
The Complex Genetic and Epigenetic Regulation of the Nrf2 Pathways: A Review

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
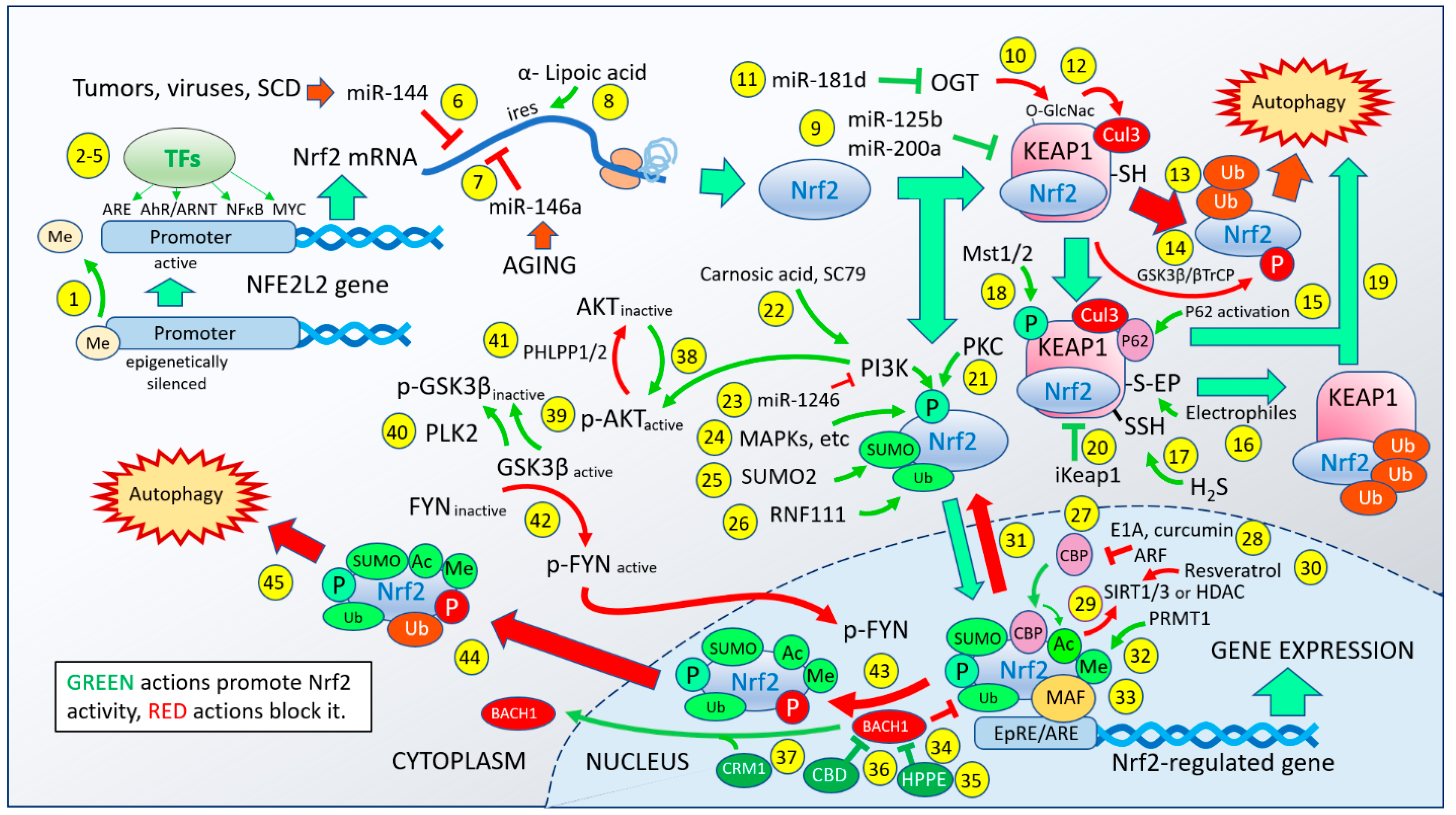
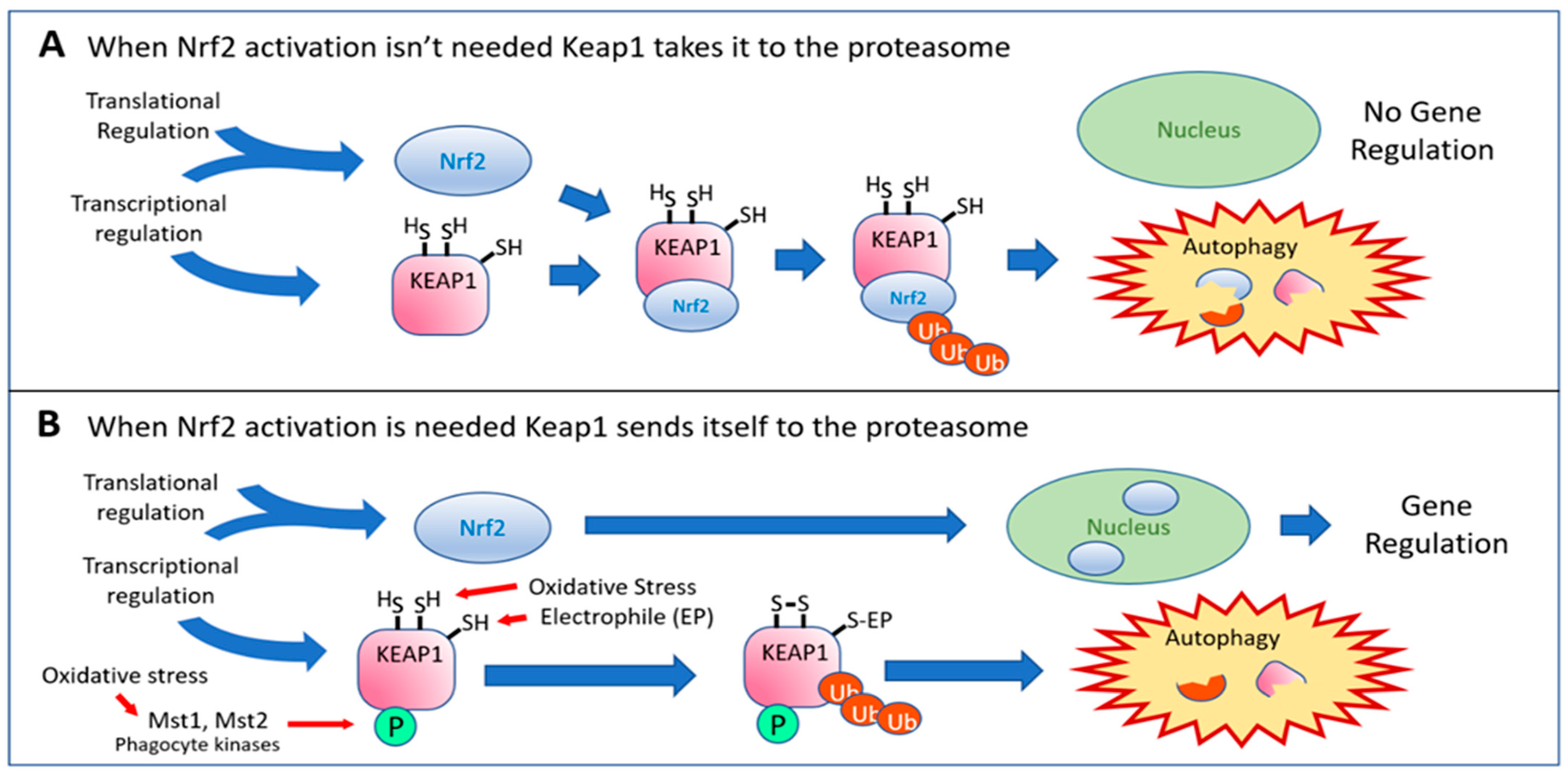
2. The Nrf2 Activation Pathway
3. The Complexity of the Nrf2 Pathway Tells Us “One Size Does Not Fit All”
4. MST1 Presents an Intriguing Alternative Route to Nrf2 Activation
5. Local Control May Selectively Regulate Nrf2 Effects That Are Context-Dependent
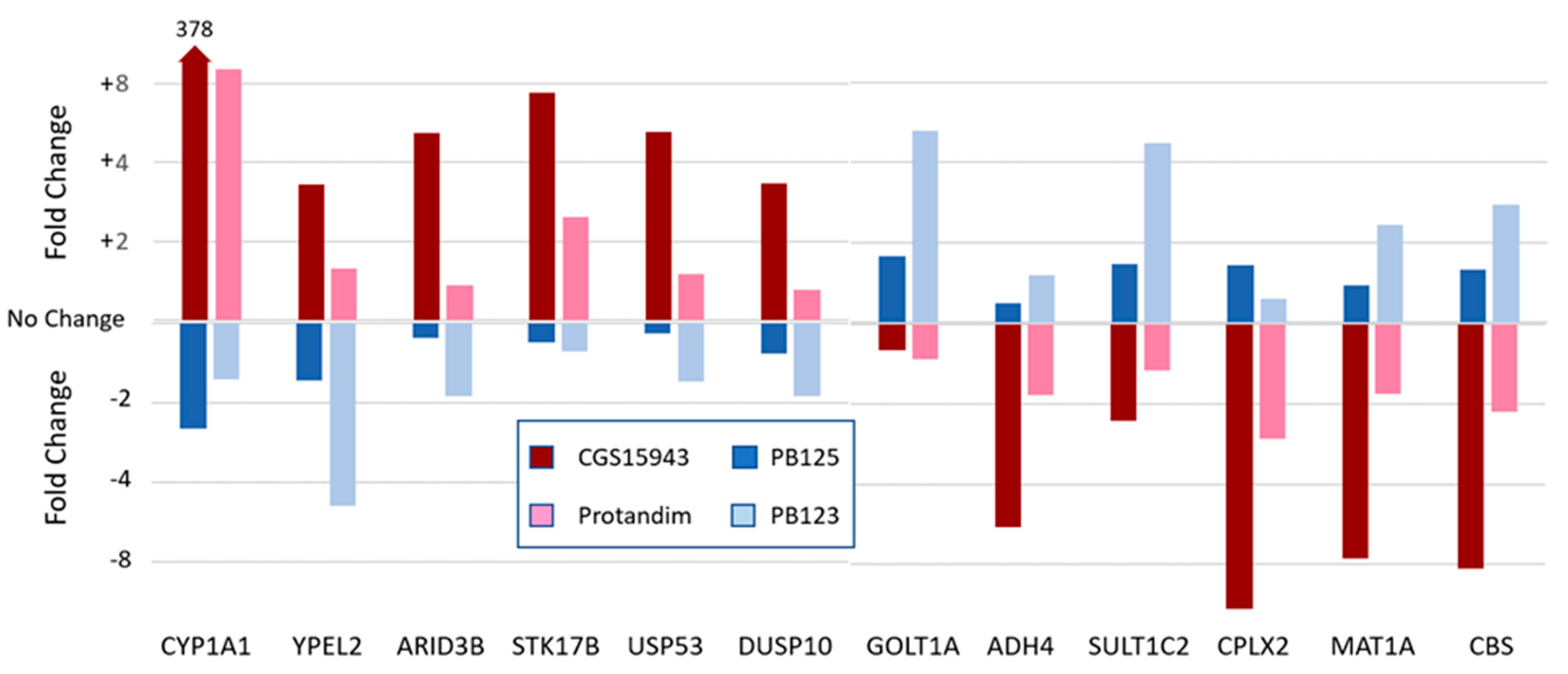
6. Can the Activation Pathway Affect the Gene Expression Profile?
7. Network Pharmacology Versus the “One Gene, One Drug, One Disease” Paradigm
8. Future Directions
9. Conclusions
Funding
Conflicts of Interest
References
- Prestera, T.; Talalay, P. Electrophile and antioxidant regulation of enzymes that detoxify carcinogens. Proc. Natl. Acad. Sci. USA 1995, 92, 8965–8969. [Google Scholar] [CrossRef] [PubMed]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Asp. Med. 2011, 32, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Gall Trošelj, K.; Tomljanović, M.; Jaganjac, M.; Matijević Glavan, T.; Čipak Gašparović, A.; Milković, L.; Borović Šunjić, S.; Buttari, B.; Profumo, E.; Saha, S.; et al. Oxidative Stress and Cancer Heterogeneity Orchestrate NRF2 Roles Relevant for Therapy Response. Molecules 2022, 27, 1468. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Fu, C.; Liu, J.; Sai, X.; Qin, C.; Di, T.; Yang, Y.; Wu, Y.; Bian, T. Hypermethylation of the Nrf2 Promoter Induces Ferroptosis by Inhibiting the Nrf2-GPX4 Axis in COPD. Int. J. Chron Obs. Pulmon. Dis. 2021, 16, 3347–3362. [Google Scholar] [CrossRef]
- Hosseini, H.; Teimouri, M.; Shabani, M.; Koushki, M.; Babaei Khorzoughi, R.; Namvarjah, F.; Izadi, P.; Meshkani, R. Resveratrol alleviates non-alcoholic fatty liver disease through epigenetic modification of the Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2020, 119, 105667. [Google Scholar] [CrossRef]
- Kukoyi, A.T.; Fan, X.; Staitieh, B.S.; Hybertson, B.M.; Gao, B.; McCord, J.M.; Guidot, D.M. MiR-144 mediates Nrf2 inhibition and alveolar epithelial dysfunction in HIV-1 transgenic rats. Am. J. Physiol. Cell Physiol. 2019, 317, C390–C397. [Google Scholar] [CrossRef]
- Li, B.; Zhu, X.; Ward, C.M.; Starlard-Davenport, A.; Takezaki, M.; Berry, A.; Ward, A.; Wilder, C.; Neunert, C.; Kutlar, A.; et al. MIR-144-mediated NRF2 gene silencing inhibits fetal hemoglobin expression in sickle cell disease. Exp. Hematol. 2019, 70, 85–96. [Google Scholar] [CrossRef]
- Azzimato, V.; Jager, J.; Chen, P.; Morgantini, C.; Levi, L.; Barreby, E.; Sulen, A.; Oses, C.; Willerbrords, J.; Xu, C.; et al. Liver macrophages inhibit the endogenous antioxidant response in obesity-associated insulin resistance. Sci. Transl. Med. 2020, 12, eaaw9709. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, Y.; Cheng, M.; Qiao, Y.; Wang, Y.; Xiong, W.; Yue, W. Suppression of microRNA-144-3p attenuates oxygen-glucose deprivation/reoxygenation-induced neuronal injury by promoting Brg1/Nrf2/ARE signaling. J. Biochem. Mol. Toxicol. 2018, 32, e22044. [Google Scholar] [CrossRef]
- Smith, E.J.; Shay, K.P.; Thomas, N.O.; Butler, J.A.; Finlay, L.F.; Hagen, T.M. Age-related loss of hepatic Nrf2 protein homeostasis: Potential role for heightened expression of miR-146a. Free Radic. Biol. Med. 2015, 89, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Ku, C.H.; Siow, R.C. Regulation of the Nrf2 antioxidant pathway by microRNAs: New players in micromanaging redox homeostasis. Free Radic. Biol. Med. 2013, 64, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Kaundal, R.K.; Datusalia, A.K.; Sharma, S.S. Posttranscriptional regulation of Nrf2 through miRNAs and their role in Alzheimer’s disease. Pharm. Res. 2022, 175, 106018. [Google Scholar] [CrossRef]
- Tian, C.; Gao, L.; Zucker, I.H. Regulation of Nrf2 signaling pathway in heart failure: Role of extracellular vesicles and non-coding RNAs. Free Radic. Biol. Med. 2021, 167, 218–231. [Google Scholar] [CrossRef]
- Padmavathi, G.; Ramkumar, K.M. MicroRNA mediated regulation of the major redox homeostasis switch, Nrf2, and its impact on oxidative stress-induced ischemic/reperfusion injury. Arch. Biochem. Biophys. 2021, 698, 108725. [Google Scholar] [CrossRef]
- Li, W.; Thakor, N.; Xu, E.Y.; Huang, Y.; Chen, C.; Yu, R.; Holcik, M.; Kong, A.N. An internal ribosomal entry site mediates redox-sensitive translation of Nrf2. Nucleic Acids Res. 2010, 38, 778–788. [Google Scholar] [CrossRef]
- Shay, K.P.; Michels, A.J.; Li, W.; Kong, A.-N.T.; Hagen, T.M. Cap-independent Nrf2 translation is part of a lipoic acid-stimulated detoxification stress response. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2012, 1823, 1102–1109. [Google Scholar] [CrossRef]
- Liu, J.X.; Ma, D.Y.; Zhi, X.Y.; Wang, M.W.; Zhao, J.Y.; Qin, Y. MiR-125b attenuates retinal pigment epithelium oxidative damage via targeting Nrf2/HIF-1alpha signal pathway. Exp. Cell Res. 2022, 410, 112955. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Wu, J.; Ma, F.; Jiang, J.; Xu, L.; Du, L.; Huang, W.; Wang, Z.; Jia, Y.; Lu, L.; et al. MicroRNA-200a improves diabetic endothelial dysfunction by targeting KEAP1/NRF2. J. Endocrinol. 2020, 245, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Smith, T.J.; Wu, J.; Siesser, P.F.; Bisnett, B.J.; Khan, F.; Hogue, M.; Soderblom, E.; Tang, F.; Marks, J.R.; et al. Glycosylation of KEAP1 links nutrient sensing to redox stress signaling. EMBO J. 2017, 36, 2233–2250. [Google Scholar] [CrossRef]
- Tao, S.; Liu, P.; Luo, G.; Rojo de la Vega, M.; Chen, H.; Wu, T.; Tillotson, J.; Chapman, E.; Zhang, D.D. p97 Negatively Regulates NRF2 by Extracting Ubiquitylated NRF2 from the KEAP1-CUL3 E3 Complex. Mol. Cell Biol. 2017, 37, e00660-16. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.-H.; Du, Y.; Sheng, Z.; Li, Y.; Qiu, X.; Tian, B.; Yao, L. OGT-Mediated KEAP1 Glycosylation Accelerates NRF2 Degradation Leading to High Phosphate-Induced Vascular Calcification in Chronic Kidney Disease. Front. Physiol. 2020, 11, 1092. [Google Scholar] [CrossRef]
- Huang, W.; Chen, L.; Zhu, K.; Wang, D. Oncogenic microRNA-181d binding to OGT contributes to resistance of ovarian cancer cells to cisplatin. Cell Death Discov. 2021, 7, 379. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/beta-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef]
- Fernández-Ginés, R.; Encinar, J.A.; Hayes, J.D.; Oliva, B.; Rodríguez-Franco, M.I.; Rojo, A.I.; Cuadrado, A. An inhibitor of interaction between the transcription factor NRF2 and the E3 ubiquitin ligase adapter β-TrCP delivers anti-inflammatory responses in mouse liver. Redox Biol. 2022, 55, 102396. [Google Scholar] [CrossRef]
- Zhou, Y.; Lan, R.; Xu, Y.; Zhou, Y.; Lin, X.; Miao, J. Resveratrol alleviates oxidative stress caused by Streptococcus uberis infection via activating the Nrf2 signaling pathway. Int. Immunopharmacol. 2020, 89, 107076. [Google Scholar] [CrossRef]
- Zhao, Y.; Song, W.; Wang, Z.; Wang, Z.; Jin, X.; Xu, J.; Bai, L.; Li, Y.; Cui, J.; Cai, L. Resveratrol attenuates testicular apoptosis in type 1 diabetic mice: Role of Akt-mediated Nrf2 activation and p62-dependent Keap1 degradation. Redox Biol. 2018, 14, 609–617. [Google Scholar] [CrossRef]
- Dai, X.Y.; Zhu, S.Y.; Chen, J.; Li, M.Z.; Zhao, Y.; Talukder, M.; Li, J.L. Lycopene alleviates di(2-ethylhexyl) phthalate-induced splenic injury by activating P62-Keap1-NRF2 signaling. Food Chem. Toxicol. 2022, 168, 113324. [Google Scholar] [CrossRef]
- Yu, M.; Wang, W.; Dang, J.; Liu, B.; Xu, J.; Li, J.; Liu, Y.; He, L.; Ying, Y.; Cai, J.; et al. Hydrogen sulfide protects retinal pigment epithelium cells against ferroptosis through the AMPK- and p62-dependent non-canonical NRF2-KEAP1 pathway. Exp. Cell Res. 2022, 422, 113436. [Google Scholar] [CrossRef]
- Lee, D.H.; Han, D.H.; Nam, K.T.; Park, J.S.; Kim, S.H.; Lee, M.; Kim, G.; Min, B.S.; Cha, B.S.; Lee, Y.S.; et al. Ezetimibe, an NPC1L1 inhibitor, is a potent Nrf2 activator that protects mice from diet-induced nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2016, 99, 520–532. [Google Scholar] [CrossRef]
- Liang, S.T.; Chen, C.; Chen, R.X.; Li, R.; Chen, W.L.; Jiang, G.H.; Du, L.L. Michael acceptor molecules in natural products and their mechanism of action. Front. Pharm. 2022, 13, 1033003. [Google Scholar] [CrossRef] [PubMed]
- Prosperini, L.; Pontecorvo, S. Dimethyl fumarate in the management of multiple sclerosis: Appropriate patient selection and special considerations. Ther. Clin. Risk Manag. 2016, 2016, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Timpani, C.A.; Rybalka, E. Calming the (Cytokine) Storm: Dimethyl Fumarate as a Therapeutic Candidate for COVID-19. Pharmaceuticals 2020, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Hammer, A.; Waschbisch, A.; Kuhbandner, K.; Bayas, A.; Lee, D.H.; Duscha, A.; Haghikia, A.; Gold, R.; Linker, R.A. The NRF2 pathway as potential biomarker for dimethyl fumarate treatment in multiple sclerosis. Ann. Clin. Transl. Neurol. 2018, 5, 668–676. [Google Scholar] [CrossRef]
- To, C.; Ringelberg, C.S.; Royce, D.B.; Williams, C.R.; Risingsong, R.; Sporn, M.B.; Liby, K.T. Dimethyl fumarate and the oleanane triterpenoids, CDDO-imidazolide and CDDO-methyl ester, both activate the Nrf2 pathway but have opposite effects in the A/J model of lung carcinogenesis. Carcinogenesis 2015, 36, 769–781. [Google Scholar] [CrossRef]
- Escartin, C.; Brouillet, E. The Nrf2 pathway as a potential therapeutic target for Huntington disease A commentary on “Triterpenoids CDDO-ethyl amide and CDDO-trifluoroethyl amide improve the behavioral phenotype and brain pathology in a transgenic mouse model of Huntington disease”. Free Radic. Biol. Med. 2010, 49, 144–146. [Google Scholar] [CrossRef]
- Yates, M.S.; Tauchi, M.; Katsuoka, F.; Flanders, K.C.; Liby, K.T.; Honda, T.; Gribble, G.W.; Johnson, D.A.; Johnson, J.A.; Burton, N.C.; et al. Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol. Cancer 2007, 6, 154–162. [Google Scholar] [CrossRef]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef]
- Balamurugan, K.; Chandra, K.; Sai Latha, S.; Swathi, M.; Joshi, M.B.; Misra, P.; Parsa, K.V.L. PHLPPs: Emerging players in metabolic disorders. Drug Discov. Today 2022, 27, 103317. [Google Scholar] [CrossRef]
- Wang, P.; Geng, J.; Gao, J.; Zhao, H.; Li, J.; Shi, Y.; Yang, B.; Xiao, C.; Linghu, Y.; Sun, X.; et al. Macrophage achieves self-protection against oxidative stress-induced ageing through the Mst-Nrf2 axis. Nat. Commun. 2019, 10, 755. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Eggler, A.L.; Liu, G.; Pezzuto, J.M.; van Breemen, R.B.; Mesecar, A.D. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc. Natl. Acad. Sci. USA 2005, 102, 10070–10075. [Google Scholar] [CrossRef]
- Turpaev, K.T. Keap1-Nrf2 signaling pathway: Mechanisms of regulation and role in protection of cells against toxicity caused by xenobiotics and electrophiles. Biochemistry 2013, 78, 111–126. [Google Scholar] [CrossRef]
- Nguyen, T.; Sherratt, P.J.; Huang, H.C.; Yang, C.S.; Pickett, C.B. Increased Protein Stability as a Mechanism That Enhances Nrf2-mediated Transcriptional Activation of the Antioxidant Response Element. J. Biol. Chem. 2003, 278, 4536–4541. [Google Scholar] [CrossRef]
- Donatus, I.A.; Sardjoko; Vermeulen, N.P.E. Cytotoxic and cytoprotective activities of curcumin. Biochem. Pharmacol. 1990, 39, 1869–1875. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Hsieh, C.-W.; Weng, Y.-C.; Chuang, S.-H.; Hsieh, C.-Y.; Wung, B.-S. Sulforaphane inhibition of monocyte adhesion via the suppression of ICAM-1 and NF-κB is dependent upon glutathione depletion in endothelial cells. Vasc. Pharmacol. 2008, 48, 54–61. [Google Scholar] [CrossRef]
- Xu, Y.; Han, X.; Li, Y.; Min, H.; Zhao, X.; Zhang, Y.; Qi, Y.; Shi, J.; Qi, S.; Bao, Y.; et al. Sulforaphane Mediates Glutathione Depletion via Polymeric Nanoparticles to Restore Cisplatin Chemosensitivity. ACS Nano 2019, 13, 13445–13455. [Google Scholar] [CrossRef]
- Brennan, M.S.; Matos, M.F.; Li, B.; Hronowski, X.; Gao, B.; Juhasz, P.; Rhodes, K.J.; Scannevin, R.H. Dimethyl fumarate and monoethyl fumarate exhibit differential effects on KEAP1, NRF2 activation, and glutathione depletion in vitro. PLoS ONE 2015, 10, e0120254. [Google Scholar] [CrossRef]
- Halegoua-DeMarzio, D.; Navarro, V.; Ahmad, J.; Avula, B.; Barnhart, H.; Barritt, A.S.; Bonkovsky, H.L.; Fontana, R.J.; Ghabril, M.S.; Hoofnagle, J.H.; et al. Liver Injury Associated with Turmeric-A Growing Problem: Ten Cases from the Drug-Induced Liver Injury Network [DILIN]. Am. J. Med. 2022, 136, 200–206. [Google Scholar] [CrossRef]
- Lombardi, N.; Crescioli, G.; Maggini, V.; Ippoliti, I.; Menniti-Ippolito, F.; Gallo, E.; Brilli, V.; Lanzi, C.; Mannaioni, G.; Firenzuoli, F.; et al. Acute liver injury following turmeric use in Tuscany: An analysis of the Italian Phytovigilance database and systematic review of case reports. Br. J. Clin. Pharm. 2021, 87, 741–753. [Google Scholar] [CrossRef]
- Samudio, I.; Konopleva, M.; Hail, N.; Shi, Y.-X.; McQueen, T.; Hsu, T.; Evans, R.; Honda, T.; Gribble, G.W.; Sporn, M.; et al. 2-Cyano-3,12-dioxooleana-1,9-dien-28-imidazolide (CDDO-Im) Directly Targets Mitochondrial Glutathione to Induce Apoptosis in Pancreatic Cancer. J. Biol. Chem. 2005, 280, 36273–36282. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Kosaka, K.; Itoh, K.; Kobayashi, A.; Yamamoto, M.; Shimojo, Y.; Kitajima, C.; Cui, J.; Kamins, J.; Okamoto, S.; et al. Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. J. Neurochem. 2008, 104, 1116–1131. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; McKercher, S.R.; Lipton, S.A. Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic. Biol. Med. 2013, 65, 645–657. [Google Scholar] [CrossRef]
- Satoh, T.; Trudler, D.; Oh, C.-K.; Lipton, S.A. Potential Therapeutic Use of the Rosemary Diterpene Carnosic Acid for Alzheimer’s Disease, Parkinson’s Disease, and Long-COVID through NRF2 Activation to Counteract the NLRP3 Inflammasome. Antioxidants 2022, 11, 124. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Hybertson, B.M.; Cota-Gomez, A.; Gao, B. Nrf2 Activator PB125® as a Carnosic Acid-Based Therapeutic Agent against Respiratory Viral Diseases, including COVID-19. Free Radic. Biol. Med. 2021, 125, 56–64. [Google Scholar] [CrossRef]
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef]
- de Oliveira, M.R.; Ferreira, G.C.; Schuck, P.F.; Dal Bosco, S.M. Role for the PI3K/Akt/Nrf2 signaling pathway in the protective effects of carnosic acid against methylglyoxal-induced neurotoxicity in SH-SY5Y neuroblastoma cells. Chem. Biol. Interact. 2015, 242, 396–406. [Google Scholar] [CrossRef]
- de Oliveira, M.R.; Ferreira, G.C.; Schuck, P.F. Protective effect of carnosic acid against paraquat-induced redox impairment and mitochondrial dysfunction in SH-SY5Y cells: Role for PI3K/Akt/Nrf2 pathway. Toxicol. In Vitro 2016, 32, 41–54. [Google Scholar] [CrossRef]
- Jo, H.; Mondal, S.; Tan, D.; Nagata, E.; Takizawa, S.; Sharma, A.K.; Hou, Q.; Shanmugasundaram, K.; Prasad, A.; Tung, J.K.; et al. Small molecule-induced cytosolic activation of protein kinase Akt rescues ischemia-elicited neuronal death. Proc. Natl. Acad. Sci. USA 2012, 109, 10581–10586. [Google Scholar] [CrossRef]
- Gopallawa, I.; Kuek, L.E.; Adappa, N.D.; Palmer, J.N.; Lee, R.J. Small-molecule Akt-activation in airway cells induces NO production and reduces IL-8 transcription through Nrf-2. Respir. Res. 2021, 22, 267. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Z.; Hu, J.; Wan, X.; Huang, W.; Zhang, H.; Jiang, N. MiR-1246 regulates the PI3K/AKT signaling pathway by targeting PIK3AP1 and inhibits thyroid cancer cell proliferation and tumor growth. Mol. Cell. Biochem. 2021, 477, 649–661. [Google Scholar] [CrossRef]
- Liu, T.; Lv, Y.F.; Zhao, J.L.; You, Q.D.; Jiang, Z.Y. Regulation of Nrf2 by phosphorylation: Consequences for biological function and therapeutic implications. Free Radic. Biol. Med. 2021, 168, 129–141. [Google Scholar] [CrossRef]
- Shen, G.; Hebbar, V.; Nair, S.; Xu, C.; Li, W.; Lin, W.; Keum, Y.S.; Han, J.; Gallo, M.A.; Kong, A.N. Regulation of Nrf2 transactivation domain activity. The differential effects of mitogen-activated protein kinase cascades and synergistic stimulatory effect of Raf and CREB-binding protein. J. Biol. Chem. 2004, 279, 23052–23060. [Google Scholar] [CrossRef]
- Walters, T.S.; McIntosh, D.J.; Ingram, S.M.; Tillery, L.; Motley, E.D.; Arinze, I.J.; Misra, S. SUMO-Modification of Human Nrf2 at K(110) and K(533) Regulates Its Nucleocytoplasmic Localization, Stability and Transcriptional Activity. Cell Physiol. Biochem. 2021, 55, 141–159. [Google Scholar] [CrossRef]
- McIntosh, D.J.; Walters, T.S.; Arinze, I.J.; Davis, J. Arkadia (RING Finger Protein 111) Mediates Sumoylation-Dependent Stabilization of Nrf2 Through K48-Linked Ubiquitination. Cell Physiol. Biochem. 2018, 46, 418–430. [Google Scholar] [CrossRef]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef]
- Wu, J.; Jiang, Z.; Zhang, H.; Liang, W.; Huang, W.; Zhang, H.; Li, Y.; Wang, Z.; Wang, J.; Jia, Y.; et al. Sodium butyrate attenuates diabetes-induced aortic endothelial dysfunction via P300-mediated transcriptional activation of Nrf2. Free Radic. Biol. Med. 2018, 124, 454–465. [Google Scholar] [CrossRef]
- Yildirim, F.; Ji, S.; Kronenberg, G.; Barco, A.; Olivares, R.; Benito, E.; Dirnagl, U.; Gertz, K.; Endres, M.; Harms, C.; et al. Histone Acetylation and CREB Binding Protein Are Required for Neuronal Resistance against Ischemic Injury. PLoS ONE 2014, 9, e95465. [Google Scholar] [CrossRef]
- Balasubramanyam, K.; Varier, R.A.; Altaf, M.; Swaminathan, V.; Siddappa, N.B.; Ranga, U.; Kundu, T.K. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J. Biol. Chem. 2004, 279, 51163–51171. [Google Scholar] [CrossRef] [Green Version]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Seto, E. Lysine acetylation: Codified crosstalk with other posttranslational modifications. Mol. Cell 2008, 31, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Tavana, O.; Gu, W. ARF–NRF2: A new checkpoint for oxidative stress responses? Mol. Amp. Cell. Oncol. 2018, 5, e1432256. [Google Scholar] [CrossRef] [PubMed]
- Kawai, Y.; Garduno, L.; Theodore, M.; Yang, J.; Arinze, I.J. Acetylation-deacetylation of the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) regulates its transcriptional activity and nucleocytoplasmic localization. J. Biol. Chem. 2011, 286, 7629–7640. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Okada, M. Quercetin, caffeic acid and resveratrol regulate circadian clock genes and aging-related genes in young and old human lung fibroblast cells. Mol. Biol. Rep. 2020, 47, 1021–1032. [Google Scholar] [CrossRef]
- Ding, Y.W.; Zhao, G.J.; Li, X.L.; Hong, G.L.; Li, M.F.; Qiu, Q.M.; Wu, B.; Lu, Z.Q. SIRT1 exerts protective effects against paraquat-induced injury in mouse type II alveolar epithelial cells by deacetylating NRF2 in vitro. Int. J. Mol. Med. 2016, 37, 1049–1058. [Google Scholar] [CrossRef]
- Mercado, N.; Thimmulappa, R.; Thomas, C.M.; Fenwick, P.S.; Chana, K.K.; Donnelly, L.E.; Biswal, S.; Ito, K.; Barnes, P.J. Decreased histone deacetylase 2 impairs Nrf2 activation by oxidative stress. Biochem. Biophys. Res. Commun. 2011, 406, 292–298. [Google Scholar] [CrossRef]
- Liu, X.; Li, H.; Liu, L.; Lu, Y.; Gao, Y.; Geng, P.; Li, X.; Huang, B.; Zhang, Y.; Lu, J. Methylation of arginine by PRMT1 regulates Nrf2 transcriptional activity during the antioxidative response. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 2093–2103. [Google Scholar] [CrossRef]
- Katsuoka, F.; Yamamoto, M. Small Maf proteins (MafF, MafG, MafK): History, structure and function. Gene 2016, 586, 197–205. [Google Scholar] [CrossRef]
- Sengoku, T.; Shiina, M.; Suzuki, K.; Hamada, K.; Sato, K.; Uchiyama, A.; Kobayashi, S.; Oguni, A.; Itaya, H.; Kasahara, K.; et al. Structural basis of transcription regulation by CNC family transcription factor, Nrf2. Nucleic Acids Res. 2022, 50, 12543–12557. [Google Scholar] [CrossRef]
- Oyake, T.; Itoh, K.; Motohashi, H.; Hayashi, N.; Hoshino, H.; Nishizawa, M.; Yamamoto, M.; Igarashi, K. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol. Cell Biol. 1996, 16, 6083–6095. [Google Scholar] [CrossRef]
- Ahuja, M.; Kaidery, N.A.; Dutta, D.; Attucks, O.C.; Kazakov, E.H.; Gazaryan, I.; Matsumoto, M.; Igarashi, K.; Sharma, S.M.; Thomas, B. Harnessing the Therapeutic Potential of the Nrf2/Bach1 Signaling Pathway in Parkinson’s Disease. Antioxidants 2022, 11, 1780. [Google Scholar] [CrossRef]
- Ahuja, M.; Ammal Kaidery, N.; Attucks, O.C.; McDade, E.; Hushpulian, D.M.; Gaisin, A.; Gaisina, I.; Ahn, Y.H.; Nikulin, S.; Poloznikov, A.; et al. Bach1 derepression is neuroprotective in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2111643118. [Google Scholar] [CrossRef]
- Juknat, A.; Pietr, M.; Kozela, E.; Rimmerman, N.; Levy, R.; Coppola, G.; Geschwind, D.; Vogel, Z. Differential transcriptional profiles mediated by exposure to the cannabinoids cannabidiol and Delta9-tetrahydrocannabinol in BV-2 microglial cells. Br. J. Pharmacol. 2012, 165, 2512–2528. [Google Scholar] [CrossRef]
- Casares, L.; Garcia, V.; Garrido-Rodriguez, M.; Millan, E.; Collado, J.A.; Garcia-Martin, A.; Penarando, J.; Calzado, M.A.; de la Vega, L.; Munoz, E. Cannabidiol induces antioxidant pathways in keratinocytes by targeting BACH1. Redox Biol. 2019, 28, 101321. [Google Scholar] [CrossRef]
- Cai, Y.; Li, B.; Peng, D.; Wang, X.; Li, P.; Huang, M.; Xing, H.; Chen, J. Crm1-Dependent Nuclear Export of Bach1 is Involved in the Protective Effect of Hyperoside on Oxidative Damage in Hepatocytes and CCl(4)-induced Acute Liver Injury. J. Inflamm. Res. 2021, 14, 551–565. [Google Scholar] [CrossRef]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef]
- Woodgett, J.R. Recent advances in the protein kinase B signaling pathway. Curr. Opin. Cell Biol. 2005, 17, 150–157. [Google Scholar] [CrossRef]
- Fan, Y.; Wang, J.; He, N.; Feng, H. PLK2 protects retinal ganglion cells from oxidative stress by potentiating Nrf2 signaling via GSK-3beta. J. Biochem. Mol. Toxicol. 2021, 35, e22815. [Google Scholar] [CrossRef]
- Guo, C.; Zhu, J.; Wang, J.; Duan, J.; Ma, S.; Yin, Y.; Quan, W.; Zhang, W.; Guan, Y.; Ding, Y.; et al. Neuroprotective effects of protocatechuic aldehyde through PLK2/p-GSK3β/Nrf2 signaling pathway in both in vivo and in vitro models of Parkinson’s disease. Aging 2019, 11, 9424–9441. [Google Scholar] [CrossRef]
- Mathur, A.; Rizvi, F.; Kakkar, P. PHLPP2 down regulation influences nuclear Nrf2 stability via Akt-1/Gsk3beta/Fyn kinase axis in acetaminophen induced oxidative renal toxicity: Protection accorded by morin. Food Chem. Toxicol. 2016, 89, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, F.; Mathur, A.; Krishna, S.; Siddiqi, M.I.; Kakkar, P. Suppression in PHLPP2 induction by morin promotes Nrf2-regulated cellular defenses against oxidative injury to primary rat hepatocytes. Redox Biol. 2015, 6, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Jaiswal, A.K. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Oh, S.; Lee, D.; Oh, H.J.; Park, J.Y.; Lee, S.B.; Lim, D.S. Mst1-FoxO signaling protects Naïve T lymphocytes from cellular oxidative stress in mice. PLoS ONE 2009, 4, e8011. [Google Scholar] [CrossRef] [PubMed]
- Nehme, N.T.; Schmid, J.P.; Debeurme, F.; Andre-Schmutz, I.; Lim, A.; Nitschke, P.; Rieux-Laucat, F.; Lutz, P.; Picard, C.; Mahlaoui, N.; et al. MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival. Blood 2012, 119, 3458–3468. [Google Scholar] [CrossRef]
- Jensen, D.M.; Hendricks, K.V.; Mason, A.T.; Tessem, J.S. Good Cop, Bad Cop: The Opposing Effects of Macrophage Activation State on Maintaining or Damaging Functional β-Cell Mass. Metabolites 2020, 10, 485. [Google Scholar] [CrossRef]
- Hybertson, B.M.; Gao, B.; Bose, S.; McCord, J.M. Phytochemical Combination PB125 Activates the Nrf2 Pathway and Induces Cellular Protection against Oxidative Injury. Antioxidants 2019, 8, 119. [Google Scholar] [CrossRef]
- Velmurugan, K.; Alam, J.; McCord, J.M.; Pugazhenthi, S. Synergistic induction of heme oxygenase-1 by the components of the antioxidant supplement Protandim. Free Radic. Biol. Med. 2009, 46, 430–440. [Google Scholar] [CrossRef]
- Hybertson, B.M.; Gao, B.; McCord, J.M. Effects of the Phytochemical Combination PB123 on Nrf2 Activation, Gene Expression, and the Cholesterol Pathway in HepG2 Cells. OBM Integr. Complement. Med. 2021, 7, 1–23. [Google Scholar] [CrossRef]
- McCord, J.M.; Hybertson, B.M.; Cota-Gomez, A.; Geraci, K.P.; Gao, B. Nrf2 Activator PB125® as a Potential Therapeutic Agent against COVID-19. Antioxidants 2020, 9, 518. [Google Scholar] [CrossRef]
- Musci, R.V.; Andrie, K.M.; Walsh, M.A.; Valenti, Z.J.; Linden, M.A.; Afzali, M.F.; Bork, S.; Campbell, M.; Johnson, T.; Kail, T.E.; et al. Phytochemical compound PB125 attenuates skeletal muscle mitochondrial dysfunction and impaired proteostasis in a model of musculoskeletal decline. J. Physiol. 2022. [Google Scholar] [CrossRef]
- Chang, W.-H.; Thai, P.; Xu, J.; Yang, D.; Wu, R.; Chen, C.-H. Cigarette Smoke Regulates the Competitive Interactions between NRF2 and BACH1 for Heme Oxygenase-1 Induction. Int. J. Mol. Sci. 2017, 18, 2386. [Google Scholar] [CrossRef]
- Wang, T.; Ashrafi, A.; Modareszadeh, P.; Deese, A.R.; Chacon Castro, M.D.C.; Alemi, P.S.; Zhang, L. An Analysis of the Multifaceted Roles of Heme in the Pathogenesis of Cancer and Related Diseases. Cancers 2021, 13, 4142. [Google Scholar] [CrossRef]
- Igarashi, K.; Katoh, Y. Metabolic aspects of epigenome: Coupling of S-adenosylmethionine synthesis and gene regulation on chromatin by SAMIT module. Subcell. Biochem. 2013, 61, 105–118. [Google Scholar] [CrossRef]
- Shu, J.; Sun, X.; Li, J.; Li, F.; Tang, J.; Shi, L. Serum homocysteine levels and their association with clinical characteristics of inflammatory arthritis. Clin. Rheumatol. 2020, 39, 3295–3302. [Google Scholar] [CrossRef]
- Luzzi, S.; Papiri, G.; Viticchi, G.; Baldinelli, S.; Fiori, C.; Silvestrini, M.; Toraldo, A. Association between homocysteine levels and cognitive profile in Alzheimer’s Disease. J. Clin. Neurosci. 2021, 94, 250–256. [Google Scholar] [CrossRef]
- Mu, Z.-J.; Fu, J.-L.; Sun, L.-N.; Chan, P.; Xiu, S.-L. Associations between homocysteine, inflammatory cytokines and sarcopenia in Chinese older adults with type 2 diabetes. BMC Geriatr. 2021, 21, 692. [Google Scholar] [CrossRef]
- Wang, C.; Xu, G.; Wen, Q.; Peng, X.; Chen, H.; Zhang, J.; Xu, S.; Zhang, C.; Zhang, M.; Ma, J.; et al. CBS promoter hypermethylation increases the risk of hypertension and stroke. Clinics 2019, 74, e630. [Google Scholar] [CrossRef]
- Talwar, S.; Prasad, S.; Kaur, L.; Mishra, J.; Puri, M.; Sachdeva, M.P.; Saraswathy, K.N. MTR, MTRR and CBS Gene Polymorphisms in Recurrent Miscarriages: A Case Control Study from North India. J. Hum. Reprod. Sci. 2022, 15, 191–196. [Google Scholar] [CrossRef]
- Kumar, M.; Sandhir, R. Neuroprotective Effect of Hydrogen Sulfide in Hyperhomocysteinemia Is Mediated Through Antioxidant Action Involving Nrf2. Neuromol. Med. 2018, 20, 475–490. [Google Scholar] [CrossRef]
- Su, Y.; Wang, Y.; Liu, M.; Chen, H. Hydrogen sulfide attenuates renal I/Rinduced activation of the inflammatory response and apoptosis via regulating Nrf2mediated NLRP3 signaling pathway inhibition. Mol. Med. Rep. 2021, 24, 518. [Google Scholar] [CrossRef] [PubMed]
- Ramani, K.; Lu, S.C. Methionine adenosyltransferases in liver health and diseases. Liver Res. 2017, 1, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Frau, M.; Feo, F.; Pascale, R.M. Pleiotropic effects of methionine adenosyltransferases deregulation as determinants of liver cancer progression and prognosis. J. Hepatol. 2013, 59, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Zapletal, O.; Tylichová, Z.; Neča, J.; Kohoutek, J.; Machala, M.; Milcová, A.; Pokorná, M.; Topinka, J.; Moyer, M.P.; Hofmanová, J.; et al. Butyrate alters expression of cytochrome P450 1A1 and metabolism of benzo[a]pyrene via its histone deacetylase activity in colon epithelial cell models. Arch. Toxicol. 2017, 91, 2135–2150. [Google Scholar] [CrossRef] [PubMed]
- Mimura, J.; Fujii-Kuriyama, Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim Biophys. Acta 2003, 1619, 263–268. [Google Scholar] [CrossRef]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef]
- Lipton, S.A. Pathologically activated therapeutics for neuroprotection. Nat. Rev. Neurosci. 2007, 8, 803–808. [Google Scholar] [CrossRef]
- Li, J.; Baker, J.; Higham, A.; Shah, R.; Montero-Fernandez, A.; Murray, C.; Cooper, N.; Lucas, C.; Fox, C.; Singh, D.; et al. COPD lung studies of Nrf2 expression and the effects of Nrf2 activators. Inflammopharmacology 2022, 30, 1431–1443. [Google Scholar] [CrossRef]
- Satoh, T.; Lipton, S. Recent advances in understanding NRF2 as a druggable target: Development of pro-electrophilic and non-covalent NRF2 activators to overcome systemic side effects of electrophilic drugs like dimethyl fumarate. F1000Research 2017, 6, 2138. [Google Scholar] [CrossRef]
- Liu, P.; Tian, W.; Tao, S.; Tillotson, J.; Wijeratne, E.M.K.; Gunatilaka, A.A.L.; Zhang, D.D.; Chapman, E. Non-covalent NRF2 Activation Confers Greater Cellular Protection than Covalent Activation. Cell Chem. Biol. 2019, 26, 1427–1435. [Google Scholar] [CrossRef]
- Mandlik, D.S.; Mandlik, S.K.; S, A. Therapeutic implications of glycogen synthase kinase-3β in Alzheimer’s disease: A novel therapeutic target. Int. J. Neurosci. 2022, 1–17. [Google Scholar] [CrossRef]
- Elmadbouh, O.H.M.; Pandol, S.J.; Edderkaoui, M. Glycogen Synthase Kinase 3β: A True Foe in Pancreatic Cancer. Int. J. Mol. Sci. 2022, 23, 14133. [Google Scholar] [CrossRef]
- Soni, D.; Kumar, P. GSK-3β-mediated regulation of Nrf2/HO-1 signaling as a new therapeutic approach in the treatment of movement disorders. Pharm. Rep. 2022, 74, 557–569. [Google Scholar] [CrossRef]
- Wei, J.; Wang, J.; Zhang, J.; Yang, J.; Wang, G.; Wang, Y. Development of inhibitors targeting glycogen synthase kinase-3beta for human diseases: Strategies to improve selectivity. Eur. J. Med. Chem. 2022, 236, 114301. [Google Scholar] [CrossRef]
- Shimizu, Y.; Yonezawa, T.; Sakamoto, J.; Furuya, T.; Osawa, M.; Ikeda, K. Identification of novel inhibitors of Keap1/Nrf2 by a promising method combining protein-protein interaction-oriented library and machine learning. Sci. Rep. 2021, 11, 7420. [Google Scholar] [CrossRef]
- Garcia, A.D.; Bach, A. A Comparative Assessment Study of Known Small-Molecule Keap1-Nrf2 Protein-Protein Interaction Inhibitors: Chemical Synthesis, Binding Properties, and Cellular Activity. J. Med. Chem. 2019, 62, 8028–8052. [Google Scholar] [CrossRef]
- Boyenle, I.D.; Divine, U.C.; Adeyemi, R.; Ayinde, K.S.; Olaoba, O.T.; Apu, C.; Du, L.; Lu, Q.; Yin, X.; Adelusi, T.I. Direct Keap1-kelch inhibitors as potential drug candidates for oxidative stress-orchestrated diseases: A review on In silico perspective. Pharm. Res. 2021, 167, 105577. [Google Scholar] [CrossRef]
- Kim, S.; Indu Viswanath, A.N.; Park, J.H.; Lee, H.E.; Park, A.Y.; Choi, J.W.; Kim, H.J.; Londhe, A.M.; Jang, B.K.; Lee, J.; et al. Nrf2 activator via interference of Nrf2-Keap1 interaction has antioxidant and anti-inflammatory properties in Parkinson’s disease animal model. Neuropharmacology 2020, 167, 107989. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCord, J.M.; Gao, B.; Hybertson, B.M. The Complex Genetic and Epigenetic Regulation of the Nrf2 Pathways: A Review. Antioxidants 2023, 12, 366. https://doi.org/10.3390/antiox12020366
McCord JM, Gao B, Hybertson BM. The Complex Genetic and Epigenetic Regulation of the Nrf2 Pathways: A Review. Antioxidants. 2023; 12(2):366. https://doi.org/10.3390/antiox12020366
Chicago/Turabian StyleMcCord, Joe M., Bifeng Gao, and Brooks M. Hybertson. 2023. "The Complex Genetic and Epigenetic Regulation of the Nrf2 Pathways: A Review" Antioxidants 12, no. 2: 366. https://doi.org/10.3390/antiox12020366