
Azadirachtin Attenuates Carcinogen Benzo(a) Pyrene-Induced DNA Damage, Cell Cycle Arrest, Apoptosis, Inflammatory, Metabolic, and Oxidative Stress in HepG2 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Cell Culture and Experimental Protocol
2.2.2. Cytotoxicity and Apoptosis Measurement
2.2.3. DNA Damage and Cell Cycle Analysis
2.2.4. Measurement of Reactive Oxygen Species (ROS)
2.2.5. Measurement of Lipid Peroxidation (LPO), Nitric Oxide (NO) Levels, and SOD/Catalase Activities
2.2.6. Measurement of GSH Metabolism and Redox Homeostasis
2.2.7. Measurement of CYP 450 Activities
2.2.8. Measurement of Mitochondrial Membrane Potential (MMP)
2.2.9. Measurement of Bioenergetics and Activities of Mitochondrial Enzymes
2.2.10. SDS-PAGE and Western Blot Analysis
2.2.11. Statistical Analysis
3. Results
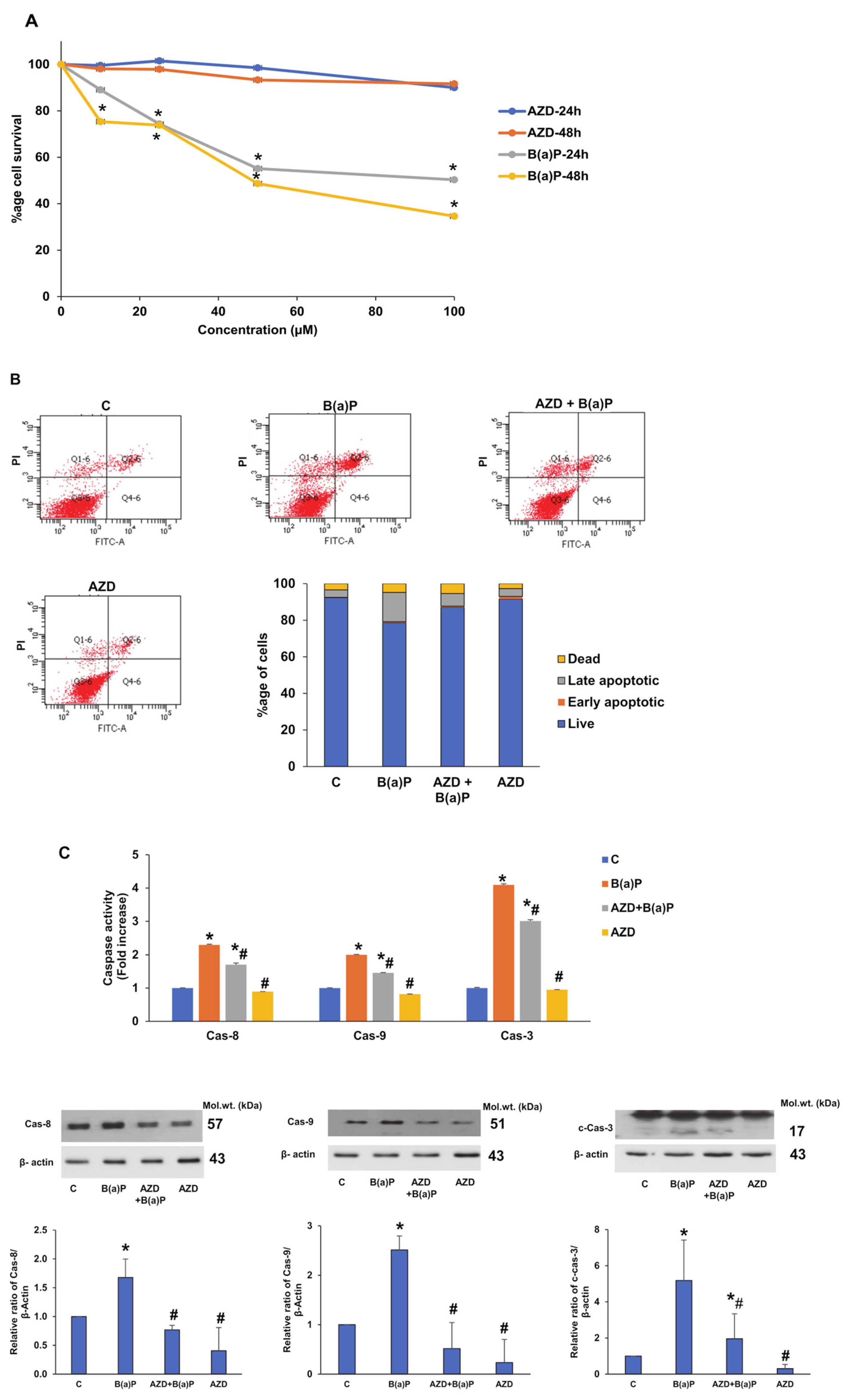
3.1. Differential Effects of LPS and AZD on Cell Viability, and Apoptosis
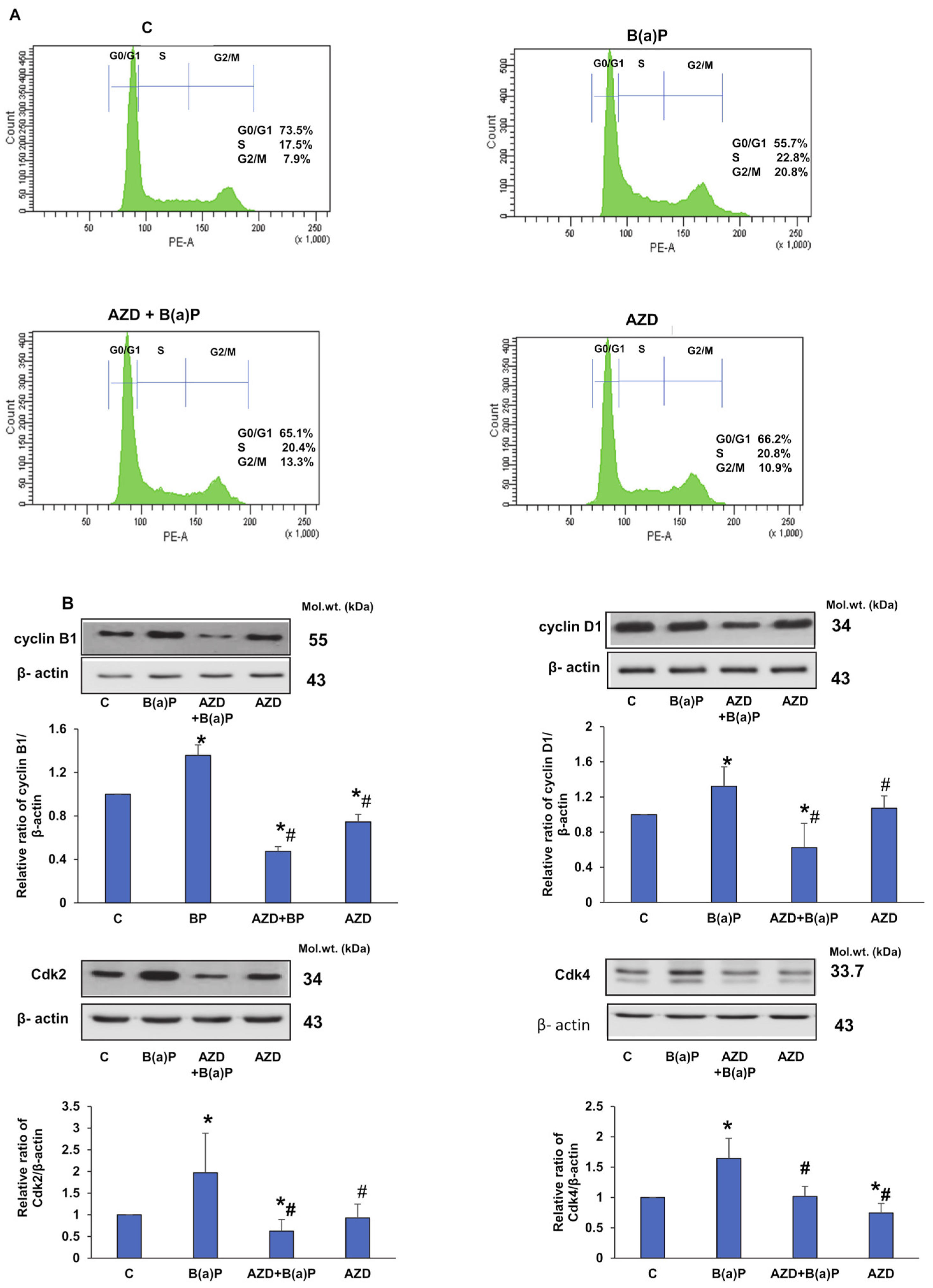
3.2. Attenuation of B(a)P-Induced DNA Damage and Cell Cycle Perturbation by AZD
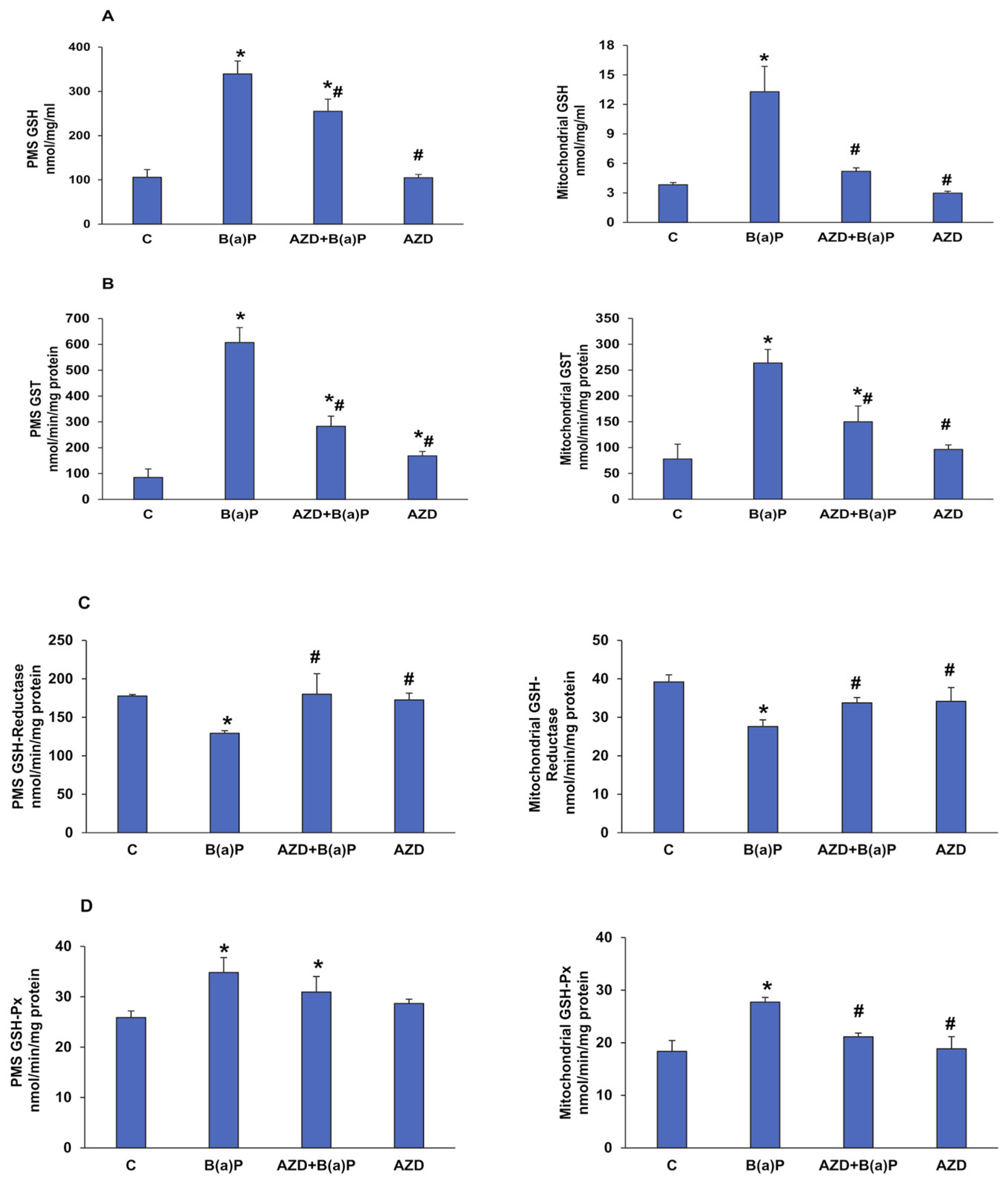
3.3. Effects of B(a)P and/or AZD on Oxidative Stress and Redox Homeostasis
3.4. Effects of B(a)P and/or AZD on Phase I Drug-Metabolizing Enzymes
3.5. Effects of B(a)P and/or AZD on Mitochondrial Functions and Bioenergetics
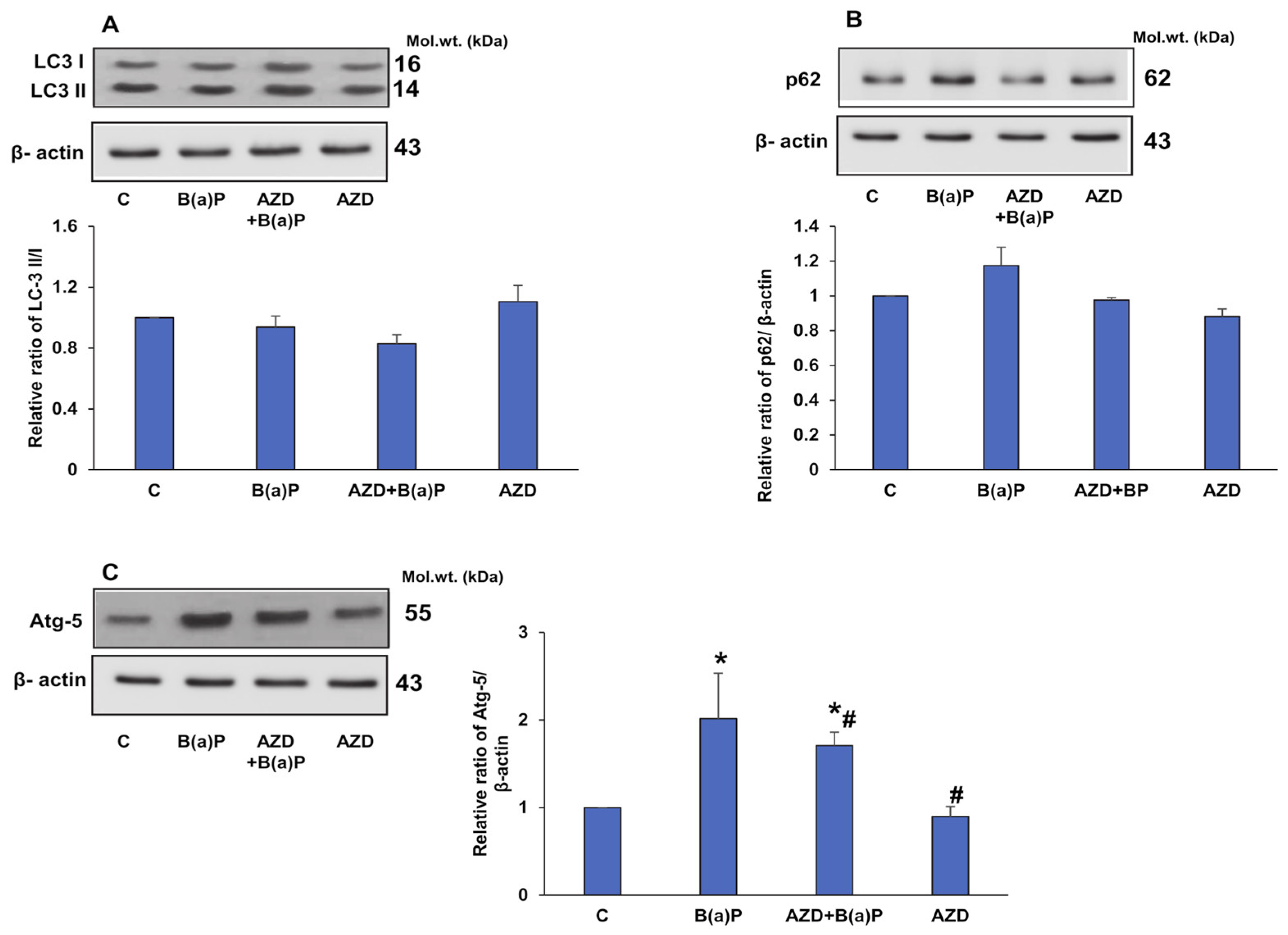
3.6. B(a)P-Induced Expression of Inflammatory, Anti-Oxidant, and Autophagy Markers
3.7. B(a)P-Induced Alterations in the Cell-Signaling Markers
4. Discussion
5. Conclusions
6. Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chhikara, B.S.; Parang, K. Global Cancer Statistics 2022: The Trends Projection Analysis. Chem. Biol. Lett. 2023, 10, 451. [Google Scholar]
- U.S. EPA. IRIS Toxicological Review of Benzo[A]Pyrene (Final Report); EPA/635/R-17/003F; U.S. Environmental Protection Agency: Washington, DC, USA, 2017. [Google Scholar]
- Liu, B.-Y.; Chiou, J.-Z.; Huang, K.-M.; Chen, T.-Y.; Hwang, D.-F. Effects of Taurine against Benzo[α]Pyrene-Induced Cell Cycle Arrest and Reactive Oxygen Species-Mediated Nuclear Factor-Kappa B Apoptosis via Reduction of Mitochondrial Stress in A549 Cells. Chin. J. Physiol. 2022, 65, 199. [Google Scholar] [CrossRef] [PubMed]
- Nemmar, A.; Raza, H.; Subramaniyan, D.; Yasin, J.; John, A.; Ali, B.H.; Kazzam, E.E. Short-Term Systemic Effects of Nose-Only Cigarette Smoke Exposure in Mice: Role of Oxidative Stress. Cell. Physiol. Biochem. 2013, 31, 15–24. [Google Scholar] [CrossRef]
