
Non-Excitatory Amino Acids, Melatonin, and Free Radicals: Examining the Role in Stroke and Aging
 , , , ,
, , , ,  and
and 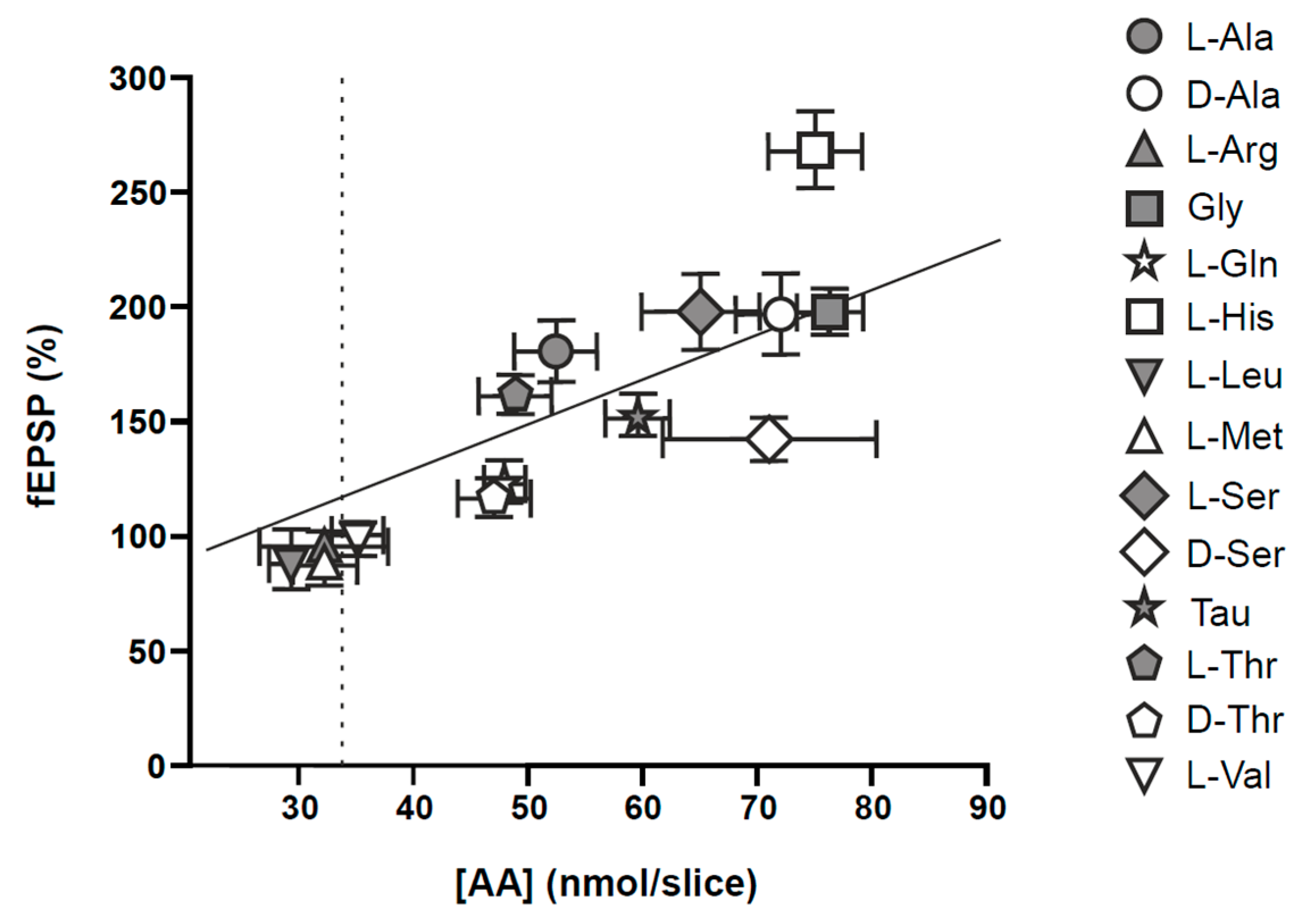{kind=link}
{kind=link}
{kind=link}
{kind=link}
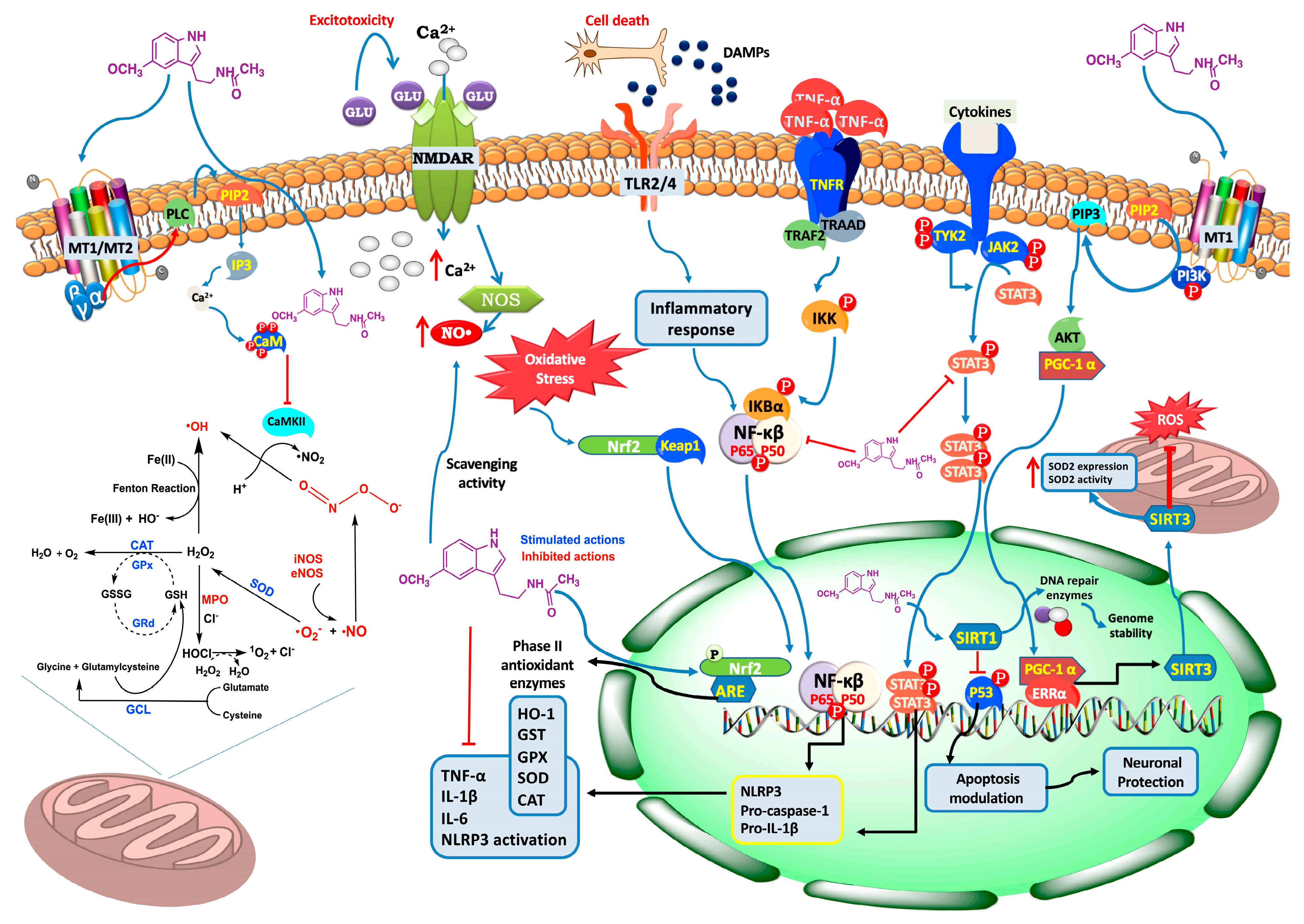{kind=link}
Abstract
:1. Introduction
Mechanisms Underlying Local Blood Flow
2. Stroke and Aging
3. Non-Excitatory Amino Acids as an Alternative Therapeutic Strategy to Reduce Ischemic Damage
4. Neuroprotective Effects of Melatonin in Ischemic Stroke
4.1. Melatonin’s Effects on Cerebral Edema
4.2. Melatonin’s Effects on the Post-Stroke Inflammatory Response
4.3. Melatonin’s Effects on Oxidative Stress
4.4. Exploring the Neuroprotective Potential of Melatonin in Stroke: Insights into Signaling Pathways
5. Melatonin as an Antioxidant and Free Radical Scavenger in Stroke: Mechanisms and Therapeutic Implications
6. Melatonin Improved Cognitive and Behavioral Performance
7. Suppressing Inflammatory Response by Melatonin: Effects on the Inflammasome
8. Melatonin, iNOS, and Calcium/Calmodulin
9. Melatonin in Microglial and Astrocyte Cell Activation
9.1. Melatonin in Astrocyte Activation
9.2. Melatonin in Microglia Activation
10. Post-Stroke Melatonin Treatment
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arendt, J.; Ravault, J.P. Suppression of melatonin secretion in Ile-de-France rams by different light intensities. J. Pineal Res. 1988, 5, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Comai, S.; Gobbi, G. Unveiling the role of melatonin MT2 receptors in sleep, anxiety and other neuropsychiatric diseases: A novel target in psychopharmacology. J. Psychiatry Neurosci. JPN 2014, 39, 6–21. [Google Scholar] [CrossRef]
- Carrascal, L.; Nunez-Abades, P.; Ayala, A.; Cano, M. Role of Melatonin in the Inflammatory Process and its Therapeutic Potential. Curr. Pharm. Des. 2018, 24, 1563–1588. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; You, W.; Shan, T. The regulatory role of melatonin in skeletal muscle. J. Muscle Res. Cell Motil. 2020, 41, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Chitimus, D.M.; Popescu, M.R.; Voiculescu, S.E.; Panaitescu, A.M.; Pavel, B.; Zagrean, L.; Zagrean, A.M. Melatonin’s Impact on Antioxidative and Anti-Inflammatory Reprogramming in Homeostasis and Disease. Biomolecules 2020, 10, 1211. [Google Scholar] [CrossRef]
- Gunata, M.; Parlakpinar, H.; Acet, H.A. Melatonin: A review of its potential functions and effects on neurological diseases. Rev. Neurol. 2020, 176, 148–165. [Google Scholar] [CrossRef]
- Lalanne, S.; Fougerou-Leurent, C.; Anderson, G.M.; Schroder, C.M.; Nir, T.; Chokron, S.; Delorme, R.; Claustrat, B.; Bellissant, E.; Kermarrec, S.; et al. Melatonin: From Pharmacokinetics to Clinical Use in Autism Spectrum Disorder. Int. J. Mol. Sci. 2021, 22, 1490. [Google Scholar] [CrossRef]
- Malakoti, F.; Zare, F.; Zarezadeh, R.; Raei Sadigh, A.; Sadeghpour, A.; Majidinia, M.; Yousefi, B.; Alemi, F. The role of melatonin in bone regeneration: A review of involved signaling pathways. Biochimie 2022, 202, 56–70. [Google Scholar] [CrossRef]
- Patel, R.; Parmar, N.; Pramanik Palit, S.; Rathwa, N.; Ramachandran, A.V.; Begum, R. Diabetes mellitus and melatonin: Where are we? Biochimie 2022, 202, 2–14. [Google Scholar] [CrossRef]
- Pivonello, C.; Negri, M.; Patalano, R.; Amatrudo, F.; Montò, T.; Liccardi, A.; Graziadio, C.; Muscogiuri, G.; Pivonello, R.; Colao, A. The role of melatonin in the molecular mechanisms underlying metaflammation and infections in obesity: A narrative review. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2022, 23, e13390. [Google Scholar] [CrossRef]
- Roy, J.; Wong, K.Y.; Aquili, L.; Uddin, M.S.; Heng, B.C.; Tipoe, G.L.; Wong, K.H.; Fung, M.L.; Lim, L.W. Role of melatonin in Alzheimer’s disease: From preclinical studies to novel melatonin-based therapies. Front. Neuroendocrinol. 2022, 65, 100986. [Google Scholar] [CrossRef]
- Skarlis, C.; Anagnostouli, M. The role of melatonin in Multiple Sclerosis. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2020, 41, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Talib, W.H. Melatonin and Cancer Hallmarks. Molecules 2018, 23, 518. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.H.; Jiang, Z.L.; Fan, X.J.; Zhang, L.; Li, X.; Ke, K.F. Neuroprotective effect of taurine against focal cerebral ischemia in rats possibly mediated by activation of both GABAA and glycine receptors. Neuropharmacology 2007, 52, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ni, L.; Di, X.; Ma, B.; Niu, S.; Rong, Z.; Liu, C. Potential Role of Melatonin as an Adjuvant for Atherosclerotic Carotid Arterial Stenosis. Molecules 2021, 26, 811. [Google Scholar] [CrossRef]
- Chen, J.; Zhuang, Y.; Zhang, Y.; Liao, H.; Liu, R.; Cheng, J.; Zhang, Z.; Sun, J.; Gao, J.; Wang, X.; et al. A synthetic BBB-permeable tripeptide GCF confers neuroprotection by increasing glycine in the ischemic brain. Front. Pharmacol. 2022, 13, 950376. [Google Scholar] [CrossRef]
- Piniella, D.; Zafra, F. Functional crosstalk of the glycine transporter GlyT1 and NMDA receptors. Neuropharmacology 2023, 232, 109514. [Google Scholar] [CrossRef]
- Surai, P.F.; Earle-Payne, K.; Kidd, M.T. Taurine as a Natural Antioxidant: From Direct Antioxidant Effects to Protective Action in Various Toxicological Models. Antioxidants 2021, 10, 1876. [Google Scholar] [CrossRef]
- Jakaria, M.; Azam, S.; Haque, M.E.; Jo, S.H.; Uddin, M.S.; Kim, I.S.; Choi, D.K. Taurine and its analogs in neurological disorders: Focus on therapeutic potential and molecular mechanisms. Redox Biol. 2019, 24, 101223. [Google Scholar] [CrossRef]
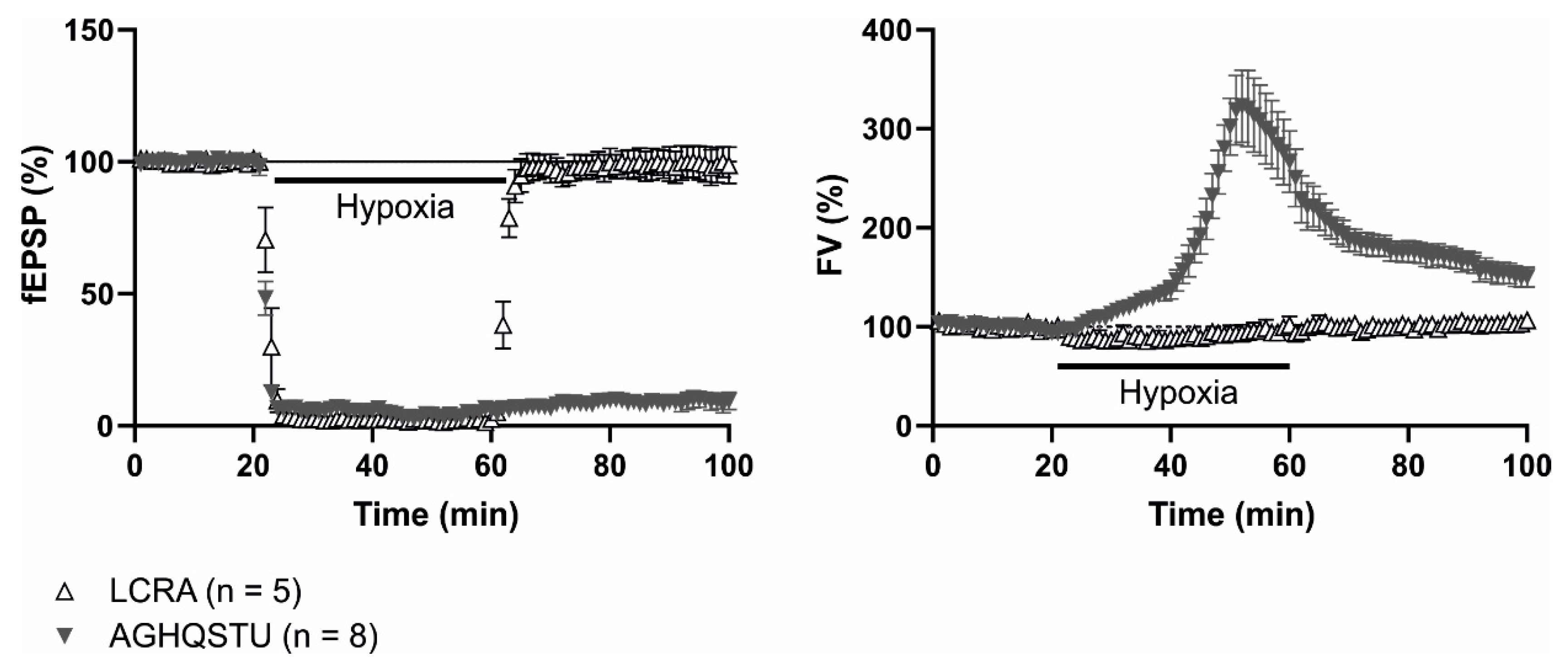
- Álvarez-Merz, I.; Luengo, J.G.; Muñoz, M.D.; Hernández-Guijo, J.M.; Solís, J.M. Hypoxia-induced depression of synaptic transmission becomes irreversible by intracellular accumulation of non-excitatory amino acids. Neuropharmacology 2021, 190, 108557. [Google Scholar] [CrossRef]
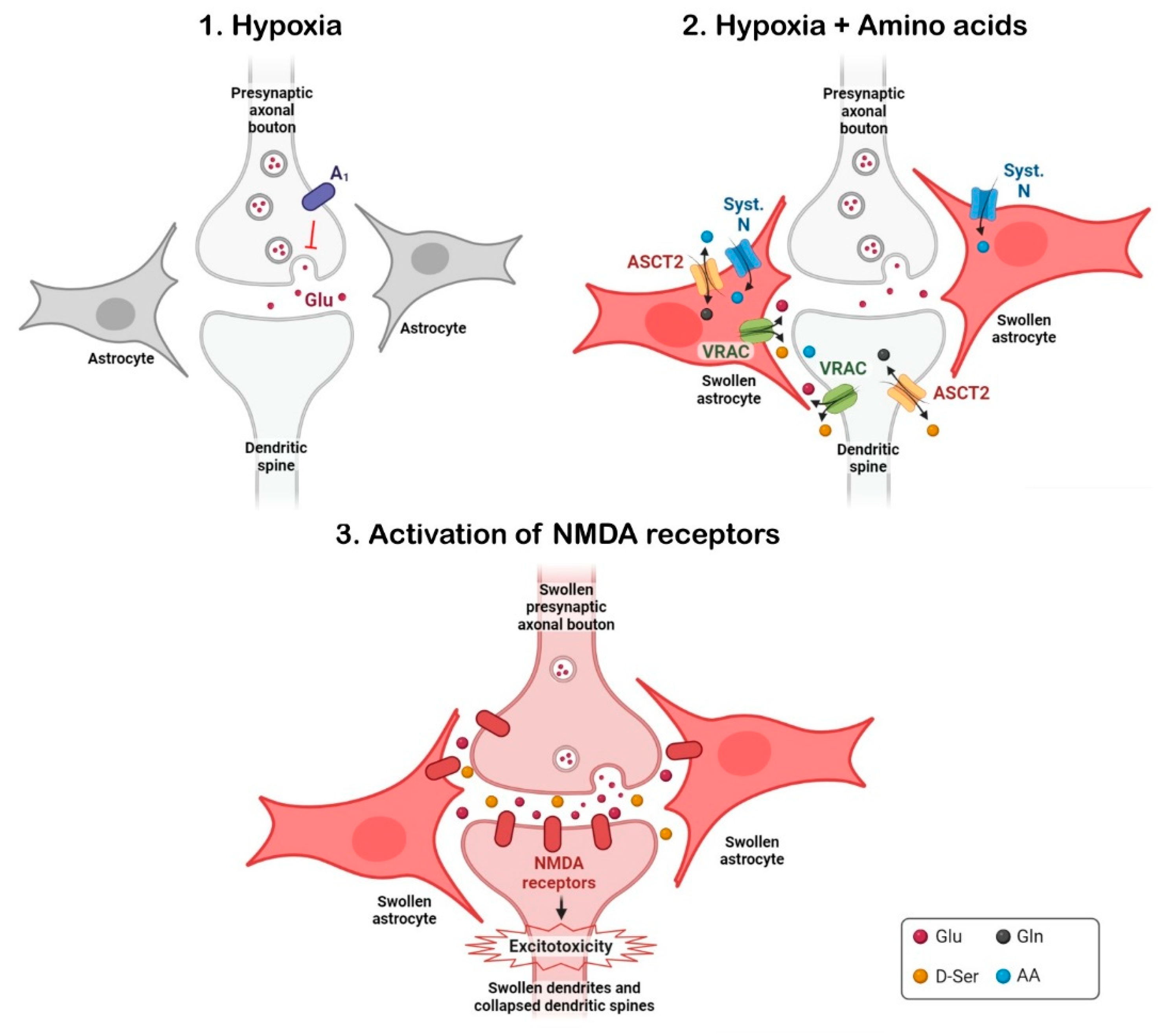
- Álvarez-Merz, I.; Fomitcheva, I.V.; Sword, J.; Hernández-Guijo, J.M.; Solís, J.M.; Kirov, S.A. Novel mechanism of hypoxic neuronal injury mediated by non-excitatory amino acids and astroglial swelling. Glia 2022, 70, 2108–2130. [Google Scholar] [CrossRef]
- Kemp, J.A.; McKernan, R.M. NMDA receptor pathways as drug targets. Nat. Neurosci. 2002, 5 (Suppl. S11), 1039–1042. [Google Scholar] [CrossRef] [PubMed]
- Ramos, E.; Patiño, P.; Reiter, R.J.; Gil-Martín, E.; Marco-Contelles, J.; Parada, E.; de Los Rios, C.; Romero, A.; Egea, J. Ischemic brain injury: New insights on the protective role of melatonin. Free Radic. Biol. Med. 2017, 104, 32–53. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, F.; Dou, Y.; Tian, X.; Liu, C.; Li, H.; Shen, H.; Chen, G. Melatonin Alleviates Intracerebral Hemorrhage-Induced Secondary Brain Injury in Rats via Suppressing Apoptosis, Inflammation, Oxidative Stress, DNA Damage, and Mitochondria Injury. Transl. Stroke Res. 2018, 9, 74–91. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, B.; Huang, J.; Xu, L.; Pan, J.; Fang, C.; Li, M.; Li, G.; Tao, Y.; Yang, X.; et al. Up-regulation of miR-325-3p suppresses pineal aralkylamine N-acetyltransferase (Aanat) after neonatal hypoxia-ischemia brain injury in rats. Brain Res. 2017, 1668, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Padial, L.; Arias, M.Á.; Martín Rodríguez, F. Real-world data on direct-acting oral anticoagulants. Med. Clin. 2018, 150, 161. [Google Scholar] [CrossRef]
- Roldán Rabadán, I.; Alonso de Leciñana, M.; Barba Martín, R.; Páramo Fernández, J.A.; por el Foro Multidisciplinar en Trombosis Security profile of direct anticoagulants. Preferred use in atrial fibrillation. Clin. E Investig. En Arterioscler. Publ. Of. Soc. Esp. Arterioscler. 2019, 31, 263–270. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Del Zar, M.M.; Vacas, M.I. The effects of melatonin in human platelets. Acta Physiol. Pharmacol. Ther. Latinoam. Organo Asoc. Latinoam. Cienc. Fisiol. Asoc. Latinoam. Farmacol. 1993, 43, 1–13. [Google Scholar]
- Otamas, A.; Grant, P.J.; Ajjan, R.A. Diabetes and atherothrombosis: The circadian rhythm and role of melatonin in vascular protection. Diab. Vasc. Dis. Res. 2020, 17, 1479164120920582. [Google Scholar] [CrossRef]
- Hosseinzadeh, A.; Bagherifard, A.; Koosha, F.; Amiri, S.; Karimi-Behnagh, A.; Reiter, R.J.; Mehrzadi, S. Melatonin effect on platelets and coagulation: Implications for a prophylactic indication in COVID-19. Life Sci. 2022, 307, 120866. [Google Scholar] [CrossRef]
- Amirzargar, M.R.; Shahriyary, F.; Shahidi, M.; Kooshari, A.; Vafajoo, M.; Nekouian, R.; Faranoush, M. Angiogenesis, coagulation, and fibrinolytic markers in acute promyelocytic leukemia (NB4): An evaluation of melatonin effects. J. Pineal Res. 2023, 75, e12901. [Google Scholar] [CrossRef] [PubMed]
- Karasek, M. Melatonin, human aging, and age-related diseases. Exp. Gerontol. 2004, 39, 1723–1729. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, M.; Piekut, T.; Prendecki, M.; Sodel, A.; Kozubski, W.; Dorszewska, J. Mitochondrial and Nuclear DNA Oxidative Damage in Physiological and Pathological Aging. DNA Cell Biol. 2020, 39, 1410–1420. [Google Scholar] [CrossRef]
- Leyane, T.S.; Jere, S.W.; Houreld, N.N. Oxidative Stress in Ageing and Chronic Degenerative Pathologies: Molecular Mechanisms Involved in Counteracting Oxidative Stress and Chronic Inflammation. Int. J. Mol. Sci. 2022, 23, 7273. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Sastre, J.; Pallardó, F.V.; García de la Asunción, J.; Viña, J. Mitochondria, oxidative stress and aging. Free Radic. Res. 2000, 32, 189–198. [Google Scholar] [CrossRef]
- Warraich, U.E.A.; Hussain, F.; Kayani, H.U.R. Aging—Oxidative stress, antioxidants and computational modeling. Heliyon 2020, 6, e04107. [Google Scholar] [CrossRef]
- Krnjević, K. Electrophysiology of cerebral ischemia. Neuropharmacology 2008, 55, 319–333. [Google Scholar] [CrossRef]
- Jelinek, M.; Jurajda, M.; Duris, K. Oxidative Stress in the Brain: Basic Concepts and Treatment Strategies in Stroke. Antioxidants 2021, 10, 1886. [Google Scholar] [CrossRef]
- Jurcau, A.; Ardelean, A.I. Oxidative Stress in Ischemia/Reperfusion Injuries following Acute Ischemic Stroke. Biomedicines 2022, 10, 574. [Google Scholar] [CrossRef] [PubMed]
- Orellana-Urzúa, S.; Rojas, I.; Líbano, L.; Rodrigo, R. Pathophysiology of Ischemic Stroke: Role of Oxidative Stress. Curr. Pharm. Des. 2020, 26, 4246–4260. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.S.; Jin, H.; Sun, X.; Huang, S.; Zhang, F.L.; Guo, Z.N.; Yang, Y. Free Radical Damage in Ischemia-Reperfusion Injury: An Obstacle in Acute Ischemic Stroke after Revascularization Therapy. Oxid. Med. Cell. Longev. 2018, 2018, 3804979. [Google Scholar] [CrossRef]
- Yang, J.L.; Mukda, S.; Chen, S.D. Diverse roles of mitochondria in ischemic stroke. Redox Biol. 2018, 16, 263–275. [Google Scholar] [CrossRef]
- Rodriguez, C.; Mayo, J.C.; Sainz, R.M.; Antolín, I.; Herrera, F.; Martín, V.; Reiter, R.J. Regulation of antioxidant enzymes: A significant role for melatonin. J. Pineal Res. 2004, 36, 1–9. [Google Scholar] [CrossRef]
- Bikjdaouene, L.; Escames, G.; León, J.; Ferrer, J.M.R.; Khaldy, H.; Vives, F.; Acuña-Castroviejo, D. Changes in brain amino acids and nitric oxide after melatonin administration in rats with pentylenetetrazole-induced seizures. J. Pineal Res. 2003, 35, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Touyz, R.M. Oxidative Stress, Inflammation, and Vascular Aging in Hypertension. Hypertension 2017, 70, 660–667. [Google Scholar] [CrossRef]
- Liu, Y.; Bloom, S.I.; Donato, A.J. The role of senescence, telomere dysfunction and shelterin in vascular aging. Microcirculation 2019, 26, e12487. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Donato, A.J.; Galvan, V.; Csiszar, A. Mechanisms of Vascular Aging. Circ. Res. 2018, 123, 849–867. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Sorond, F.; Merkely, B.; Csiszar, A. Mechanisms of Vascular Aging, A Geroscience Perspective: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 931–941. [Google Scholar] [CrossRef]
- Du, S.; Ling, H.; Guo, Z.; Cao, Q.; Song, C. Roles of exosomal miRNA in vascular aging. Pharmacol. Res. 2021, 165, 105278. [Google Scholar] [CrossRef] [PubMed]
- Barbu, E.; Popescu, M.R.; Popescu, A.C.; Balanescu, S.M. Inflammation as A Precursor of Atherothrombosis, Diabetes and Early Vascular Aging. Int. J. Mol. Sci. 2022, 23, 963. [Google Scholar] [CrossRef] [PubMed]
- Tarantini, S.; Tran, C.H.T.; Gordon, G.R.; Ungvari, Z.; Csiszar, A. Impaired neurovascular coupling in aging and Alzheimer’s disease: Contribution of astrocyte dysfunction and endothelial impairment to cognitive decline. Exp. Gerontol. 2017, 94, 52–58. [Google Scholar] [CrossRef]
- Jakovljevic, D.G. Physical activity and cardiovascular aging: Physiological and molecular insights. Exp. Gerontol. 2018, 109, 67–74. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Iadecola, C. Alzheimer’s Disease and Vascular Aging: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 942–951. [Google Scholar] [CrossRef]
- Harraz, O.F.; Jensen, L.J. Aging, calcium channel signaling and vascular tone. Mech. Ageing Dev. 2020, 191, 111336. [Google Scholar] [CrossRef]
- Durante, W. Carbon monoxide and bile pigments: Surprising mediators of vascular function. Vasc. Med. 2002, 7, 195–202. [Google Scholar] [CrossRef]
- Perry, S.; Kumai, Y.; Porteus, C.S.; Tzaneva, V.; Kwong, R.W.M. An emerging role for gasotransmitters in the control of breathing and ionic regulation in fish. J. Comp. Physiol. B 2016, 186, 145–159. [Google Scholar] [CrossRef]
- Wareham, L.K.; Southam, H.M.; Poole, R.K. Do nitric oxide, carbon monoxide and hydrogen sulfide really qualify as “gasotransmitters” in bacteria? Biochem. Soc. Trans. 2018, 46, 1107–1118. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N. The NOS/NO System in Renal Programming and Reprogramming. Antioxidants 2023, 12, 1629. [Google Scholar] [CrossRef]
- Wang, Y.; Hong, F.; Yang, S. Roles of Nitric Oxide in Brain Ischemia and Reperfusion. Int. J. Mol. Sci. 2022, 23, 4243. [Google Scholar] [CrossRef]
- Barbagallo, I.; Marrazzo, G.; Frigiola, A.; Zappala, A.; Li Volti, G. Role of carbon monoxide in vascular diseases. Curr. Pharm. Biotechnol. 2012, 13, 787–796. [Google Scholar] [CrossRef]
- Durante, W.; Schafer, A.I. Carbon monoxide and vascular cell function (review). Int. J. Mol. Med. 1998, 2, 255–262. [Google Scholar] [CrossRef]
- Wang, R. Resurgence of carbon monoxide: An endogenous gaseous vasorelaxing factor. Can. J. Physiol. Pharmacol. 1998, 76, 1. [Google Scholar] [CrossRef]
- Leffler, C.W.; Parfenova, H.; Jaggar, J.H. Carbon monoxide as an endogenous vascular modulator. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1–H11. [Google Scholar] [CrossRef]
- Marazioti, A.; Bucci, M.; Coletta, C.; Vellecco, V.; Baskaran, P.; Szabó, C.; Cirino, G.; Marques, A.R.; Guerreiro, B.; Gonçalves, A.M.L.; et al. Inhibition of nitric oxide-stimulated vasorelaxation by carbon monoxide-releasing molecules. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2570–2576. [Google Scholar] [CrossRef]
- Failli, P.; Vannacci, A.; Di Cesare Mannelli, L.; Motterlini, R.; Masini, E. Relaxant effect of a water soluble carbon monoxide-releasing molecule (CORM-3) on spontaneously hypertensive rat aortas. Cardiovasc. Drugs Ther. 2012, 26, 285–292. [Google Scholar] [CrossRef]
- Li, B.; Xiong, J.; Liu, H.X.; Li, D.; Chen, G. Devil or angel: Two roles of carbon monoxide in stroke. Med. Gas Res. 2022, 12, 125–130. [Google Scholar] [CrossRef]
- Cirino, G.; Szabo, C.; Papapetropoulos, A. Physiological roles of hydrogen sulfide in mammalian cells, tissues, and organs. Physiol. Rev. 2023, 103, 31–276. [Google Scholar] [CrossRef]
- Hsu, C.N.; Tain, Y.L. Gasotransmitters for the Therapeutic Prevention of Hypertension and Kidney Disease. Int. J. Mol. Sci. 2021, 22, 7808. [Google Scholar] [CrossRef]
- Iova, O.M.; Marin, G.E.; Lazar, I.; Stanescu, I.; Dogaru, G.; Nicula, C.A.; Bulboacă, A.E. Nitric Oxide/Nitric Oxide Synthase System in the Pathogenesis of Neurodegenerative Disorders-An Overview. Antioxidants 2023, 12, 753. [Google Scholar] [CrossRef]
- Rengarajan, A.; Mauro, A.K.; Boeldt, D.S. Maternal disease and gasotransmitters. Nitric Oxide Biol. Chem. 2020, 96, 1–12. [Google Scholar] [CrossRef]
- Ali, A.; Wang, Y.; Wu, L.; Yang, G. Gasotransmitter signaling in energy homeostasis and metabolic disorders. Free Radic. Res. 2021, 55, 83–105. [Google Scholar] [CrossRef]
- Juin, S.K.; Ouseph, R.; Gondim, D.D.; Jala, V.R.; Sen, U. Diabetic Nephropathy and Gaseous Modulators. Antioxidants 2023, 12, 1088. [Google Scholar] [CrossRef]
- Murad, F.; Waldman, S.; Molina, C.; Bennett, B.; Leitman, D. Regulation and role of guanylate cyclase-cyclic GMP in vascular relaxation. Prog. Clin. Biol. Res. 1987, 249, 65–76. [Google Scholar] [PubMed]
- Vesely, D.L. Melatonin enhances guanylate cyclase activity in a variety of tissues. Mol. Cell. Biochem. 1981, 35, 55–58. [Google Scholar] [CrossRef]
- Zhu, Y.; Gao, H.; Lu, M.; Hao, C.; Pu, Z.; Guo, M.; Hou, D.; Chen, L.Y.; Huang, X. Melatonin-Nitric Oxide Crosstalk and Their Roles in the Redox Network in Plants. Int. J. Mol. Sci. 2019, 20, 6200. [Google Scholar] [CrossRef] [PubMed]
- Bettahi, I.; Pozo, D.; Osuna, C.; Reiter, R.J.; Acuña-Castroviejo, D.; Guerrero, J.M. Melatonin reduces nitric oxide synthase activity in rat hypothalamus. J. Pineal Res. 1996, 20, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Pozo, D.; Reiter, R.J.; Calvo, J.R.; Guerrero, J.M. Inhibition of cerebellar nitric oxide synthase and cyclic GMP production by melatonin via complex formation with calmodulin. J. Cell. Biochem. 1997, 65, 430–442. [Google Scholar] [CrossRef]
- León, J.; Macías, M.; Escames, G.; Camacho, E.; Khaldy, H.; Martín, M.; Espinosa, A.; Gallo, M.A.; Acuña-Castroviejo, D. Structure-related inhibition of calmodulin-dependent neuronal nitric-oxide synthase activity by melatonin and synthetic kynurenines. Mol. Pharmacol. 2000, 58, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Camacho, M.E.; Carrion, M.D.; Lopez-Cara, L.C.; Entrena, A.; Gallo, M.A.; Espinosa, A.; Escames, G.; Acuna-Castroviejo, D. Melatonin synthetic analogs as nitric oxide synthase inhibitors. Mini Rev. Med. Chem. 2012, 12, 600–617. [Google Scholar] [CrossRef]
- Genade, S.; Genis, A.; Ytrehus, K.; Huisamen, B.; Lochner, A. Melatonin receptor-mediated protection against myocardial ischaemia/reperfusion injury: Role of its anti-adrenergic actions. J. Pineal Res. 2008, 45, 449–458. [Google Scholar] [CrossRef]
- Paulis, L.; Simko, F.; Laudon, M. Cardiovascular effects of melatonin receptor agonists. Expert Opin. Investig. Drugs 2012, 21, 1661–1678. [Google Scholar] [CrossRef]
- Tobeiha, M.; Jafari, A.; Fadaei, S.; Mirazimi, S.M.A.; Dashti, F.; Amiri, A.; Khan, H.; Asemi, Z.; Reiter, R.J.; Hamblin, M.R.; et al. Evidence for the Benefits of Melatonin in Cardiovascular Disease. Front. Cardiovasc. Med. 2022, 9, 888319. [Google Scholar] [CrossRef] [PubMed]
- Bastin, G.; Heximer, S.P. Intracellular regulation of heterotrimeric G-protein signaling modulates vascular smooth muscle cell contraction. Arch. Biochem. Biophys. 2011, 510, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Brozovich, F.V.; Nicholson, C.J.; Degen, C.V.; Gao, Y.Z.; Aggarwal, M.; Morgan, K.G. Mechanisms of Vascular Smooth Muscle Contraction and the Basis for Pharmacologic Treatment of Smooth Muscle Disorders. Pharmacol. Rev. 2016, 68, 476–532. [Google Scholar] [CrossRef]
- Lacolley, P.; Regnault, V.; Segers, P.; Laurent, S. Vascular Smooth Muscle Cells and Arterial Stiffening: Relevance in Development, Aging, and Disease. Physiol. Rev. 2017, 97, 1555–1617. [Google Scholar] [CrossRef]
- Liu, Z.; Khalil, R.A. Evolving mechanisms of vascular smooth muscle contraction highlight key targets in vascular disease. Biochem. Pharmacol. 2018, 153, 91–122. [Google Scholar] [CrossRef]
- Shimokawa, H.; Sunamura, S.; Satoh, K. RhoA/Rho-Kinase in the Cardiovascular System. Circ. Res. 2016, 118, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Xu, C.; Carraway, M.S.; Piantadosi, C.A.; Whorton, A.R.; Li, S. RhoA inactivation by S-nitrosylation regulates vascular smooth muscle contractive signaling. Nitric Oxide Biol. Chem. 2018, 74, 56–64. [Google Scholar] [CrossRef]
- Nunes, K.P.; Webb, R.C. New insights into RhoA/Rho-kinase signaling: A key regulator of vascular contraction. Small GTPases 2021, 12, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Ringvold, H.C.; Khalil, R.A. Protein Kinase C as Regulator of Vascular Smooth Muscle Function and Potential Target in Vascular Disorders. Adv. Pharmacol. 2017, 78, 203–301. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.M.; Throckmorton, D.; Brophy, C.M. Mitogen-activated protein kinase activation: An alternate signaling pathway for sustained vascular smooth muscle contraction. J. Vasc. Surg. 1997, 26, 327–332. [Google Scholar] [CrossRef]
- Touyz, R.M.; El Mabrouk, M.; He, G.; Wu, X.H.; Schiffrin, E.L. Mitogen-activated protein/extracellular signal-regulated kinase inhibition attenuates angiotensin II-mediated signaling and contraction in spontaneously hypertensive rat vascular smooth muscle cells. Circ. Res. 1999, 84, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Trebak, M.; Ginnan, R.; Singer, H.A.; Jourd’heuil, D. Interplay between calcium and reactive oxygen/nitrogen species: An essential paradigm for vascular smooth muscle signaling. Antioxid. Redox Signal. 2010, 12, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.H.; Jiang, M.J. Reactive oxygen species are involved in regulating alpha1-adrenoceptor-activated vascular smooth muscle contraction. J. Biomed. Sci. 2010, 17, 67. [Google Scholar] [CrossRef]
- MacKay, C.E.; Knock, G.A. Control of vascular smooth muscle function by Src-family kinases and reactive oxygen species in health and disease. J. Physiol. 2015, 593, 3815–3828. [Google Scholar] [CrossRef]
- Tang, D.D.; Anfinogenova, Y. Physiologic properties and regulation of the actin cytoskeleton in vascular smooth muscle. J. Cardiovasc. Pharmacol. Ther. 2008, 13, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Ohanian, J.; Pieri, M.; Ohanian, V. Non-receptor tyrosine kinases and the actin cytoskeleton in contractile vascular smooth muscle. J. Physiol. 2015, 593, 3807–3814. [Google Scholar] [CrossRef]
- Tang, D.D. The Dynamic Actin Cytoskeleton in Smooth Muscle. Adv. Pharmacol. 2018, 81, 1–38. [Google Scholar] [CrossRef]
- Touyz, R.M.; Alves-Lopes, R.; Rios, F.J.; Camargo, L.L.; Anagnostopoulou, A.; Arner, A.; Montezano, A.C. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 2018, 114, 529–539. [Google Scholar] [CrossRef]
- Sacharidou, A.; Stratman, A.N.; Davis, G.E. Molecular mechanisms controlling vascular lumen formation in three-dimensional extracellular matrices. Cells Tissues Organs 2012, 195, 122–143. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Appel, S.; Vetterkind, S.; Gangopadhyay, S.S.; Morgan, K.G. Smooth muscle signalling pathways in health and disease. J. Cell. Mol. Med. 2008, 12, 2165–2180. [Google Scholar] [CrossRef]
- Matchkov, V.V.; Kudryavtseva, O.; Aalkjaer, C. Intracellular Ca2+ signalling and phenotype of vascular smooth muscle cells. Basic Clin. Pharmacol. Toxicol. 2012, 110, 42–48. [Google Scholar] [CrossRef]
- Antón-Tay, F.; Ramírez, G.; Martínez, I.; Benítez-King, G. In vitro stimulation of protein kinase C by melatonin. Neurochem. Res. 1998, 23, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Pandi-Perumal, S.R.; Trakht, I.; Srinivasan, V.; Spence, D.W.; Maestroni, G.J.M.; Zisapel, N.; Cardinali, D.P. Physiological effects of melatonin: Role of melatonin receptors and signal transduction pathways. Prog. Neurobiol. 2008, 85, 335–353. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Rodríguez, G.; Ortiz-López, L.; Benítez-King, G. Melatonin increases stress fibers and focal adhesions in MDCK cells: Participation of Rho-associated kinase and protein kinase C. J. Pineal Res. 2007, 42, 180–190. [Google Scholar] [CrossRef]
- Xu, Y.; Cui, K.; Li, J.; Tang, X.; Lin, J.; Lu, X.; Huang, R.; Yang, B.; Shi, Y.; Ye, D.; et al. Melatonin attenuates choroidal neovascularization by regulating macrophage/microglia polarization via inhibition of RhoA/ROCK signaling pathway. J. Pineal Res. 2020, 69, e12660. [Google Scholar] [CrossRef]
- Gomez-Pinilla, P.J.; Gomez, M.F.; Swärd, K.; Hedlund, P.; Hellstrand, P.; Camello, P.J.; Andersson, K.E.; Pozo, M.J. Melatonin restores impaired contractility in aged guinea pig urinary bladder. J. Pineal Res. 2008, 44, 416–425. [Google Scholar] [CrossRef]
- Li, W.; Wang, Z.; Chen, Y.; Wang, K.; Lu, T.; Ying, F.; Fan, M.; Li, Z.; Wu, J. Melatonin treatment induces apoptosis through regulating the nuclear factor-κB and mitogen-activated protein kinase signaling pathways in human gastric cancer SGC7901 cells. Oncol. Lett. 2017, 13, 2737–2744. [Google Scholar] [CrossRef]
- Lai, C.P.; Chen, Y.S.; Ying, T.H.; Kao, C.Y.; Chiou, H.L.; Kao, S.H.; Hsieh, Y.H. Melatonin acts synergistically with pazopanib against renal cell carcinoma cells through p38 mitogen-activated protein kinase-mediated mitochondrial and autophagic apoptosis. Kidney Res. Clin. Pract. 2023, 42, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Gim, S.A.; Koh, P.O. Melatonin attenuates hepatic ischemia through mitogen-activated protein kinase signaling. J. Surg. Res. 2015, 198, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Koh, P.O. Melatonin attenuates the cerebral ischemic injury via the MEK/ERK/p90RSK/bad signaling cascade. J. Vet. Med. Sci. 2008, 70, 1219–1223. [Google Scholar] [CrossRef]
- Leal Denis, M.F.; Incicco, J.J.; Espelt, M.V.; Verstraeten, S.V.; Pignataro, O.P.; Lazarowski, E.R.; Schwarzbaum, P.J. Kinetics of extracellular ATP in mastoparan 7-activated human erythrocytes. Biochim. Biophys. Acta 2013, 1830, 4692–4707. [Google Scholar] [CrossRef]
- Ramírez, V.T.; Ramos-Fernández, E.; Inestrosa, N.C. The Gαo Activator Mastoparan-7 Promotes Dendritic Spine Formation in Hippocampal Neurons. Neural Plast. 2016, 2016, 4258171. [Google Scholar] [CrossRef] [PubMed]
- Grześk, G.; Malinowski, B.; Grześk, E.; Wiciński, M.; Szadujkis-Szadurska, K. Direct regulation of vascular smooth muscle contraction by mastoparan-7. Biomed. Rep. 2014, 2, 34–38. [Google Scholar] [CrossRef]
- Grześk, E.; Darwish, N.; Stolarek, W.; Wiciński, M.; Malinowski, B.; Burdziński, I.; Grześk, G. Effect of reperfusion on vascular smooth muscle reactivity in three contraction models. Microvasc. Res. 2019, 121, 24–29. [Google Scholar] [CrossRef]
- Grześk, E.; Mackiewicz-Milewska, M.; Mackiewicz-Nartowicz, H.; Wiciński, M.; Burdziński, I.; Korsak, M.; Kopczyńska, A.; Hagner, W.; Grześk, G. Modulatory effect of laser irradiation on mastoparan-7-induced contraction. Biomed. Rep. 2020, 12, 23–29. [Google Scholar] [CrossRef]
- Hernández-Guijo, J.M.; Gandía, L.; Lara, B.; García, A.G. Autocrine/paracrine modulation of calcium channels in bovine chromaffin cells. Pflug. Arch. 1998, 437, 104–113. [Google Scholar] [CrossRef]
- Hernández-Guijo, J.M.; Carabelli, V.; Gandía, L.; García, A.G.; Carbone, E. Voltage-independent autocrine modulation of L-type channels mediated by ATP, opioids and catecholamines in rat chromaffin cells. Eur. J. Neurosci. 1999, 11, 3574–3584. [Google Scholar] [CrossRef]
- Alhakamy, N.A.; Ahmed, O.A.A.; Md, S.; Fahmy, U.A. Mastoparan, a Peptide Toxin from Wasp Venom Conjugated Fluvastatin Nanocomplex for Suppression of Lung Cancer Cell Growth. Polymers 2021, 13, 4225. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Kitani, S. Inhibitory effect of ganglioside on mastoparan-induced cytotoxicity and degranulation in lipid raft of connective tissue type mast cell. J. Biochem. Mol. Toxicol. 2011, 25, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.M. Fashioning blood vessels by ROS signalling and metabolism. Semin. Cell Dev. Biol. 2018, 80, 35–42. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Cross-Talk between NADPH Oxidase and Mitochondria: Role in ROS Signaling and Angiogenesis. Cells 2020, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zou, M.H. Measurement of Reactive Oxygen Species (ROS) and Mitochondrial ROS in AMPK Knockout Mice Blood Vessels. Methods Mol. Biol. 2018, 1732, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Pugsley, M.K.; Tabrizchi, R. The vascular system. An overview of structure and function. J. Pharmacol. Toxicol. Methods 2000, 44, 333–340. [Google Scholar] [CrossRef]
- Shimokawa, H.; Satoh, K. Vascular function. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2359–2362. [Google Scholar] [CrossRef]
- Feske, S.K. Ischemic Stroke. Am. J. Med. 2021, 134, 1457–1464. [Google Scholar] [CrossRef]
- Paul, S.; Candelario-Jalil, E. Emerging neuroprotective strategies for the treatment of ischemic stroke: An overview of clinical and preclinical studies. Exp. Neurol. 2021, 335, 113518. [Google Scholar] [CrossRef]
- Saini, V.; Guada, L.; Yavagal, D.R. Global Epidemiology of Stroke and Access to Acute Ischemic Stroke Interventions. Neurology 2021, 97 (Suppl. S2), S6–S16. [Google Scholar] [CrossRef]
- Rutten-Jacobs, L.C.; Larsson, S.C.; Malik, R.; Rannikmäe, K.; MEGASTROKE consortium; International Stroke Genetics Consortium; Sudlow, C.L.; Dichgans, M.; Markus, H.S.; Traylor, M. Genetic risk, incident stroke, and the benefits of adhering to a healthy lifestyle: Cohort study of 306 473 UK Biobank participants. BMJ 2018, 363, k4168. [Google Scholar] [CrossRef] [PubMed]
- GBD 2019 Stroke Collaborators Global, regional, and national burden of stroke and its risk factors, 1990-2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021, 20, 795–820. [CrossRef] [PubMed]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Beaton, A.Z.; Boehme, A.K.; Buxton, A.E.; et al. Heart Disease and Stroke Statistics-2023 Update: A Report From the American Heart Association. Circulation 2023, 147, e93–e621. [Google Scholar] [CrossRef]
- Cai, W.; Zhang, K.; Li, P.; Zhu, L.; Xu, J.; Yang, B.; Hu, X.; Lu, Z.; Chen, J. Dysfunction of the neurovascular unit in ischemic stroke and neurodegenerative diseases: An aging effect. Ageing Res. Rev. 2017, 34, 77–87. [Google Scholar] [CrossRef]
- Drozdowska, B.A.; Singh, S.; Quinn, T.J. Thinking About the Future: A Review of Prognostic Scales Used in Acute Stroke. Front. Neurol. 2019, 10, 274. [Google Scholar] [CrossRef]
- Bangad, A.; Abbasi, M.; de Havenon, A. Secondary Ischemic Stroke Prevention. Neurother. J. Am. Soc. Exp. Neurother. 2023, 20, 721–731. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Donnan, G.A.; Fisher, M.; Macleod, M.; Davis, S.M. Stroke. Lancet 2008, 371, 1612–1623. [Google Scholar] [CrossRef]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Muoio, V.; Persson, P.B.; Sendeski, M.M. The neurovascular unit–concept review. Acta Physiol. 2014, 210, 790–798. [Google Scholar] [CrossRef]
- Knobel, P.; Litke, R.; Mobbs, C.V. Biological age and environmental risk factors for dementia and stroke: Molecular mechanisms. Front. Aging Neurosci. 2022, 14, 1042488. [Google Scholar] [CrossRef]
- Soriano-Tárraga, C.; Mola-Caminal, M.; Giralt-Steinhauer, E.; Ois, A.; Rodríguez-Campello, A.; Cuadrado-Godia, E.; Gómez-González, A.; Vivanco-Hidalgo, R.M.; Fernández-Cadenas, I.; Cullell, N.; et al. Biological age is better than chronological as predictor of 3-month outcome in ischemic stroke. Neurology 2017, 89, 830–836. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Koya, D. Autophagy in metabolic disease and ageing. Nat. Rev. Endocrinol. 2021, 17, 647–661. [Google Scholar] [CrossRef]
- Pluta, R.; Januszewski, S.; Czuczwar, S.J. The Role of Gut Microbiota in an Ischemic Stroke. Int. J. Mol. Sci. 2021, 22, 915. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A. Biol. Sci. Med. Sci. 2014, 69 (Suppl. S1), S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Gallizioli, M.; Arbaizar-Rovirosa, M.; Brea, D.; Planas, A.M. Differences in the post-stroke innate immune response between young and old. Semin. Immunopathol. 2023, 45, 367–376. [Google Scholar] [CrossRef]
- Broman, J.; Rinvik, E.; Sassoe-Pognetto, M.; Shandiz, H.K.; Ottersen, O.P. CHAPTER 36—Glutamate. In The Rat Nervous System, 3rd ed.; Paxinos, G., Ed.; Academic Press: Burlington, NJ, USA, 2004; pp. 1269–1292. Available online: https://www.sciencedirect.com/science/article/pii/B9780125476386500377 (accessed on 22 September 2023).
- Charych, E.I.; Liu, F.; Moss, S.J.; Brandon, N.J. GABA(A) receptors and their associated proteins: Implications in the etiology and treatment of schizophrenia and related disorders. Neuropharmacology 2009, 57, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W. Excitatory transmitter neurotoxicity. Neurobiol. Aging 1994, 15, 259–260. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front. Neurosci. 2020, 14, 579953. [Google Scholar] [CrossRef]
- Szydlowska, K.; Tymianski, M. Calcium, ischemia and excitotoxicity. Cell Calcium 2010, 47, 122–129. [Google Scholar] [CrossRef]
- Bode, B.P.; Kilberg, M.S. Amino acid-dependent increase in hepatic system N activity is linked to cell swelling. J. Biol. Chem. 1991, 266, 7376–7381. [Google Scholar] [CrossRef] [PubMed]
- Häussinger, D.; Schliess, F. Glutamine metabolism and signaling in the liver. Front. Biosci. J. Virtual Libr. 2007, 12, 371–391. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.O. Associations between transports of alanine and cations across cell membrane in rat hepatocytes. Am. J. Physiol. 1986, 251 Pt 1, G575–G584. [Google Scholar] [CrossRef]
- Matsumoto, K.; Lo, E.H.; Pierce, A.R.; Halpern, E.F.; Newcomb, R. Secondary elevation of extracellular neurotransmitter amino acids in the reperfusion phase following focal cerebral ischemia. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 1996, 16, 114–124. [Google Scholar] [CrossRef]
- Luengo, J.G.; Muñoz, M.D.; Álvarez-Merz, I.; Herranz, A.S.; González, J.C.; Martín Del Río, R.; Hernández-Guijo, J.M.; Solís, J.M. Intracellular accumulation of amino acids increases synaptic potentials in rat hippocampal slices. Amino Acids 2019, 51, 1337–1351. [Google Scholar] [CrossRef] [PubMed]
- Shimada, N.; Graf, R.; Rosner, G.; Heiss, W.D. Ischemia-induced accumulation of extracellular amino acids in cerebral cortex, white matter, and cerebrospinal fluid. J. Neurochem. 1993, 60, 66–71. [Google Scholar] [CrossRef]
- Uchiyama-Tsuyuki, Y.; Araki, H.; Yae, T.; Otomo, S. Changes in the extracellular concentrations of amino acids in the rat striatum during transient focal cerebral ischemia. J. Neurochem. 1994, 62, 1074–1078. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, C.; Yan, Z.Y.; Yang, J.; Chen, H. Simultaneous monitoring multiple neurotransmitters and neuromodulators during cerebral ischemia/reperfusion in rats by microdialysis and capillary electrophoresis. J. Neurosci. Methods 2010, 189, 162–168. [Google Scholar] [CrossRef]
- Hutchinson, P.J.; O’Connell, M.T.; Al-Rawi, P.G.; Kett-White, C.R.; Gupta, A.K.; Maskell, L.B.; Pickard, J.D.; Kirkpatrick, P.J. Increases in GABA concentrations during cerebral ischaemia: A microdialysis study of extracellular amino acids. J. Neurol. Neurosurg. Psychiatry 2002, 72, 99–105. [Google Scholar] [CrossRef]
- Zetterling, M.; Hillered, L.; Samuelsson, C.; Karlsson, T.; Enblad, P.; Ronne-Engström, E. Temporal patterns of interstitial pyruvate and amino acids after subarachnoid haemorrhage are related to the level of consciousness--a clinical microdialysis study. Acta Neurochir. 2009, 151, 771–780, discussion 780. [Google Scholar] [CrossRef]
- Jung, C.S.; Lange, B.; Zimmermann, M.; Seifert, V. CSF and Serum Biomarkers Focusing on Cerebral Vasospasm and Ischemia after Subarachnoid Hemorrhage. Stroke Res. Treat. 2013, 2013, 560305. [Google Scholar] [CrossRef]
- Kanthan, R.; Shuaib, A.; Griebel, R.; Miyashita, H. Intracerebral human microdialysis. In vivo study of an acute focal ischemic model of the human brain. Stroke 1995, 26, 870–873. [Google Scholar] [CrossRef] [PubMed]
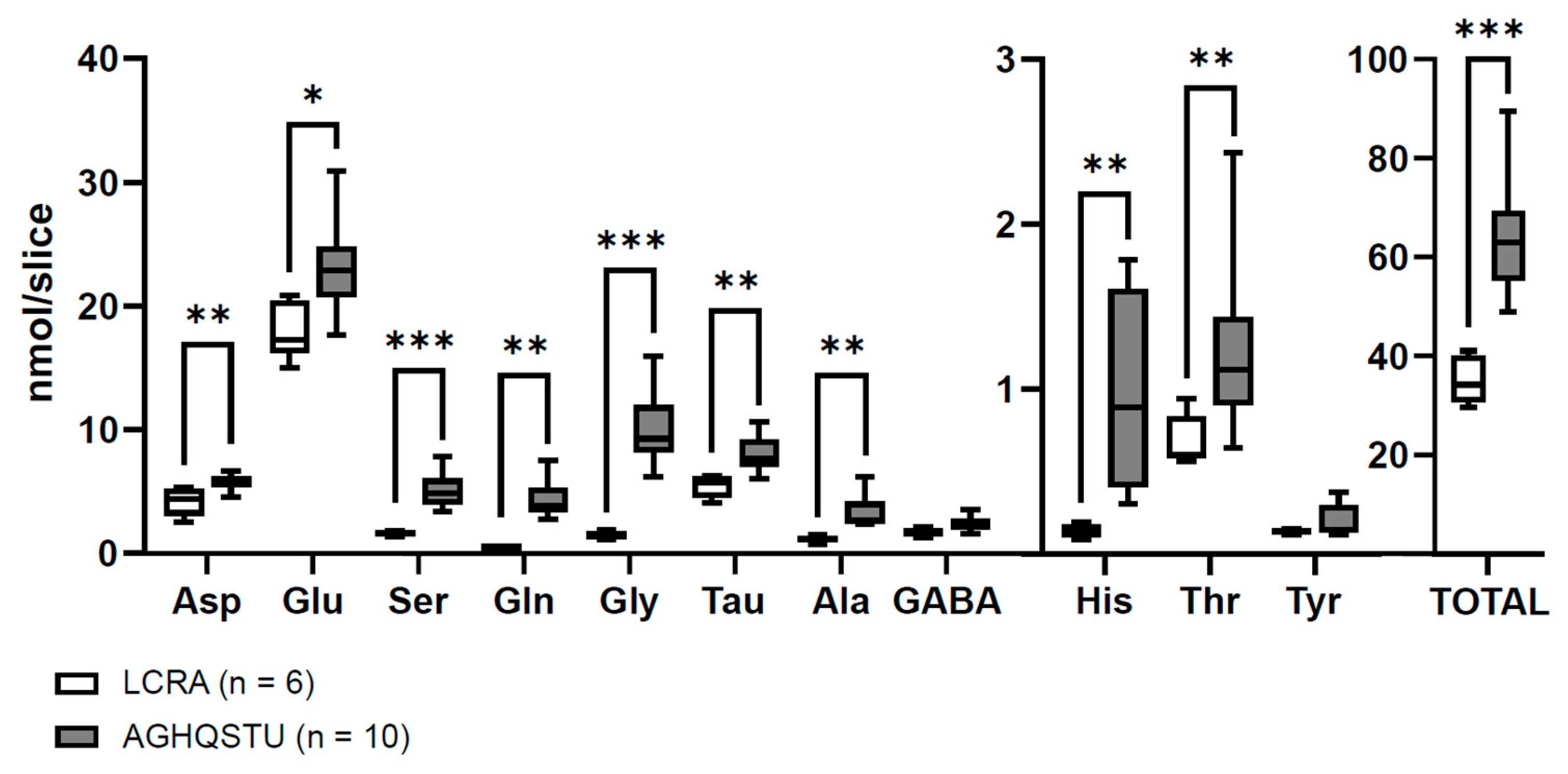
- Álvarez-Merz, I.; Muñoz, M.; Hernández-Guijo, J.M.; Solís, J.M. Identification of non-excitatory amino acids and transporters mediating the irreversible synaptic silencing after hypoxia. Transl. Stroke Res. 2023, in press. [Google Scholar] [CrossRef]
- Dale, N.; Pearson, T.; Frenguelli, B.G. Direct measurement of adenosine release during hypoxia in the CA1 region of the rat hippocampal slice. J. Physiol. 2000, 526 Pt 1, 143–155. [Google Scholar] [CrossRef]
- Duarte, J.M.N.; Cunha, R.A.; Carvalho, R.A. Adenosine A1 receptors control the metabolic recovery after hypoxia in rat hippocampal slices. J. Neurochem. 2016, 136, 947–957. [Google Scholar] [CrossRef]
- Chebabo, S.R.; Hester, M.A.; Jing, J.; Aitken, P.G.; Somjen, G.G. Interstitial space, electrical resistance and ion concentrations during hypotonia of rat hippocampal slices. J. Physiol. 1995, 487 Pt 3, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Dingledine, R. Role of extracellular space in hyperosmotic suppression of potassium-induced electrographic seizures. J. Neurophysiol. 1989, 61, 927–938. [Google Scholar] [CrossRef]
- Wilson, C.S.; Bach, M.D.; Ashkavand, Z.; Norman, K.R.; Martino, N.; Adam, A.P.; Mongin, A.A. Metabolic constraints of swelling-activated glutamate release in astrocytes and their implication for ischemic tissue damage. J. Neurochem. 2019, 151, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, A.R.; Rao, K.V.R.; Murthy, C.R.K.; Norenberg, M.D. Glutamine in the mechanism of ammonia-induced astrocyte swelling. Neurochem. Int. 2006, 48, 623–628. [Google Scholar] [CrossRef]
- Utsunomiya-Tate, N.; Endou, H.; Kanai, Y. Cloning and functional characterization of a system ASC-like Na+-dependent neutral amino acid transporter. J. Biol. Chem. 1996, 271, 14883–14890. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.R.; López-Corcuera, B.; Mandiyan, S.; Nelson, H.; Nelson, N. Molecular characterization of four pharmacologically distinct gamma-aminobutyric acid transporters in mouse brain [corrected]. J. Biol. Chem. 1993, 268, 2106–2112. [Google Scholar] [CrossRef] [PubMed]
- Bröer, A.; Wagner, C.; Lang, F.; Bröer, S. Neutral amino acid transporter ASCT2 displays substrate-induced Na+ exchange and a substrate-gated anion conductance. Biochem. J. 2000, 346 Pt 3, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The glutamate/GABA-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.; Fernández, E.; Torrents, D.; Estévez, R.; López, C.; Camps, M.; Lloberas, J.; Zorzano, A.; Palacín, M. Identification of a membrane protein, LAT-2, that Co-expresses with 4F2 heavy chain, an L-type amino acid transport activity with broad specificity for small and large zwitterionic amino acids. J. Biol. Chem. 1999, 274, 19738–19744. [Google Scholar] [CrossRef]
- Yamakami, J.; Sakurai, E.; Sakurada, T.; Maeda, K.; Hikichi, N. Stereoselective blood-brain barrier transport of histidine in rats. Brain Res. 1998, 812, 105–112. [Google Scholar] [CrossRef]
- Ennis, S.R.; Kawai, N.; Ren, X.D.; Abdelkarim, G.E.; Keep, R.F. Glutamine uptake at the blood-brain barrier is mediated by N-system transport. J. Neurochem. 1998, 71, 2565–2573. [Google Scholar] [CrossRef]
- Keep, R.F.; Xiang, J. N-system amino acid transport at the blood--CSF barrier. J. Neurochem. 1995, 65, 2571–2576. [Google Scholar] [CrossRef]
- Meier, C.; Ristic, Z.; Klauser, S.; Verrey, F. Activation of system L heterodimeric amino acid exchangers by intracellular substrates. EMBO J. 2002, 21, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Kittl, M.; Dobias, H.; Beyreis, M.; Kiesslich, T.; Mayr, C.; Gaisberger, M.; Ritter, M.; Kerschbaum, H.H.; Jakab, M. Glycine Induces Migration of Microglial BV-2 Cells via SNAT-Mediated Cell Swelling. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 50, 1460–1473. [Google Scholar] [CrossRef]
- Baird, F.E.; Beattie, K.J.; Hyde, A.R.; Ganapathy, V.; Rennie, M.J.; Taylor, P.M. Bidirectional substrate fluxes through the system N (SNAT5) glutamine transporter may determine net glutamine flux in rat liver. J. Physiol. 2004, 559 Pt 2, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, F.A.; Krizaj, D.; Larsson, P.; Reimer, R.J.; Wreden, C.; Storm-Mathisen, J.; Copenhagen, D.; Kavanaugh, M.; Edwards, R.H. Coupled and uncoupled proton movement by amino acid transport system N. EMBO J. 2001, 20, 7041–7051. [Google Scholar] [CrossRef]
- Kaplan, E.; Zubedat, S.; Radzishevsky, I.; Valenta, A.C.; Rechnitz, O.; Sason, H.; Sajrawi, C.; Bodner, O.; Konno, K.; Esaki, K.; et al. ASCT1 (Slc1a4) transporter is a physiologic regulator of brain d-serine and neurodevelopment. Proc. Natl. Acad. Sci. USA 2018, 115, 9628–9633. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.L.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 2018, 163–164, 144–171. [Google Scholar] [CrossRef]
- Jia, S.W.; Liu, X.Y.; Wang, S.C.; Wang, Y.F. Vasopressin Hypersecretion-Associated Brain Edema Formation in Ischemic Stroke: Underlying Mechanisms. J. Stroke Cerebrovasc. Dis. Off. J. Natl. Stroke Assoc. 2016, 25, 1289–1300. [Google Scholar] [CrossRef]
- Wu, S.; Yuan, R.; Wang, Y.; Wei, C.; Zhang, S.; Yang, X.; Wu, B.; Liu, M. Early Prediction of Malignant Brain Edema After Ischemic Stroke. Stroke 2018, 49, 2918–2927. [Google Scholar] [CrossRef]
- Rathnasamy, G.; Ling, E.A.; Kaur, C. Therapeutic implications of melatonin in cerebral edema. Histol. Histopathol. 2014, 29, 1525–1538. [Google Scholar] [CrossRef]
- Tordjman, S.; Chokron, S.; Delorme, R.; Charrier, A.; Bellissant, E.; Jaafari, N.; Fougerou, C. Melatonin: Pharmacology, Functions and Therapeutic Benefits. Curr. Neuropharmacol. 2017, 15, 434–443. [Google Scholar] [CrossRef]
- Kondoh, T.; Uneyama, H.; Nishino, H.; Torii, K. Melatonin reduces cerebral edema formation caused by transient forebrain ischemia in rats. Life Sci. 2002, 72, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.Y.; Lee, M.Y.; Chen, H.Y.; Kuo, Y.L.; Lin, S.C.; Wu, T.S.; Lee, E.J. Melatonin attenuates the postischemic increase in blood-brain barrier permeability and decreases hemorrhagic transformation of tissue-plasminogen activator therapy following ischemic stroke in mice. J. Pineal Res. 2006, 40, 242–250. [Google Scholar] [CrossRef]
- Kaur, C.; Sivakumar, V.; Zhang, Y.; Ling, E.A. Hypoxia-induced astrocytic reaction and increased vascular permeability in the rat cerebellum. Glia 2006, 54, 826–839. [Google Scholar] [CrossRef]
- Toklu, H.; Deniz, M.; Yüksel, M.; Keyer-Uysal, M.; Sener, G. The protective effect of melatonin and amlodipine against cerebral ischemia/reperfusion-induced oxidative brain injury in rats. Marmara Med. J. 2009, 22, 34–44. [Google Scholar]
- Lotufo, C.M.C.; Yamashita, C.E.; Farsky, S.H.P.; Markus, R.P. Melatonin effect on endothelial cells reduces vascular permeability increase induced by leukotriene B4. Eur. J. Pharmacol. 2006, 534, 258–263. [Google Scholar] [CrossRef]
- Sivakumar, V.; Lu, J.; Ling, E.A.; Kaur, C. Vascular endothelial growth factor and nitric oxide production in response to hypoxia in the choroid plexus in neonatal brain. Brain Pathol. 2008, 18, 71–85. [Google Scholar] [CrossRef]
- Qin, W.; Li, J.; Zhu, R.; Gao, S.; Fan, J.; Xia, M.; Zhao, R.C.; Zhang, J. Melatonin protects blood-brain barrier integrity and permeability by inhibiting matrix metalloproteinase-9 via the NOTCH3/NF-κB pathway. Aging 2019, 11, 11391–11415. [Google Scholar] [CrossRef]
- Kawabori, M.; Yenari, M.A. Inflammatory responses in brain ischemia. Curr. Med. Chem. 2015, 22, 1258–1277. [Google Scholar] [CrossRef]
- Yawoot, N.; Govitrapong, P.; Tocharus, C.; Tocharus, J. Ischemic stroke, obesity, and the anti-inflammatory role of melatonin. Biofactors 2021, 47, 41–58. [Google Scholar] [CrossRef]
- Kumar, J.; Haldar, C.; Verma, R. Melatonin Ameliorates LPS-Induced Testicular Nitro-oxidative Stress (iNOS/TNFα) and Inflammation (NF-kB/COX-2) via Modulation of SIRT-1. Reprod. Sci. 2021, 28, 3417–3430. [Google Scholar] [CrossRef]
- Tang, L.; Zhang, C.; Lu, L.; Tian, H.; Liu, K.; Luo, D.; Qiu, Q.; Xu, G.T.; Zhang, J. Melatonin Maintains Inner Blood-Retinal Barrier by Regulating Microglia via Inhibition of PI3K/Akt/Stat3/NF-κB Signaling Pathways in Experimental Diabetic Retinopathy. Front. Immunol. 2022, 13, 831660. [Google Scholar] [CrossRef]
- Zhi, S.M.; Fang, G.X.; Xie, X.M.; Liu, L.H.; Yan, J.; Liu, D.B.; Yu, H.Y. Melatonin reduces OGD/R-induced neuron injury by regulating redox/inflammation/apoptosis signaling. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 1524–1536. [Google Scholar] [CrossRef]
- Ali, T.; Rahman, S.U.; Hao, Q.; Li, W.; Liu, Z.; Ali Shah, F.; Murtaza, I.; Zhang, Z.; Yang, X.; Liu, G.; et al. Melatonin prevents neuroinflammation and relieves depression by attenuating autophagy impairment through FOXO3a regulation. J. Pineal Res. 2020, 69, e12667. [Google Scholar] [CrossRef] [PubMed]
- Arioz, B.I.; Tastan, B.; Tarakcioglu, E.; Tufekci, K.U.; Olcum, M.; Ersoy, N.; Bagriyanik, A.; Genc, K.; Genc, S. Melatonin Attenuates LPS-Induced Acute Depressive-Like Behaviors and Microglial NLRP3 Inflammasome Activation Through the SIRT1/Nrf2 Pathway. Front. Immunol. 2019, 10, 1511. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Ran, Y.Y.; Qie, S.Y.; Gong, W.J.; Gao, F.H.; Ding, Z.T.; Xi, J.N. Melatonin protects against ischemic stroke by modulating microglia/macrophage polarization toward anti-inflammatory phenotype through STAT3 pathway. CNS Neurosci. Ther. 2019, 25, 1353–1362. [Google Scholar] [CrossRef]
- Xu, D.; Liu, L.; Zhao, Y.; Yang, L.; Cheng, J.; Hua, R.; Zhang, Z.; Li, Q. Melatonin protects mouse testes from palmitic acid-induced lipotoxicity by attenuating oxidative stress and DNA damage in a SIRT1-dependent manner. J. Pineal Res. 2020, 69, e12690. [Google Scholar] [CrossRef]
- Hung, Y.C.; Chen, T.Y.; Lee, E.J.; Chen, W.L.; Huang, S.Y.; Lee, W.T.; Lee, M.Y.; Chen, H.Y.; Wu, T.S. Melatonin decreases matrix metalloproteinase-9 activation and expression and attenuates reperfusion-induced hemorrhage following transient focal cerebral ischemia in rats. J. Pineal Res. 2008, 45, 459–467. [Google Scholar] [CrossRef]
- Song, J.; Kang, S.M.; Lee, W.T.; Park, K.A.; Lee, K.M.; Lee, J.E. The beneficial effect of melatonin in brain endothelial cells against oxygen-glucose deprivation followed by reperfusion-induced injury. Oxid. Med. Cell. Longev. 2014, 2014, 639531. [Google Scholar] [CrossRef]
- Lee, M.Y.; Kuan, Y.H.; Chen, H.Y.; Chen, T.Y.; Chen, S.T.; Huang, C.C.; Yang, I.P.; Hsu, Y.S.; Wu, T.S.; Lee, E.J. Intravenous administration of melatonin reduces the intracerebral cellular inflammatory response following transient focal cerebral ischemia in rats. J. Pineal Res. 2007, 42, 297–309. [Google Scholar] [CrossRef]
- Feng, J.; Chen, X.; Shen, J. Reactive nitrogen species as therapeutic targets for autophagy: Implication for ischemic stroke. Expert Opin. Ther. Targets 2017, 21, 305–317. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Ning, N.; Zhou, Q.; Khoshnam, S.E.; Farzaneh, M. Mitochondria as a therapeutic target for ischemic stroke. Free Radic. Biol. Med. 2020, 146, 45–58. [Google Scholar] [CrossRef]
- Hoye, A.T.; Davoren, J.E.; Wipf, P.; Fink, M.P.; Kagan, V.E. Targeting mitochondria. Acc. Chem. Res. 2008, 41, 87–97. [Google Scholar] [CrossRef]
- Pawluk, H.; Kołodziejska, R.; Grześk, G.; Woźniak, A.; Kozakiewicz, M.; Kosinska, A.; Pawluk, M.; Grzechowiak, E.; Wojtasik, J.; Kozera, G. Increased Oxidative Stress Markers in Acute Ischemic Stroke Patients Treated with Thrombolytics. Int. J. Mol. Sci. 2022, 23, 15625. [Google Scholar] [CrossRef]
- He, R.; Cui, M.; Lin, H.; Zhao, L.; Wang, J.; Chen, S.; Shao, Z. Melatonin resists oxidative stress-induced apoptosis in nucleus pulposus cells. Life Sci. 2018, 199, 122–130. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Ghosal, I.; Das, D.; Chakraborty, S.B. Melatonin ameliorates H2O2-induced oxidative stress through modulation of Erk/Akt/NFkB pathway. Biol. Res. 2018, 51, 17. [Google Scholar] [CrossRef]
- Zhao, F.; Whiting, S.; Lambourne, S.; Aitken, R.J.; Sun, Y.P. Melatonin alleviates heat stress-induced oxidative stress and apoptosis in human spermatozoa. Free Radic. Biol. Med. 2021, 164, 410–416. [Google Scholar] [CrossRef]
- Farhood, B.; Goradel, N.H.; Mortezaee, K.; Khanlarkhani, N.; Najafi, M.; Sahebkar, A. Melatonin and cancer: From the promotion of genomic stability to use in cancer treatment. J. Cell. Physiol. 2019, 234, 5613–5627. [Google Scholar] [CrossRef]
- Rao, V.K.; Carlson, E.A.; Yan, S.S. Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim. Biophys. Acta 2014, 1842, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Kılıç, E.; Çağlayan, B.; Caglar Beker, M. Physiological and pharmacological roles of melatonin in the pathophysiological components of cellular injury after ischemic stroke. Turk. J. Med. Sci. 2020, 50 (Suppl. S2), 1655–1664. [Google Scholar] [CrossRef]
- Xie, L.L.; Rui, C.; Li, Z.Z.; Li, S.S.; Fan, Y.J.; Qi, M.M. Melatonin mitigates traumatic brain injury-induced depression-like behaviors through HO-1/CREB signal in rats. Neurosci. Lett. 2022, 784, 136754. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wu, C.; Muhammad, J.S.; Yan, D.; Tsuneyama, K.; Hatta, H.; Cui, Z.G.; Inadera, H. Melatonin sensitises shikonin-induced cancer cell death mediated by oxidative stress via inhibition of the SIRT3/SOD2-AKT pathway. Redox Biol. 2020, 36, 101632. [Google Scholar] [CrossRef]
- Liu, L.; Cao, Q.; Gao, W.; Li, B.Y.; Zeng, C.; Xia, Z.; Zhao, B. Melatonin ameliorates cerebral ischemia-reperfusion injury in diabetic mice by enhancing autophagy via the SIRT1-BMAL1 pathway. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e22040. [Google Scholar] [CrossRef] [PubMed]
- Wei, N.; Pu, Y.; Yang, Z.; Pan, Y.; Liu, L. Therapeutic effects of melatonin on cerebral ischemia reperfusion injury: Role of Yap-OPA1 signaling pathway and mitochondrial fusion. Biomed. Pharmacother. 2019, 110, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Shah, F.A.; Liu, G.; Al Kury, L.T.; Zeb, A.; Abbas, M.; Li, T.; Yang, X.; Liu, F.; Jiang, Y.; Li, S.; et al. Melatonin Protects MCAO-Induced Neuronal Loss via NR2A Mediated Prosurvival Pathways. Front. Pharmacol. 2019, 10, 297. [Google Scholar] [CrossRef]
- Briyal, S.; Ranjan, A.K.; Gulati, A. Oxidative stress: A target to treat Alzheimer’s disease and stroke. Neurochem. Int. 2023, 165, 105509. [Google Scholar] [CrossRef]
- Andrabi, S.S.; Parvez, S.; Tabassum, H. Ischemic stroke and mitochondria: Mechanisms and targets. Protoplasma 2020, 257, 335–343. [Google Scholar] [CrossRef]
- Shen, X.; Li, M.; Shao, K.; Li, Y.; Ge, Z. Post-ischemic inflammatory response in the brain: Targeting immune cell in ischemic stroke therapy. Front. Mol. Neurosci. 2023, 16, 1076016. [Google Scholar] [CrossRef]
- Wang, P.; Ren, Q.; Shi, M.; Liu, Y.; Bai, H.; Chang, Y.Z. Overexpression of Mitochondrial Ferritin Enhances Blood-Brain Barrier Integrity Following Ischemic Stroke in Mice by Maintaining Iron Homeostasis in Endothelial Cells. Antioxidants 2022, 11, 1257. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Yang, S.; Chu, Y.H.; Zhang, H.; Pang, X.W.; Chen, L.; Zhou, L.Q.; Chen, M.; Tian, D.S.; Wang, W. Signaling pathways involved in ischemic stroke: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2022, 7, 215. [Google Scholar] [CrossRef] [PubMed]
- Cheung, R.T.F. The utility of melatonin in reducing cerebral damage resulting from ischemia and reperfusion. J. Pineal Res. 2003, 34, 153–160. [Google Scholar] [CrossRef]
- Vitte, P.A.; Harthe, C.; Lestage, P.; Claustrat, B.; Bobillier, P. Plasma, cerebrospinal fluid, and brain distribution of 14C-melatonin in rat: A biochemical and autoradiographic study. J. Pineal Res. 1988, 5, 437–453. [Google Scholar] [CrossRef]
- Kilic, U.; Kilic, E.; Reiter, R.J.; Bassetti, C.L.; Hermann, D.M. Signal transduction pathways involved in melatonin-induced neuroprotection after focal cerebral ischemia in mice. J. Pineal Res. 2005, 38, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Parada, E.; Buendia, I.; León, R.; Negredo, P.; Romero, A.; Cuadrado, A.; López, M.G.; Egea, J. Neuroprotective effect of melatonin against ischemia is partially mediated by alpha-7 nicotinic receptor modulation and HO-1 overexpression. J. Pineal Res. 2014, 56, 204–212. [Google Scholar] [CrossRef]
- Chen, K.H.; Lin, K.C.; Ko, S.F.; Chiang, J.Y.; Guo, J.; Yip, H.K. Melatonin against acute ischaemic stroke dependently via suppressing both inflammatory and oxidative stress downstream signallings. J. Cell. Mol. Med. 2020, 24, 10402–10419. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, X.; Xiong, X.; Zhu, H.; Chen, R.; Zhang, S.; Chen, G.; Jian, Z. Nrf2 Regulates Oxidative Stress and Its Role in Cerebral Ischemic Stroke. Antioxidants 2022, 11, 2377. [Google Scholar] [CrossRef] [PubMed]
- Vriend, J.; Reiter, R.J. The Keap1-Nrf2-antioxidant response element pathway: A review of its regulation by melatonin and the proteasome. Mol. Cell. Endocrinol. 2015, 401, 213–220. [Google Scholar] [CrossRef]
- Bright, R.; Mochly-Rosen, D. The role of protein kinase C in cerebral ischemic and reperfusion injury. Stroke 2005, 36, 2781–2790. [Google Scholar] [CrossRef]
- Weiss, M.D.; Carloni, S.; Vanzolini, T.; Coppari, S.; Balduini, W.; Buonocore, G.; Longini, M.; Perrone, S.; Sura, L.; Mohammadi, A.; et al. Human-rat integrated microRNAs profiling identified a new neonatal cerebral hypoxic-ischemic pathway melatonin-sensitive. J. Pineal Res. 2022, 73, e12818. [Google Scholar] [CrossRef]
- Fan, X.; Kavelaars, A.; Heijnen, C.J.; Groenendaal, F.; van Bel, F. Pharmacological neuroprotection after perinatal hypoxic-ischemic brain injury. Curr. Neuropharmacol. 2010, 8, 324–334. [Google Scholar] [CrossRef]
- Kilic, U.; Yilmaz, B.; Ugur, M.; Yüksel, A.; Reiter, R.J.; Hermann, D.M.; Kilic, E. Evidence that membrane-bound G protein-coupled melatonin receptors MT1 and MT2 are not involved in the neuroprotective effects of melatonin in focal cerebral ischemia. J. Pineal Res. 2012, 52, 228–235. [Google Scholar] [CrossRef]
- Kilic, U.; Yilmaz, B.; Reiter, R.J.; Yüksel, A.; Kilic, E. Effects of memantine and melatonin on signal transduction pathways vascular leakage and brain injury after focal cerebral ischemia in mice. Neuroscience 2013, 237, 268–276. [Google Scholar] [CrossRef]
- Kilic, U.; Elibol, B.; Caglayan, A.B.; Beker, M.C.; Beker, M.; Altug-Tasa, B.; Uysal, O.; Yilmaz, B.; Kilic, E. Delayed Therapeutic Administration of Melatonin Enhances Neuronal Survival Through AKT and MAPK Signaling Pathways Following Focal Brain Ischemia in Mice. J. Mol. Neurosci. 2022, 72, 994–1007. [Google Scholar] [CrossRef]
- Kryl’skii, E.D.; Popova, T.N.; Safonova, O.A.; Stolyarova, A.O.; Razuvaev, G.A.; de Carvalho, M.A.P. Transcriptional Regulation of Antioxidant Enzymes Activity and Modulation of Oxidative Stress by Melatonin in Rats Under Cerebral Ischemia/Reperfusion Conditions. Neuroscience 2019, 406, 653–666. [Google Scholar] [CrossRef]
- Ran, Y.; Ye, L.; Ding, Z.; Gao, F.; Yang, S.; Fang, B.; Liu, Z.; Xi, J. Melatonin Protects Against Ischemic Brain Injury by Modulating PI3K/AKT Signaling Pathway via Suppression of PTEN Activity. ASN Neuro 2021, 13, 17590914211022888. [Google Scholar] [CrossRef] [PubMed]
- Kilic, U.; Caglayan, A.B.; Beker, M.C.; Gunal, M.Y.; Caglayan, B.; Yalcin, E.; Kelestemur, T.; Gundogdu, R.Z.; Yulug, B.; Yılmaz, B.; et al. Particular phosphorylation of PI3K/Akt on Thr308 via PDK-1 and PTEN mediates melatonin’s neuroprotective activity after focal cerebral ischemia in mice. Redox Biol. 2017, 12, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Galano, A. Melatonin: Exceeding expectations. Physiology 2014, 29, 325–333. [Google Scholar] [CrossRef]
- Mayo, J.C.; Sainz, R.M.; González-Menéndez, P.; Hevia, D.; Cernuda-Cernuda, R. Melatonin transport into mitochondria. Cell. Mol. Life Sci. CMLS 2017, 74, 3927–3940. [Google Scholar] [CrossRef]
- Reiter, R.J.; Rosales-Corral, S.; Tan, D.X.; Jou, M.J.; Galano, A.; Xu, B. Melatonin as a mitochondria-targeted antioxidant: One of evolution’s best ideas. Cell. Mol. Life Sci. CMLS 2017, 74, 3863–3881. [Google Scholar] [CrossRef]
- Iadecola, C.; Alexander, M. Cerebral ischemia and inflammation. Curr. Opin. Neurol. 2001, 14, 89–94. [Google Scholar] [CrossRef]
- Przykaza, Ł. Understanding the Connection Between Common Stroke Comorbidities, Their Associated Inflammation, and the Course of the Cerebral Ischemia/Reperfusion Cascade. Front. Immunol. 2021, 12, 782569. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin and inflammation-Story of a double-edged blade. J. Pineal Res. 2018, 65, e12525. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ru, J.; Zhang, H.; Chen, J.; Lin, X.; Lin, Z.; Wen, M.; Huang, L.; Ni, H.; Zhuge, Q.; et al. Melatonin Enhances the Therapeutic Effect of Plasma Exosomes Against Cerebral Ischemia-Induced Pyroptosis through the TLR4/NF-κB Pathway. Front. Neurosci. 2020, 14, 848. [Google Scholar] [CrossRef]
- Manchester, L.C.; Coto-Montes, A.; Boga, J.A.; Andersen, L.P.H.; Zhou, Z.; Galano, A.; Vriend, J.; Tan, D.X.; Reiter, R.J. Melatonin: An ancient molecule that makes oxygen metabolically tolerable. J. Pineal Res. 2015, 59, 403–419. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Poeggeler, B.; Menendez-Pelaez, A.; Chen, L.D.; Saarela, S. Melatonin as a free radical scavenger: Implications for aging and age-related diseases. Ann. N. Y. Acad. Sci. 1994, 719, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Takasaki, A.; Taketani, T.; Tanabe, M.; Kizuka, F.; Lee, L.; Tamura, I.; Maekawa, R.; Asada, H.; Yamagata, Y.; et al. Melatonin as a free radical scavenger in the ovarian follicle. Endocr. J. 2013, 60, 1–13. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Esteban-Zubero, E.; Zhou, Z.; Reiter, R.J. Melatonin as a Potent and Inducible Endogenous Antioxidant: Synthesis and Metabolism. Molecules 2015, 20, 18886–18906. [Google Scholar] [CrossRef]
- Galano, A.; Reiter, R.J. Melatonin and its metabolites vs oxidative stress: From individual actions to collective protection. J. Pineal Res. 2018, 65, e12514. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R. Melatonin, Its Metabolites and Their Interference with Reactive Nitrogen Compounds. Molecules 2021, 26, 4105. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.; Terron, M.P.; Flores, L.J.; Czarnocki, Z. Melatonin and its metabolites: New findings regarding their production and their radical scavenging actions. Acta Biochim. Pol. 2007, 54, 1–9. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Burkhardt, S.; Sainz, R.M.; Mayo, J.C.; Kohen, R.; Shohami, E.; Huo, Y.S.; Hardeland, R.; Reiter, R.J. N1-acetyl-N2-formyl-5-methoxykynuramine, a biogenic amine and melatonin metabolite, functions as a potent antioxidant. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2001, 15, 2294–2296. [Google Scholar] [CrossRef]
- Ressmeyer, A.R.; Mayo, J.C.; Zelosko, V.; Sáinz, R.M.; Tan, D.X.; Poeggeler, B.; Antolín, I.; Zsizsik, B.K.; Reiter, R.J.; Hardeland, R. Antioxidant properties of the melatonin metabolite N1-acetyl-5-methoxykynuramine (AMK): Scavenging of free radicals and prevention of protein destruction. Redox Rep. Commun. Free Radic. Res. 2003, 8, 205–213. [Google Scholar] [CrossRef]
- Cambiaghi, M.; Cherchi, L.; Comai, S. Photothrombotic Mouse Models for the Study of Melatonin as a Therapeutic Tool After Ischemic Stroke. Methods Mol. Biol. 2022, 2550, 433–441. [Google Scholar] [CrossRef]
- Kaur, T.; Huang, A.C.W.; Shyu, B.C. Modulation of Melatonin in Pain Behaviors Associated with Oxidative Stress and Neuroinflammation Responses in an Animal Model of Central Post-Stroke Pain. Int. J. Mol. Sci. 2023, 24, 5413. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.; Alattar, A.; Tan, Z.; Shah, F.A.; Ali, T.; Alshaman, R.; Koh, P.O.; Li, S. A Potent Antioxidant Endogenous Neurohormone Melatonin, Rescued MCAO by Attenuating Oxidative Stress-Associated Neuroinflammation. Front. Pharmacol. 2020, 11, 1220. [Google Scholar] [CrossRef]
- Candia, A.A.; Arias, P.V.; González-Candia, C.; Navarrete, A.; Ebensperger, G.; Reyes, R.V.; Llanos, A.J.; González-Candia, A.; Herrera, E.A. Melatonin treatment during chronic hypoxic gestation improves neonatal cerebrovascular function. Vascul. Pharmacol. 2022, 144, 106971. [Google Scholar] [CrossRef] [PubMed]
- Saleh, D.O.; Jaleel, G.A.A.; Al-Awdan, S.W.; Hassan, A.; Asaad, G.F. Melatonin suppresses the brain injury after cerebral ischemia/reperfusion in hyperglycaemic rats. Res. Pharm. Sci. 2020, 15, 418–428. [Google Scholar] [CrossRef]
- Zheng, Y.; Gao, N.; Zhang, W.; Ma, R.; Chi, F.; Gao, Z.; Cong, N. Melatonin Alleviates the Oxygen-Glucose Deprivation/Reperfusion-Induced Pyroptosis of HEI-OC1 Cells and Cochlear Hair Cells via MT-1,2/Nrf2 (NFE2L2)/ROS/NLRP3 Pathway. Mol. Neurobiol. 2023, 60, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Mehrpooya, M.; Mazdeh, M.; Rahmani, E.; Khazaie, M.; Ahmadimoghaddam, D. Melatonin supplementation may benefit patients with acute ischemic stroke not eligible for reperfusion therapies: Results of a pilot study. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2022, 106, 66–75. [Google Scholar] [CrossRef]
- Zhao, Z.; Lu, C.; Li, T.; Wang, W.; Ye, W.; Zeng, R.; Ni, L.; Lai, Z.; Wang, X.; Liu, C. The protective effect of melatonin on brain ischemia and reperfusion in rats and humans: In vivo assessment and a randomized controlled trial. J. Pineal Res. 2018, 65, e12521. [Google Scholar] [CrossRef]
- McArthur, A.J.; Lewy, A.J.; Sack, R.L. Non-24-h sleep-wake syndrome in a sighted man: Circadian rhythm studies and efficacy of melatonin treatment. Sleep 1996, 19, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, M.; Okawa, M.; Shibui, K.; Kim, K.; Tagaya, H.; Kudo, Y.; Kamei, Y.; Hayakawa, T.; Urata, J.; Takahashi, K. Altered phase relation between sleep timing and core body temperature rhythm in delayed sleep phase syndrome and non-24-h sleep-wake syndrome in humans. Neurosci. Lett. 2000, 294, 101–104. [Google Scholar] [CrossRef]
- Moran, M.; Lynch, C.A.; Walsh, C.; Coen, R.; Coakley, D.; Lawlor, B.A. Sleep disturbance in mild to moderate Alzheimer’s disease. Sleep Med. 2005, 6, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Socaciu, A.I.; Ionuţ, R.; Socaciu, M.A.; Ungur, A.P.; Bârsan, M.; Chiorean, A.; Socaciu, C.; Râjnoveanu, A.G. Melatonin, an ubiquitous metabolic regulator: Functions, mechanisms and effects on circadian disruption and degenerative diseases. Rev. Endocr. Metab. Disord. 2020, 21, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S. Physiological and pharmacological perspectives of melatonin. Arch. Physiol. Biochem. 2022, 128, 1346–1367. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.V.; Madsen, M.T.; Andersen, L.T.; Hageman, I.; Rasmussen, L.S.; Bokmand, S.; Rosenberg, J.; Gögenur, I. Effect of Melatonin on Cognitive Function and Sleep in relation to Breast Cancer Surgery: A Randomized, Double-Blind, Placebo-Controlled Trial. Int. J. Breast Cancer 2014, 2014, 416531. [Google Scholar] [CrossRef]
- Baandrup, L.; Fagerlund, B.; Glenthoj, B. Neurocognitive performance, subjective well-being, and psychosocial functioning after benzodiazepine withdrawal in patients with schizophrenia or bipolar disorder: A randomized clinical trial of add-on melatonin versus placebo. Eur. Arch. Psychiatry Clin. Neurosci. 2017, 267, 163–171. [Google Scholar] [CrossRef]
- Wade, A.G.; Farmer, M.; Harari, G.; Fund, N.; Laudon, M.; Nir, T.; Frydman-Marom, A.; Zisapel, N. Add-on prolonged-release melatonin for cognitive function and sleep in mild to moderate Alzheimer’s disease: A 6-month, randomized, placebo-controlled, multicenter trial. Clin. Interv. Aging 2014, 9, 947–961. [Google Scholar] [CrossRef]
- Ahmad, S.B.; Ali, A.; Bilal, M.; Rashid, S.M.; Wani, A.B.; Bhat, R.R.; Rehman, M.U. Melatonin and Health: Insights of Melatonin Action, Biological Functions, and Associated Disorders. Cell. Mol. Neurobiol. 2023, 43, 2437–2458. [Google Scholar] [CrossRef]
- Brydon, L.; Petit, L.; de Coppet, P.; Barrett, P.; Morgan, P.J.; Strosberg, A.D.; Jockers, R. Polymorphism and signalling of melatonin receptors. Reprod. Nutr. Dev. 1999, 39, 315–324. [Google Scholar] [CrossRef]
- Jarzynka, M.J.; Passey, D.K.; Ignatius, P.F.; Melan, M.A.; Radio, N.M.; Jockers, R.; Rasenick, M.M.; Brydon, L.; Witt-Enderby, P.A. Modulation of melatonin receptors and G-protein function by microtubules. J. Pineal Res. 2006, 41, 324–336. [Google Scholar] [CrossRef]
- Nosjean, O.; Ferro, M.; Coge, F.; Beauverger, P.; Henlin, J.M.; Lefoulon, F.; Fauchere, J.L.; Delagrange, P.; Canet, E.; Boutin, J.A. Identification of the melatonin-binding site MT3 as the quinone reductase 2. J. Biol. Chem. 2000, 275, 31311–31317. [Google Scholar] [CrossRef]
- Carrillo-Vico, A.; García-Pergañeda, A.; Naji, L.; Calvo, J.R.; Romero, M.P.; Guerrero, J.M. Expression of membrane and nuclear melatonin receptor mRNA and protein in the mouse immune system. Cell. Mol. Life Sci. CMLS 2003, 60, 2272–2278. [Google Scholar] [CrossRef]
- Galano, A.; Medina, M.E.; Tan, D.X.; Reiter, R.J. Melatonin and its metabolites as copper chelating agents and their role in inhibiting oxidative stress: A physicochemical analysis. J. Pineal Res. 2015, 58, 107–116. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Terron, M.P.; Flores, L.J.; Reiter, R.J. One molecule, many derivatives: A never-ending interaction of melatonin with reactive oxygen and nitrogen species? J. Pineal Res. 2007, 42, 28–42. [Google Scholar] [CrossRef] [PubMed]
- García, J.J.; López-Pingarrón, L.; Almeida-Souza, P.; Tres, A.; Escudero, P.; García-Gil, F.A.; Tan, D.X.; Reiter, R.J.; Ramírez, J.M.; Bernal-Pérez, M. Protective effects of melatonin in reducing oxidative stress and in preserving the fluidity of biological membranes: A review. J. Pineal Res. 2014, 56, 225–237. [Google Scholar] [CrossRef]
- Jou, M.J.; Jou, S.B.; Chen, H.M.; Lin, C.H.; Peng, T.I. Critical role of mitochondrial reactive oxygen species formation in visible laser irradiation-induced apoptosis in rat brain astrocytes (RBA-1). J. Biomed. Sci. 2002, 9 Pt 1, 507–516. [Google Scholar] [CrossRef]
- Benítez-King, G.; Ríos, A.; Martínez, A.; Antón-Tay, F. In vitro inhibition of Ca2+/calmodulin-dependent kinase II activity by melatonin. Biochim. Biophys. Acta 1996, 1290, 191–196. [Google Scholar] [CrossRef]
- Liu, J.; Clough, S.J.; Hutchinson, A.J.; Adamah-Biassi, E.B.; Popovska-Gorevski, M.; Dubocovich, M.L. MT1 and MT2 Melatonin Receptors: A Therapeutic Perspective. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 361–383. [Google Scholar] [CrossRef]
- Tricoire, H.; Locatelli, A.; Chemineau, P.; Malpaux, B. Melatonin enters the cerebrospinal fluid through the pineal recess. Endocrinology 2002, 143, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Tricoire, H.; Malpaux, B.; Møller, M. Cellular lining of the sheep pineal recess studied by light-, transmission-, and scanning electron microscopy: Morphologic indications for a direct secretion of melatonin from the pineal gland to the cerebrospinal fluid. J. Comp. Neurol. 2003, 456, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Longatti, P.; Perin, A.; Rizzo, V.; Comai, S.; Giusti, P.; Costa, C.V.L. Ventricular cerebrospinal fluid melatonin concentrations investigated with an endoscopic technique. J. Pineal Res. 2007, 42, 113–118. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Sanchez-Barcelo, E.; Mediavilla, M.D.; Reiter, R.J. Significance of High Levels of Endogenous Melatonin in Mammalian Cerebrospinal Fluid and in the Central Nervous System. Curr. Neuropharmacol. 2010, 8, 162–167. [Google Scholar] [CrossRef]
- Vorhees, C.V.; Williams, M.T. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef]
- Leger, M.; Quiedeville, A.; Bouet, V.; Haelewyn, B.; Boulouard, M.; Schumann-Bard, P.; Freret, T. Object recognition test in mice. Nat. Protoc. 2013, 8, 2531–2537. [Google Scholar] [CrossRef]
- Kraeuter, A.K.; Guest, P.C.; Sarnyai, Z. The Y-Maze for Assessment of Spatial Working and Reference Memory in Mice. Methods Mol. Biol. 2019, 1916, 105–111. [Google Scholar] [CrossRef]
- Haeger, P.; Bouchet, A.; Ossandon, C.; Bresky, G. Treatment with Melatonin Improves Cognitive Behavior and Motor Skills in a Rat Model of Liver Fibrosis. Ann. Hepatol. 2019, 18, 101–108. [Google Scholar] [CrossRef]
- Mansouri, S.; Salari, A.A.; Abedi, A.; Mohammadi, P.; Amani, M. Melatonin treatment improves cognitive deficits by altering inflammatory and neurotrophic factors in the hippocampus of obese mice. Physiol. Behav. 2022, 254, 113919. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Yang, M.; Wang, Y.; Ren, J.; Lin, P.; Cui, C.; Song, J.; He, Q.; Hu, H.; Wang, K.; et al. Melatonin prevents diabetes-associated cognitive dysfunction from microglia-mediated neuroinflammation by activating autophagy via TLR4/Akt/mTOR pathway. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21485. [Google Scholar] [CrossRef]
- Lin, X.J.; Liu, R.; Li, C.; Yi, X.; Fu, B.; Walker, M.J.; Xu, X.M.; Sun, G.; Lin, C.H. Melatonin ameliorates spatial memory and motor deficits via preserving the integrity of cortical and hippocampal dendritic spine morphology in mice with neurotrauma. Inflammopharmacology 2020, 28, 1553–1566. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Li, L.; Zhang, W.; Lu, J.; Wang, Y.; Chen, Q.; Zhang, W.; Chen, M.; Sheng, L.; Cai, K.; et al. Melatonin attenuates repeated mild traumatic brain injury-induced cognitive deficits by inhibiting astrocyte reactivation. Biochem. Biophys. Res. Commun. 2021, 580, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.J.; Wang, M.; Chen, W.H.; Zhu, D.M.; She, J.Q.; Ruan, D.Y. Effects of chronic administration of melatonin on spatial learning ability and long-term potentiation in lead-exposed and control rats. Biomed. Environ. Sci. BES 2009, 22, 70–75. [Google Scholar] [CrossRef]
- Musshoff, U.; Riewenherm, D.; Berger, E.; Fauteck, J.D.; Speckmann, E.J. Melatonin receptors in rat hippocampus: Molecular and functional investigations. Hippocampus 2002, 12, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.; Man, H.Y.; Liu, F.; Braunton, J.; Niznik, H.B.; Pang, S.F.; Brown, G.M.; Wang, Y.T. Differential modulation of GABAA receptor function by Mel1a and Mel1b receptors. Nat. Neurosci. 1999, 2, 401–403. [Google Scholar] [CrossRef] [PubMed]
- Hogan, M.V.; El-Sherif, Y.; Wieraszko, A. The modulation of neuronal activity by melatonin: In vitro studies on mouse hippocampal slices. J. Pineal Res. 2001, 30, 87–96. [Google Scholar] [CrossRef] [PubMed]
- El-Sherif, Y.; Tesoriero, J.; Hogan, M.V.; Wieraszko, A. Melatonin regulates neuronal plasticity in the hippocampus. J. Neurosci. Res. 2003, 72, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Zeise, M.L.; Semm, P. Melatonin lowers excitability of guinea pig hippocampal neurons in vitro. J. Comp. Physiol. A 1985, 157, 23–29. [Google Scholar] [CrossRef]
- Wang, L.M.; Suthana, N.A.; Chaudhury, D.; Weaver, D.R.; Colwell, C.S. Melatonin inhibits hippocampal long-term potentiation. Eur. J. Neurosci. 2005, 22, 2231–2237. [Google Scholar] [CrossRef]
- Pulsinelli, W. Pathophysiology of acute ischaemic stroke. Lancet 1992, 339, 533–536. [Google Scholar] [CrossRef]
- Nelson, C.W.; Wei, E.P.; Povlishock, J.T.; Kontos, H.A.; Moskowitz, M.A. Oxygen radicals in cerebral ischemia. Am. J. Physiol. 1992, 263, H1356–H1362. [Google Scholar] [CrossRef]
- Banjara, M.; Ghosh, C. Sterile Neuroinflammation and Strategies for Therapeutic Intervention. Int. J. Inflamm. 2017, 2017, 8385961. [Google Scholar] [CrossRef]
- Gülke, E.; Gelderblom, M.; Magnus, T. Danger signals in stroke and their role on microglia activation after ischemia. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418774254. [Google Scholar] [CrossRef]
- Chamorro, Á.; Meisel, A.; Planas, A.M.; Urra, X.; van de Beek, D.; Veltkamp, R. The immunology of acute stroke. Nat. Rev. Neurol. 2012, 8, 401–410. [Google Scholar] [CrossRef]
- Wang, Q.; Tang, X.N.; Yenari, M.A. The inflammatory response in stroke. J. Neuroimmunol. 2007, 184, 53–68. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Liu, G.; Tsuruta, Y.; Gao, Z.; Park, Y.J.; Abraham, E. Variant IL-1 receptor-associated kinase-1 mediates increased NF-kappa B activity. J. Immunol. 2007, 179, 4125–4134. [Google Scholar] [CrossRef]
- Chaparro-Huerta, V.; Rivera-Cervantes, M.C.; Flores-Soto, M.E.; Gómez-Pinedo, U.; Beas-Zárate, C. Proinflammatory cytokines and apoptosis following glutamate-induced excitotoxicity mediated by p38 MAPK in the hippocampus of neonatal rats. J. Neuroimmunol. 2005, 165, 53–62. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Alishahi, M.; Farzaneh, M.; Ghaedrahmati, F.; Nejabatdoust, A.; Sarkaki, A.; Khoshnam, S.E. NLRP3 inflammasome in ischemic stroke: As possible therapeutic target. Int. J. Stroke Off. J. Int. Stroke Soc. 2019, 14, 574–591. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef]
- De Vasconcelos, N.M.; Van Opdenbosch, N.; Van Gorp, H.; Parthoens, E.; Lamkanfi, M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. 2019, 26, 146–161. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, D.; Wang, J.; Guo, J.; Li, Y.; Cao, Y.; Zhang, N.; Fu, Y. Melatonin inhibits endoplasmic reticulum stress-associated TXNIP/NLRP3 inflammasome activation in lipopolysaccharide-induced endometritis in mice. Int. Immunopharmacol. 2018, 64, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Stone, W.M.; van Heerden, J.A.; Zimmerman, D. An 8-year-old patient with complicated primary hyperparathyroidism. J. Pediatr. Surg. 1989, 24, 1113–1114. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Shrestha, S.; Li, J.; Yu, X.; Chen, J.; Yan, F.; Ying, G.; Gu, C.; Wang, L.; Chen, G. Melatonin-mediated mitophagy protects against early brain injury after subarachnoid hemorrhage through inhibition of NLRP3 inflammasome activation. Sci. Rep. 2017, 7, 2417. [Google Scholar] [CrossRef] [PubMed]
- Farré-Alins, V.; Narros-Fernández, P.; Palomino-Antolín, A.; Decouty-Pérez, C.; Lopez-Rodriguez, A.B.; Parada, E.; Muñoz-Montero, A.; Gómez-Rangel, V.; López-Muñoz, F.; Ramos, E.; et al. Melatonin Reduces NLRP3 Inflammasome Activation by Increasing α7 nAChR-Mediated Autophagic Flux. Antioxidants 2020, 9, 1299. [Google Scholar] [CrossRef]
- García, J.A.; Volt, H.; Venegas, C.; Doerrier, C.; Escames, G.; López, L.C.; Acuña-Castroviejo, D. Disruption of the NF-κB/NLRP3 connection by melatonin requires retinoid-related orphan receptor-α and blocks the septic response in mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 3863–3875. [Google Scholar] [CrossRef]
- Che, H.; Wang, Y.; Li, H.; