacent cysteine motif as a new substrate of the selenoperoxidase. J. Biol. Chem. 2005, 280, 38395–38402. [Google Scholar] [CrossRef]
- Azuma, K.; Koumura, T.; Iwamoto, R.; Matsuoka, M.; Terauchi, R.; Yasuda, S.; Shiraya, T.; Watanabe, S.; Aihara, M.; Imai, H. Mitochondrial glutathione peroxidase 4 is indispensable for photoreceptor development and survival in mice. J. Biol. Chem. 2022, 298, 4. [Google Scholar] [CrossRef] [PubMed]
- Flohé, L.; Toppo, S.; Cozza, G.; Ursini, F. A comparison of thiol peroxidase mechanisms. Antioxid. Redox Signal. 2011, 15, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.-K.; Yun, C.-H.; Kim, I.-H. Interaction of human thiol-specific antioxidant protein 1 with erythrocyte plasma membrane. Biochemistry 2000, 39, 6944–6950. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Benipal, B.; Zhou, S.; Dodia, C.; Chatterjee, S.; Tao, J.-Q.; Sorokina, E.M.; Raabe, T.; Feinstein, S.I.; Fisher, A.B. Critical role of peroxiredoxin 6 in the repair of peroxidized cell membranes following oxidative stress. Free. Radic. Biol. Med. 2015, 87, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Schlecker, T.; Schmidt, A.; Dirdjaja, N.; Voncken, F.; Clayton, C.; Krauth-Siegel, R.L. Substrate specificity, localization, and essential role of the glutathione peroxidase-type tryparedoxin peroxidases in Trypanosoma brucei. J. Biol. Chem. 2005, 280, 14385–14394. [Google Scholar] [CrossRef]
- Budde, H.; Flohé, L.; Hecht, H.-J.; Hofmann, B.; Stehr, M.; Wissing, J.; Lünsdorf, H. Kinetics and redox-sensitive oligomerisation reveal negative subunit cooperativity in tryparedoxin peroxidase of Trypanosoma brucei brucei. De Gruyter 2003. [Google Scholar] [CrossRef]
- Jaeger, T.; Budde, H.; Flohé, L.; Menge, U.; Singh, M.; Trujillo, M.; Radi, R. Multiple thioredoxin-mediated routes to detoxify hydroperoxides in Mycobacterium tuberculosis. Arch. Biochem. Biophys. 2004, 423, 182–191. [Google Scholar] [CrossRef]
- Reyes, A.M.; Hugo, M.; Trostchansky, A.; Capece, L.; Radi, R.; Trujillo, M. Oxidizing substrate specificity of Mycobacterium tuberculosis alkyl hydroperoxide reductase E: Kinetics and mechanisms of oxidation and overoxidation. Free. Radic. Biol. Med. 2011, 51, 464–473. [Google Scholar] [CrossRef]
- Reyes, A.M.; Vazquez, D.S.; Zeida, A.; Hugo, M.; Piñeyro, M.D.; De Armas, M.I.; Estrin, D.; Radi, R.; Santos, J.; Trujillo, M. PrxQ B from Mycobacterium tuberculosis is a monomeric, thioredoxin-dependent and highly efficient fatty acid hydroperoxide reductase. Free. Radic. Biol. Med. 2016, 101, 249–260. [Google Scholar] [CrossRef]
- Zeida, A.; Reyes, A.M.; Lichtig, P.; Hugo, M.; Vazquez, D.S.; Santos, J.; González Flecha, F.L.; Radi, R.; Estrin, D.A.; Trujillo, M. Molecular basis of hydroperoxide specificity in peroxiredoxins: The case of AhpE from Mycobacterium tuberculosis. Biochemistry 2015, 54, 7237–7247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Park, J.; Han, S.-J.; Lim, Y.; Park, I.; Kim, J.-S.; Woo, H.; Lee, S.-R. Peroxiredoxin III protects tumor suppressor PTEN from oxidation by 15-Hydroperoxy-eicosatetraenoic acid. Oxidative Med. Cell. Longev. 2019, 2019, 2828493. [Google Scholar] [CrossRef] [PubMed]
- Cordray, P.; Doyle, K.; Edes, K.; Moos, P.J.; Fitzpatrick, F.A. Oxidation of 2-Cys-peroxiredoxins by arachidonic acid peroxide metabolites of lipoxygenases and cyclooxygenase-2. J. Biol. Chem. 2007, 282, 32623–32629. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Lustgarten, M.; Shi, Y.; Liu, Y.; Jang, Y.C.; Pulliam, D.; Jernigan, A.L.; Van Remmen, H. Increased mitochondrial matrix-directed superoxide production by fatty acid hydroperoxides in skeletal muscle mitochondria. Free. Radic. Biol. Med. 2011, 50, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, H.; Patel, D.; Howat, W.; Warren, A.; Kay, J.; Sangan, T.; Marioni, J.; Mitchell, J.; Aldridge, S.; Luxton, H. Peroxiredoxin-3 is overexpressed in prostate cancer and promotes cancer cell survival by protecting cells from oxidative stress. Br. J. Cancer 2013, 109, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, X.; Li, J.; Zhang, R.; Yu, C.; Zhou, Z.; Liu, J.; Liao, S.; Klionsky, D.J.; Kroemer, G. Identification of HPCAL1 as a specific autophagy receptor involved in ferroptosis. Autophagy 2023, 19, 54–74. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Freitas, F.P.; Seibt, T. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 2018, 172, 409–422.e421. [Google Scholar] [CrossRef]
- Evans, R.C.; Chen, L.; Na, R.; Yoo, K.; Ran, Q. The Gpx4NIKO Mouse Is a Versatile Model for Testing Interventions Targeting Ferroptotic Cell Death of Spinal Motor Neurons. Neurotox. Res. 2022, 40, 373–383. [Google Scholar] [CrossRef]
- Musicco, C.; Capelli, V.; Pesce, V.; Timperio, A.M.; Calvani, M.; Mosconi, L.; Zolla, L.; Cantatore, P.; Gadaleta, M.N. Accumulation of overoxidized Peroxiredoxin III in aged rat liver mitochondria. Biochim. Et Biophys. Acta BBA Bioenerg. 2009, 1787, 890–896. [Google Scholar] [CrossRef]
- Kil, I.S.; Lee, S.K.; Ryu, K.W.; Woo, H.A.; Hu, M.-C.; Bae, S.H.; Rhee, S.G. Feedback control of adrenal steroidogenesis via H2O2-dependent, reversible inactivation of peroxiredoxin III in mitochondria. Mol. Cell 2012, 46, 584–594. [Google Scholar] [CrossRef] [Green Version]
- Claiborne, A.; Miller, H.; Parsonage, D.; Ross, R.P. Protein-sulfenic acid stabilization and function in enzyme catalysis and gene regulation. FASEB J. 1993, 7, 1483–1490. [Google Scholar] [CrossRef]
- Jiang, Z.-Y.; Hunt, J.V.; Wolff, S.P. Ferrous ion oxidation in the presence of xylenol orange for detection of lipid hydroperoxide in low density lipoprotein. Anal. Biochem. 1992, 202, 384–389. [Google Scholar] [CrossRef]
- Cuevasanta, E.; Reyes, A.M.; Zeida, A.; Mastrogiovanni, M.; De Armas, M.I.; Radi, R.; Alvarez, B.; Trujillo, M. Kinetics of formation and reactivity of the persulfide in the one-cysteine peroxiredoxin from Mycobacterium tuberculosis. J. Biol. Chem. 2019, 294, 13593–13605. [Google Scholar] [CrossRef] [PubMed]
- Hanschmann, E.-M.; Lönn, M.E.; Schütte, L.D.; Funke, M.; Godoy, J.R.; Eitner, S.; Hudemann, C.; Lillig, C.H. Both thioredoxin 2 and glutaredoxin 2 contribute to the reduction of the mitochondrial 2-Cys peroxiredoxin Prx3. J. Biol. Chem. 2010, 285, 40699–40705. [Google Scholar] [CrossRef] [PubMed]
- Manta, B.; Hugo, M.; Ortiz, C.; Ferrer-Sueta, G.; Trujillo, M.; Denicola, A. The peroxidase and peroxynitrite reductase activity of human erythrocyte peroxiredoxin 2. Arch. Biochem. Biophys. 2009, 484, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, M.; Denicola, A.; Novoselov, S.V.; Turanov, A.A.; Protasio, A.; Izmendi, D.; Gladyshev, V.N.; Salinas, G. Platyhelminth mitochondrial and cytosolic redox homeostasis is controlled by a single thioredoxin glutathione reductase and dependent on selenium and glutathione. J. Biol. Chem. 2008, 283, 17898–17907. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.J.; Parsonage, D. Measurement of peroxiredoxin activity. Curr. Protoc. Toxicol. 2011, 49, 7.10.1–7.10.28. [Google Scholar] [CrossRef]
- Peskin, A.V.; Meotti, F.C.; de Souza, L.F.; Anderson, R.F.; Winterbourn, C.C.; Salvador, A. Intra-dimer cooperativity between the active site cysteines during the oxidation of peroxiredoxin 2. Free Radic. Biol. Med. 2020, 158, 115–125. [Google Scholar] [CrossRef]
- Portillo-Ledesma, S.; Randall, L.M.; Parsonage, D.; Dalla Rizza, J.; Karplus, P.A.; Poole, L.B.; Denicola, A.; Ferrer-Sueta, G. Differential kinetics of two-cysteine peroxiredoxin disulfide formation reveal a novel model for peroxide sensing. Biochemistry 2018, 57, 3416–3424. [Google Scholar] [CrossRef] [PubMed]
- Parsonage, D.; Nelson, K.J.; Ferrer-Sueta, G.; Alley, S.; Karplus, P.A.; Furdui, C.M.; Poole, L.B. Dissecting peroxiredoxin catalysis: Separating binding, peroxidation, and resolution for a bacterial AhpC. Biochemistry 2015, 54, 1567–1575. [Google Scholar] [CrossRef] [Green Version]
- Cardamone, M.; Puri, N. Spectrofluorimetric assessment of the surface hydrophobicity of proteins. Biochem. J. 1992, 282, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Matulis, D.; Lovrien, R. 1-Anilino-8-naphthalene sulfonate anion-protein binding depends primarily on ion pair formation. Biophys. J. 1998, 74, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.-s.; Wen, E.-y.; Hung, J.-j.; Chang, C.-c. Energy transfer from tryptophan residues of proteins to 8-anilinonaphthalene-1-sulfonate. J. Protein Chem. 1994, 13, 635–640. [Google Scholar] [CrossRef]
- Das, A.; Basak, P.; Pattanayak, R.; Kar, T.; Majumder, R.; Pal, D.; Bhattacharya, A.; Bhattacharyya, M.; Banik, S.P. Trehalose induced structural modulation of Bovine Serum Albumin at ambient temperature. Int. J. Biol. Macromol. 2017, 105, 645–655. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Suttapitugsakul, S.; Xiao, H.; Smeekens, J.; Wu, R. Evaluation and optimization of reduction and alkylation methods to maximize peptide identification with MS-based proteomics. Mol. BioSyst. 2017, 13, 2574–2582. [Google Scholar] [CrossRef] [PubMed]
- Messineo, F.; Pinto, P.; Katz, A. Palmitic acid enhances calcium sequestration by isolated sarcoplasmic reticulum. J. Mol. Cell. Cardiol. 1980, 12, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Pompeia, C.; Lima, T.; Curi, R. Arachidonic acid cytotoxicity: Can arachidonic acid be a physiological mediator of cell death? Cell Biochem. Funct. Cell. Biochem. Its Modul. Act. Agents Dis. 2003, 21, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Serth, J.; Lautwein, A.; Frech, M.; Wittinghofer, A.; Pingoud, A. The inhibition of the GTPase activating protein-Ha-ras interaction by acidic lipids is due to physical association of the C-terminal domain of the GTPase activating protein with micellar structures. EMBO J. 1991, 10, 1325–1330. [Google Scholar] [CrossRef]
- Scott, D.E.; Spry, C.; Abell, C. Differential scanning fluorimetry as part of a biophysical screening cascade. In Fragment-Based Drug Discovery: Lessons and Outlook; Wiley-VCH: Weinheim, Germany, 2016. [Google Scholar]
- Peskin, A.V.; Low, F.M.; Paton, L.N.; Maghzal, G.J.; Hampton, M.B.; Winterbourn, C.C. The high reactivity of peroxiredoxin 2 with H2O2 is not reflected in its reaction with other oxidants and thiol reagents. J. Biol. Chem. 2007, 282, 11885–11892. [Google Scholar] [CrossRef] [PubMed]
- Yewdall, N.A.; Venugopal, H.; Desfosses, A.; Abrishami, V.; Yosaatmadja, Y.; Hampton, M.B.; Gerrard, J.A.; Goldstone, D.C.; Mitra, A.K.; Radjainia, M. Structures of human peroxiredoxin 3 suggest self-chaperoning assembly that maintains catalytic state. Structure 2016, 24, 1120–1129. [Google Scholar] [CrossRef]
- Poynton, R.A.; Peskin, A.V.; Haynes, A.C.; Lowther, W.T.; Hampton, M.B.; Winterbourn, C.C. Kinetic analysis of structural influences on the susceptibility of peroxiredoxins 2 and 3 to hyperoxidation. Biochem. J. 2016, 473, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Palleros, D.R.; Fink, A.L. Protein conformational changes induced by 1,1’-bis(4-anilino-5-naphthalenesulfonic acid): Preferential binding to the molten globule of DnaK. Biochemistry 1994, 33, 7536–7546. [Google Scholar] [CrossRef] [PubMed]
- Jencks, W.P. Binding energy, specificity, and enzymic catalysis: The circe effect. Adv. Enzymol. Relat. Areas Mol. Biol. 1975, 43, 219–410. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, A.J. Dispelling the effects of a sorceress in enzyme catalysis. Proc. Natl. Acad. Sci. USA 2016, 113, 2328–2330. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, M.; Clippe, A.; Manta, B.; Ferrer-Sueta, G.; Smeets, A.; Declercq, J.-P.; Knoops, B.; Radi, R. Pre-steady state kinetic characterization of human peroxiredoxin 5: Taking advantage of Trp84 fluorescence increase upon oxidation. Arch. Biochem. Biophys. 2007, 467, 95–106. [Google Scholar] [CrossRef] [PubMed]
- De Simoni, S.; Linard, D.; Hermans, E.; Knoops, B.; Goemaere, J. Mitochondrial peroxiredoxin-5 as potential modulator of mitochondria-ER crosstalk in MPP+-induced cell death. J. Neurochem. 2013, 125, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Quijano, C.; Castro, L.; Peluffo, G.; Valez, V.; Radi, R. Enhanced mitochondrial superoxide in hyperglycemic endothelial cells: Direct measurements and formation of hydrogen peroxide and peroxynitrite. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3404–H3414. [Google Scholar] [CrossRef] [PubMed]
- Antunes, F.; Salvador, A.; Marinho, H.S.; Alves, R.; Pinto, R.E. Lipid peroxidation in mitochondrial inner membranes. I. An integrative kinetic model. Free Radic. Biol. Med. 1996, 21, 917–943. [Google Scholar] [CrossRef] [PubMed]
- Noh, Y.H.; Baek, J.Y.; Jeong, W.; Rhee, S.G.; Chang, T.-S. Sulfiredoxin translocation into mitochondria plays a crucial role in reducing hyperoxidized peroxiredoxin III. J. Biol. Chem. 2009, 284, 8470–8477. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lu, D.; He, F.; Zhou, H.; Liu, Q.; Wang, Y.; Shao, C.; Gong, Y. Cullin 4B protein ubiquitin ligase targets peroxiredoxin III for degradation. J. Biol. Chem. 2011, 286, 32344–32354. [Google Scholar] [CrossRef]
- Belikova, N.A.; Tyurina, Y.Y.; Borisenko, G.; Tyurin, V.; Samhan Arias, A.K.; Yanamala, N.; Furtmüller, P.G.; Klein-Seetharaman, J.; Obinger, C.; Kagan, V.E. Heterolytic reduction of fatty acid hydroperoxides by cytochrome c/cardiolipin complexes: Antioxidant function in mitochondria. J. Am. Chem. Soc. 2009, 131, 11288–11289. [Google Scholar] [CrossRef]
- Khailova, L.S.; Prikhodko, E.A.; Dedukhova, V.I.; Mokhova, E.N.; Popov, V.N.; Skulachev, V.P. Participation of ATP/ADP antiporter in oleate-and oleate hydroperoxide-induced uncoupling suppressed by GDP and carboxyatractylate. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2006, 1757, 1324–1329. [Google Scholar] [CrossRef]
- Jabůrek, M.; Miyamoto, S.; Di Mascio, P.; Garlid, K.D.; Ježek, P. Hydroperoxy fatty acid cycling mediated by mitochondrial uncoupling protein UCP2. J. Biol. Chem. 2004, 279, 53097–53102. [Google Scholar] [CrossRef]
- De Simoni, S.; Goemaere, J.; Knoops, B. Silencing of peroxiredoxin 3 and peroxiredoxin 5 reveals the role of mitochondrial peroxiredoxins in the protection of human neuroblastoma SH-SY5Y cells toward MPP+. Neurosci. Lett. 2008, 433, 219–224. [Google Scholar] [CrossRef]
- Portillo, S. Peroxirredoxinas: Eficientes Reductoras de Peróxidos y Eficientemente Reducidas. Función de los Aminoácidos Conservados en Ambas Reacciones. Ph.D. Thesis, Universidad de la República, Montevideo, Uruguay, 2018. [Google Scholar]
- Arteel, G.E.; Briviba, K.; Sies, H. Protection against peroxynitrite. FEBS Lett. 1999, 445, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.H.; Woo, H.A.; Sung, S.H.; Lee, H.E.; Lee, S.K.; Kil, I.S.; Rhee, S.G. Induction of sulfiredoxin via an Nrf2-dependent pathway and hyperoxidation of peroxiredoxin III in the lungs of mice exposed to hyperoxia. Antioxid. Redox Signal. 2009, 11, 937–948. [Google Scholar] [CrossRef]
- Collins, J.A.; Wood, S.T.; Nelson, K.J.; Rowe, M.A.; Carlson, C.S.; Chubinskaya, S.; Poole, L.B.; Furdui, C.M.; Loeser, R.F. Oxidative stress promotes peroxiredoxin hyperoxidation and attenuates pro-survival signaling in aging chondrocytes. J. Biol. Chem. 2016, 291, 6641–6654. [Google Scholar] [CrossRef] [PubMed]
- Kameritsch, P.; Singer, M.; Nuernbergk, C.; Rios, N.; Reyes, A.M.; Schmidt, K.; Kirsch, J.; Schneider, H.; Müller, S.; Pogoda, K. The mitochondrial thioredoxin reductase system (TrxR2) in vascular endothelium controls peroxynitrite levels and tissue integrity. Proc. Natl. Acad. Sci. USA 2021, 118, e1921828118. [Google Scholar] [CrossRef] [PubMed]
- Dalla Rizza, J.; Randall, L.M.; Santos, J.; Ferrer-Sueta, G.; Denicola, A. Differential parameters between cytosolic 2-Cys peroxiredoxins, PRDX1 and PRDX2. Protein Sci. 2019, 28, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Peskin, A.V.; Dickerhof, N.; Poynton, R.A.; Paton, L.N.; Pace, P.E.; Hampton, M.B.; Winterbourn, C.C. Hyperoxidation of peroxiredoxins 2 and 3: Rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. J. Biol. Chem. 2013, 288, 14170–14177. [Google Scholar] [CrossRef]
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653. [Google Scholar] [CrossRef]
- Zeida, A.; Trujillo, M.; Ferrer-Sueta, G.; Denicola, A.; Estrin, D.A.; Radi, R. Catalysis of peroxide reduction by fast reacting protein thiols: Focus review. Chem. Rev. 2019, 119, 10829–10855. [Google Scholar] [CrossRef] [PubMed]
- Talwar, D.; Dick, T.P. Thiol peroxidase-based redox relays. In Redox Chemistry and Biology of Thiols; Elsevier: Amsterdam, The Netherlands, 2022; pp. 307–320. [Google Scholar]
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Measuring the redox state of cellular peroxiredoxins by immunoblotting. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2010; Volume 474, pp. 51–66. [Google Scholar]
- Watabe, S.; Kohno, H.; Kouyama, H.; Hiroi, T.; Yago, N.; Nakazawa, T. Purification and characterization of a substrate protein for mitochondrial ATP-dependent protease in bovine adrenal cortex. J. Biochem. 1994, 115, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Nesmachnow, S.; Iturriaga, S. Cluster-UY Collaborative Scientific High Performance Computing in Uruguay. In Supercomputing. Communications in Computer and Information Science; Torres, M., Klapp, J., Eds.; Springer: Mexico City, Mexico, 2019; Volume 1151, pp. 188–202. [Google Scholar]







| Oxidizing Substrate | vo 1 (µM/s) | vo after Subsequent Addition of 20 µM H2O2 (µM/s) |
|---|---|---|
| H2O2 | 0.55 | 0.60 |
| 15(S)-HpETE | 0.07 | 0.03 |
| 15(S)-HpEPE | 0.08 | 0.02 |
| PGG2 | 0.09 | 0.08 |
| Treatment | Mass (Da) | Peak Description |
|---|---|---|
| Reduced HsPrx3 | 21,910 | HsPrx3-2CAM |
| 21,967 | HsPrx3-3CAM | |
| 22,025 | HsPrx3-4CAM | |
| HsPrx3 + H2O2 | 43,591 | HsPrx3 dimer |
| 43,648 | HsPrx3-CAM dimer | |
| 43,705 | HsPrx3-2CAM dimer | |
| 43,762 | HsPrx3-3CAM dimer | |
| 43,816 | HsPrx3-4CAM dimer | |
| HsPrx3 + 15(S)-HpEPE | 21,886 | CAM-HsPrx3-SO2 |
| 43,696 | CAM-HsPrx3-SO3 dimer | |
| 21,901 | CAM-HsPrx3-SO3 | |
| CPSHsPrx3 * + 15(S)-HpEPE | 21,838 | CPSHsPrx3-CAM |
| 21,894 | CPSHsPrx3-2CAM |
| Hydroperoxide | kox. (M−1s−1) | khyperox. (M−1s−1) | Reference |
|---|---|---|---|
| H2O2 | 2.0 × 107 (1) | 6 × 103 (1); 1.1 × 103 (2) | [12,13,15] |
| Peroxynitrite | 1.0 × 107 (3) | Not determined | [13] |
| 15(S)-HpEPE | ˃3.5 × 107 (4) | (2.6 ± 0.4) × 107 (4) | This work |
| 15(S)-HpETE | ˃3.5 × 107 (4) | (1.7 ± 0.1) × 107 (4) | This work |
| PGG2 | 2.4 × 107 (4) | (3.1 ± 0.7) × 105 (4) | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardozo, G.; Mastrogiovanni, M.; Zeida, A.; Viera, N.; Radi, R.; Reyes, A.M.; Trujillo, M. Mitochondrial Peroxiredoxin 3 Is Rapidly Oxidized and Hyperoxidized by Fatty Acid Hydroperoxides. Antioxidants 2023, 12, 408. https://doi.org/10.3390/antiox12020408
Cardozo G, Mastrogiovanni M, Zeida A, Viera N, Radi R, Reyes AM, Trujillo M. Mitochondrial Peroxiredoxin 3 Is Rapidly Oxidized and Hyperoxidized by Fatty Acid Hydroperoxides. Antioxidants. 2023; 12(2):408. https://doi.org/10.3390/antiox12020408
Chicago/Turabian StyleCardozo, Giuliana, Mauricio Mastrogiovanni, Ari Zeida, Nicolás Viera, Rafael Radi, Aníbal M. Reyes, and Madia Trujillo. 2023. "Mitochondrial Peroxiredoxin 3 Is Rapidly Oxidized and Hyperoxidized by Fatty Acid Hydroperoxides" Antioxidants 12, no. 2: 408. https://doi.org/10.3390/antiox12020408