- Raza, H.; John, A.; Nemmar, A. Short-Term Effects of Nose-Only Cigarette Smoke Exposure on Glutathione Redox Homeostasis, Cytochrome P450 1A1/2 and Respiratory Enzyme Activities in Mice Tissues. Cell. Physiol. Biochem. 2013, 31, 683–692. [Google Scholar] [CrossRef]
- Wang, C.; Zhao, F.; Bai, Y.; Li, C.; Xu, X.; Kristiansen, K.; Zhou, G. Proteomic Analysis of the Protective Effect of Eriodictyol on Benzo(a)Pyrene-Induced Caco-2 Cytotoxicity. Front. Nutr. 2022, 9, 839364. [Google Scholar] [CrossRef]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Some Non-Heterocyclic Polycyclic Aromatic Hydrocarbons and Some Related Exposures. IARC Monogr. Eval. Carcinog. Risks Hum. 2010, 92, 1–853. [Google Scholar]
- He, J.; Pang, Q.; Huang, C.; Xie, J.; Hu, J.; Wang, L.; Wang, C.; Meng, L.; Fan, R. Environmental Dose of 16 Priority-Controlled PAHs Mixture Induce Damages of Vascular Endothelial Cells Involved in Oxidative Stress and Inflammation. Toxicol. Vitr. 2022, 79, 105296. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jin, M.; Meng, Y.; Dai, Y.; Chen, S.; Zhou, Y.; Li, Y.; Tang, L. Involvement and Targeted Intervention of Benzo(a)Pyrene-Regulated Apoptosis Related Proteome Modification and Muti-Drug Resistance in Hepatocellular Carcinoma. Cell Death Dis. 2023, 14, 265. [Google Scholar] [CrossRef] [PubMed]
- Barangi, S.; Mehri, S.; Moosavi, Z.; Hayesd, A.W.; Reiter, R.J.; Cardinali, D.P.; Karimi, G. Melatonin Inhibits Benzo(a)Pyrene-Induced Apoptosis through Activation of the Mir-34a/Sirt1/Autophagy Pathway in Mouse Liver. Ecotoxicol. Environ. Saf. 2020, 196, 110556. [Google Scholar] [CrossRef]
- Ba, Q.; Li, J.; Huang, C.; Qiu, H.; Li, J.; Chu, R.; Zhang, W.; Xie, D.; Wu, Y.; Wang, H. Effects of Benzo[a]Pyrene Exposure on Human Hepatocellular Carcinoma Cell Angiogenesis, Metastasis, and NF-ΚB Signaling. Environ. Health Perspect. 2015, 123, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Bucher, S.; Tête, A.; Podechard, N.; Liamin, M.; Le Guillou, D.; Chevanne, M.; Coulouarn, C.; Imran, M.; Gallais, I.; Fernier, M.; et al. Co-Exposure to Benzo[a]Pyrene and Ethanol Induces a Pathological Progression of Liver Steatosis In Vitro and In Vivo. Sci. Rep. 2018, 8, 5963. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-C.; Jee, S.-C.; Kim, M.; Kim, S.; Shin, M.K.; Kim, Y.; Sung, J.-S. Curcumin Suppresses the Lipid Accumulation and Oxidative Stress Induced by Benzo[a]Pyrene Toxicity in HepG2 Cells. Antioxidants 2021, 10, 1314. [Google Scholar] [CrossRef] [PubMed]
- Bukowska, B.; Duchnowicz, P. Molecular Mechanisms of Action of Selected Substances Involved in the Reduction of Benzo[a]Pyrene-Induced Oxidative Stress. Molecules 2022, 27, 1379. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Cromie, M.M.; Cai, Q.; Lv, T.; Singh, K.; Gao, W. Curcumin and Vitamin E Protect against Adverse Effects of Benzo[a]Pyrene in Lung Epithelial Cells. PLoS ONE 2014, 9, e92992. [Google Scholar] [CrossRef]
- Kumar, M.; Sharma, V.L.; Sehgal, A.; Jain, M. Protective Effects of Green and White Tea Against Benzo(a)Pyrene Induced Oxidative Stress and DNA Damage in Murine Model. Nutr. Cancer 2012, 64, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.; Cerella, C.; Reuter, S.; Dicato, M.; Diederich, M. Anti-Inflammatory, pro-Apoptotic, and Anti-Proliferative Effects of a Methanolic Neem (Azadirachta Indica) Leaf Extract Are Mediated via Modulation of the Nuclear Factor-ΚB Pathway. Genes Nutr. 2011, 6, 149–160. [Google Scholar] [CrossRef]
- Fernandes, S.R.; Barreiros, L.; Oliveira, R.F.; Cruz, A.; Prudêncio, C.; Oliveira, A.I.; Pinho, C.; Santos, N.; Morgado, J. Chemistry, Bioactivities, Extraction and Analysis of Azadirachtin: State-of-the-Art. Fitoterapia 2019, 134, 141–150. [Google Scholar] [CrossRef]
- Dubey, R.; Patil, K.; Dantu, S.C.; Sardesai, D.M.; Bhatia, P.; Malik, N.; Acharya, J.D.; Sarkar, S.; Ghosh, S.; Chakrabarti, R.; et al. Azadirachtin Inhibits Amyloid Formation, Disaggregates Pre-Formed Fibrils and Protects Pancreatic β-Cells from Human Islet Amyloid Polypeptide/Amylin-Induced Cytotoxicity. Biochem. J. 2019, 476, 889–907. [Google Scholar] [CrossRef] [PubMed]
- John, A.; Raza, H. Azadirachtin Attenuates Lipopolysaccharide-Induced ROS Production, DNA Damage, and Apoptosis by Regulating JNK/Akt and AMPK/MTOR-Dependent Pathways in Rin-5F Pancreatic Beta Cells. Biomedicines 2021, 9, 1943. [Google Scholar] [CrossRef]
- John, A.; Raza, H. Alterations in Inflammatory Cytokines and Redox Homeostasis in LPS-Induced Pancreatic Beta-Cell Toxicity and Mitochondrial Stress: Protection by Azadirachtin. Front. Cell. Dev. Biol. 2022, 10, 867608. [Google Scholar] [CrossRef]
- Siddavaram, N.; Palrasu, M.; Bishayee, A. Limonoids from Neem (Azadirachta indica A. Juss.) Are Potential Anticancer Drug Candidates. Med. Res. Rev. 2023, in press. [Google Scholar] [CrossRef]
- Gangar, S.C.; Koul, A. Azadirachta Indica Leaf Extract Modulates Initiation Phase of Murine Forestomach Tumorigenesis. Indian J. Biochem. Biophys. 2007, 44, 209–215. [Google Scholar] [PubMed]
- Gangar, S.C.; Koul, A. Azadirachta Indica Modulates Carcinogen Biotransformation and Reduced Glutathione at Peri-Initiation Phase of Benzo(a)Pyrene Induced Murine Forestomach Tumorigenesis. Phytother. Res. 2008, 22, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- Gangar, S.C.; Koul, A. Histochemical, Ultrastructural, and Biochemical Evidences for Azadirachta Indica- Induced Apoptosis in Benzo (a) Pyrene- Induced Murine Forestomach Tumors. J. Environ. Pathol. Toxicol. Oncol. 2008, 27, 219–232. [Google Scholar] [CrossRef]
- Gangar, S.C.; Sandhir, R.; Rai, D.V.; Koul, A. Modulatory Effects of Azadirachta Indica on Benzo(a)Pyrene-Induced Forestomach Tumorigenesis in Mice. World J. Gastroenterol. 2006, 12, 2749–2755. [Google Scholar] [CrossRef] [PubMed]
- van Delft, J.H.M.; Mathijs, K.; Staal, Y.C.M.; van Herwijnen, M.H.M.; Brauers, K.J.J.; Boorsma, A.; Kleinjans, J.C.S. Time Series Analysis of Benzo[A]Pyrene-Induced Transcriptome Changes Suggests That a Network of Transcription Factors Regulates the Effects on Functional Gene Sets. Toxicol. Sci. 2010, 117, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Yang, M.S. Benzo[a]Pyrene-Induced Elevation of GSH Level Protects against Oxidative Stress and Enhances Xenobiotic Detoxification in Human HepG2 Cells. Toxicology 2007, 235, 1–10. [Google Scholar] [CrossRef]
- Alnahdi, A.; John, A.; Raza, H. Mitigation of Glucolipotoxicity-Induced Apoptosis, Mitochondrial Dysfunction, and Metabolic Stress by N-Acetyl Cysteine in Pancreatic β-Cells. Biomolecules 2020, 10, 239. [Google Scholar] [CrossRef]
- Raza, H.; John, A. Differential Cytotoxicity of Acetaminophen in Mouse Macrophage J774.2 and Human Hepatoma HepG2 Cells: Protection by Diallyl Sulfide. PLoS ONE 2015, 10, e0145965. [Google Scholar] [CrossRef] [PubMed]
- Alnahdi, A.; John, A.; Raza, H. N-Acetyl Cysteine Attenuates Oxidative Stress and Glutathione-Dependent Redox Imbalance Caused by High Glucose/High Palmitic Acid Treatment in Pancreatic Rin-5F Cells. PLoS ONE 2019, 14, e0226696. [Google Scholar] [CrossRef]
- John, A.; Howarth, F.C.; Raza, H. Exercise Alleviates Diabetic Complications by Inhibiting Oxidative Stress-Mediated Signaling Cascade and Mitochondrial Metabolic Stress in GK Diabetic Rat Tissues. Front. Physiol. 2022, 13, 1052608. [Google Scholar] [CrossRef]
- Birch-Machin, M.A.; Turnbull, D.M. Assaying Mitochondrial Respiratory Complex Activity in Mitochondria Isolated from Human Cells and Tissues. Methods Cell Biol. 2001, 65, 97–117. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Dang, F.; Gao, J.; Zhao, H.; Qi, S.; Gao, M. Acute Benzo[a]Pyrene Treatment Causes Different Antioxidant Response and DNA Damage in Liver, Lung, Brain, Stomach and Kidney. Heliyon 2018, 4, e00898. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Zhu, Y.; Mallah, M.A.; Lu, P.; Yang, L.; He, X.; Shang, P.; Chen, Y.; Zhou, X.; Feng, F.; et al. The Activation of SIRT1 Ameliorates BPDE-Induced Inflammatory Damage in BEAS-2B Cells via HMGB1/TLR4/NF-ΚB Pathway. Environ. Toxicol. 2023, 38, 2429–2439. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhang, M.; Huang, Z.; Sun, S.; Zhang, Y.; Zhang, L.; Peng, L.; Ma, A.; Ji, P.; Dai, J.; et al. SIRT1 in B[a]P-Induced Lung Tumorigenesis. Oncotarget 2015, 6, 27113–27129. [Google Scholar] [CrossRef]
- Yagishita, Y.; Chartoumpekis, D.V.; Kensler, T.W.; Wakabayashi, N. NRF2 and the Moirai: Life and Death Decisions on Cell Fates. Antioxid. Redox Signal. 2023, 38, 684–708. [Google Scholar] [CrossRef]
- Nguyen, P.M.; Park, M.S.; Chow, M.; Chang, J.H.; Wrischnik, L.; Chan, W.K. Benzo[a]Pyrene Increases the Nrf2 Content by Downregulating the Keap1 Message. Toxicol. Sci. 2010, 116, 549–561. [Google Scholar] [CrossRef]
- Washimkar, K.R.; Tomar, M.S.; Ishteyaque, S.; Kumar, A.; Shrivastava, A.; Mugale, M.N. Benzo[a]Pyrene Treatment Modulates Nrf2/Keap1 Axis and Changes the Metabolic Profile in Rat Lung Cancer. Chem. Biol. Interact. 2023, 373, 110373. [Google Scholar] [CrossRef]
- Lyu, Y.; Yang, J.; Cheng, L.; Li, Z.; Zheng, J. Benzo(a)Pyrene-Induced Mitochondrial Respiration and Glycolysis Disturbance in Human Neuroblastoma Cells. J. Toxicol. Sci. 2023, 48, 87–97. [Google Scholar] [CrossRef]
- Ji, K.; Xing, C.; Jiang, F.; Wang, X.; Guo, H.; Nan, J.; Qian, L.; Yang, P.; Lin, J.; Li, M.; et al. Benzo[a]Pyrene Induces Oxidative Stress and Endothelial Progenitor Cell Dysfunction via the Activation of the NF-ΚB Pathway. Int. J. Mol. Med. 2013, 31, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Nithya, G.; Santhanasabapathy, R.; Vanitha, M.K.; Anandakumar, P.; Sakthisekaran, D. Antioxidant, Antiproliferative, and Apoptotic Activity of Thymoquinone against Benzo(a)Pyrene-Induced Experimental Lung Cancer. J. Biochem. Mol. Toxicol. 2023, 37, e23230. [Google Scholar] [CrossRef]
- Banerjee, B.; Chakraborty, S.; Ghosh, D.; Raha, S.; Sen, P.C.; Jana, K. Benzo(a)Pyrene Induced P53 Mediated Male Germ Cell Apoptosis: Synergistic Protective Effects of Curcumin and Resveratrol. Front. Pharmacol. 2016, 7, 245. [Google Scholar] [CrossRef]
- Dutta, K.; Ghosh, D.; Nazmi, A.; Kumawat, K.L.; Basu, A. A Common Carcinogen Benzo[a]Pyrene Causes Neuronal Death in Mouse via Microglial Activation. PLoS ONE 2010, 5, e9984. [Google Scholar] [CrossRef] [PubMed]
- Holme, J.A.; Gorria, M.; Arlt, V.M.; Øvrebø, S.; Solhaug, A.; Tekpli, X.; Landvik, N.E.; Huc, L.; Fardel, O.; Lagadic-Gossmann, D. Different Mechanisms Involved in Apoptosis Following Exposure to Benzo[a]Pyrene in F258 and Hepa1c1c7 Cells. Chem. Biol. Interact. 2007, 167, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Chung, J.-Y.; Park, J.-E.; Lee, S.G.; Kim, Y.-J.; Cha, M.-S.; Han, M.S.; Lee, H.-J.; Yoo, Y.H.; Kim, J.-M. Benzo[a]Pyrene Induces Apoptosis in RL95-2 Human Endometrial Cancer Cells by Cytochrome P450 1A1 Activation. Endocrinology 2007, 148, 5112–5122. [Google Scholar] [CrossRef] [PubMed]
- Khattab, S.A.; Hussien, W.F.; Raafat, N.; Ahmed Alaa El-Din, E. Effects of Catechin Hydrate in Benzo[a]Pyrene-Induced Lung Toxicity: Roles of Oxidative Stress, Apoptosis, and DNA Damage. Toxicol. Mech. Methods 2021, 31, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Chen, S. The Role of the Ah Receptor and P38 in Benzo[a]Pyrene-7,8-Dihydrodiol and Benzo[a]Pyrene-7,8-Dihydrodiol-9,10-Epoxide-Induced Apoptosis. J. Biol. Chem. 2003, 278, 19526–19533. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.-S.; Zhang, H.-M.; Zhao, J.; Liu, H.-J.; Niu, Q. Involvement of Mitochondrial Pathway in Benzo[a]Pyrene-Induced Neuron Apoptosis. Hum. Exp. Toxicol. 2014, 33, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Hardonnière, K.; Saunier, E.; Lemarié, A.; Fernier, M.; Gallais, I.; Héliès-Toussaint, C.; Mograbi, B.; Antonio, S.; Bénit, P.; Rustin, P.; et al. The Environmental Carcinogen Benzo[a]Pyrene Induces a Warburg-like Metabolic Reprogramming Dependent on NHE1 and Associated with Cell Survival. Sci. Rep. 2016, 6, 30776. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; Guo, Z.; Zhu, Y.; Kong, M.; Zhang, R.; Lu, L.; Wu, F.; Liu, Z.; Wu, J. Houttuynia Cordata Thunb. and Its Bioactive Compound 2-Undecanone Significantly Suppress Benzo(a)Pyrene-Induced Lung Tumorigenesis by Activating the Nrf2-HO-1/NQO-1 Signaling Pathway. J. Exp. Clin. Cancer Res. 2019, 38, 242. [Google Scholar] [CrossRef]
- Jee, S.-C.; Kim, M.; Sung, J.-S. Modulatory Effects of Silymarin on Benzo[a]Pyrene-Induced Hepatotoxicity. Int. J. Mol. Sci. 2020, 21, 2369. [Google Scholar] [CrossRef]
- Tasdemir, E.; Maiuri, M.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Gao, C.; Deng, H.; Song, Q.; Yuan, L. Benzo(a)pyrene induces pyroptic and autophagic death through inhibiting PI3K/Akt signaling pathway in HL-7702 human normal liver cells. J. Toxicol. Sci. 2019, 44, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Genies, C.; Maître, A.; Lefèbvre, E.; Jullien, A.; Chopard-Lallier, M.; Douki, T. The Extreme Variety of Genotoxic Response to Benzo[a]Pyrene in Three Different Human Cell Lines from Three Different Organs. PLoS ONE 2013, 8, e78356. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Lee, S.-M.; Ye, S.-K.; Yoon, S.-H.; Chung, M.-H.; Choi, J. Benzo[a]Pyrene-Induced DNA Damage and P53 Modulation in Human Hepatoma HepG2 Cells for the Identification of Potential Biomarkers for PAH Monitoring and Risk Assessment. Toxicol. Lett. 2006, 167, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Singh, S.V. P53 Regulates Cellular Responses to Environmental Carcinogen Benzo[a]Pyrene-7,8-Diol-9,10-Epoxide in Human Lung Cancer Cells. Cell Cycle 2007, 6, 1753–1761. [Google Scholar] [CrossRef] [PubMed]
- Asweto, C.O.; Wu, J.; Hu, H.; Feng, L.; Yang, X.; Duan, J.; Sun, Z. Combined Effect of Silica Nanoparticles and Benzo[a]Pyrene on Cell Cycle Arrest Induction and Apoptosis in Human Umbilical Vein Endothelial Cells. Int. J. Environ. Res. Public Health 2017, 14, 289. [Google Scholar] [CrossRef]
- Hamouchene, H.; Arlt, V.M.; Giddings, I.; Phillips, D.H. Influence of Cell Cycle on Responses of MCF-7 Cells to Benzo[a]Pyrene. BMC Genom. 2011, 12, 333. [Google Scholar] [CrossRef]
- Jeffy, B.D.; Chen, E.J.; Gudas, J.M.; Romagnolo, D.F. Disruption of Cell Cycle Kinetics by Benzo[a]Pyrene: Inverse Expression Patterns of BRCA-1 and P53 in MCF-7 Cells Arrested in S and G2. Neoplasia 2000, 2, 460–470. [Google Scholar] [CrossRef]
- Gao, M.; Zheng, A.; Chen, L.; Dang, F.; Liu, X.; Gao, J. Benzo(a)Pyrene Affects Proliferation with Reference to Metabolic Genes and ROS/HIF-1α/HO-1 Signaling in A549 and MCF-7 Cancer Cells. Drug Chem. Toxicol. 2022, 45, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Khan, Q.A.; Dipple, A.; Anderson, L.M. Protease Inhibitor-Induced Stabilization of P21(Waf1/Cip1) and Cell-Cycle Arrest in Chemical Carcinogen-Exposed Mammary and Lung Cells. Mol. Carcinog. 2002, 33, 1–8. [Google Scholar] [CrossRef]
- Xu, H.; Yi, T.; Liu, M.; Gao, R.; Liu, X.; He, J.; Ding, Y.; Geng, Y.; Mu, X.; Wang, Y.; et al. Exposure to Benzo(a)Pyrene Promotes Proliferation and Inhibits Differentiation of Stromal Cells in Mice during Decidualization. Ecotoxicol. Environ. Saf. 2023, 251, 114531. [Google Scholar] [CrossRef] [PubMed]
- Du, H.J.; Tang, N.; Liu, B.C.; You, B.R.; Shen, F.H.; Ye, M.; Gao, A.; Huang, C.S. Benzo[a]Pyrene-Induced Cell Cycle Progression Is through ERKs/Cyclin D1 Pathway and Requires the Activation of JNKs and P38 Mapk in Human Diploid Lung Fibroblasts. Mol. Cell Biochem. 2006, 287, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Ning, B.; Gong, W.; Wen, W.; Wu, K.; Liang, J.; He, G.; Huang, S.; Sun, W.; Han, T.; et al. Cyclin D1 Induction by Benzo[a]Pyrene-7,8-Diol-9,10-Epoxide via the Phosphatidylinositol 3-Kinase/Akt/MAPK- and P70s6k-Dependent Pathway Promotes Cell Transformation and Tumorigenesis. J. Biol. Chem. 2009, 284, 33311–33319. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Ouyang, Z. Targeting CDK4/6 for Anticancer Therapy. Biomedicines 2022, 10, 685. [Google Scholar] [CrossRef]
- Du, Q.; Guo, X.; Wang, M.; Li, Y.; Sun, X.; Li, Q. The Application and Prospect of CDK4/6 Inhibitors in Malignant Solid Tumors. J. Hematol. Oncol. 2020, 13, 41. [Google Scholar] [CrossRef] [PubMed]















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
John, A.; Raza, H. Azadirachtin Attenuates Carcinogen Benzo(a) Pyrene-Induced DNA Damage, Cell Cycle Arrest, Apoptosis, Inflammatory, Metabolic, and Oxidative Stress in HepG2 Cells. Antioxidants 2023, 12, 2001. https://doi.org/10.3390/antiox12112001
John A, Raza H. Azadirachtin Attenuates Carcinogen Benzo(a) Pyrene-Induced DNA Damage, Cell Cycle Arrest, Apoptosis, Inflammatory, Metabolic, and Oxidative Stress in HepG2 Cells. Antioxidants. 2023; 12(11):2001. https://doi.org/10.3390/antiox12112001
Chicago/Turabian StyleJohn, Annie, and Haider Raza. 2023. "Azadirachtin Attenuates Carcinogen Benzo(a) Pyrene-Induced DNA Damage, Cell Cycle Arrest, Apoptosis, Inflammatory, Metabolic, and Oxidative Stress in HepG2 Cells" Antioxidants 12, no. 11: 2001. https://doi.org/10.3390/antiox12112001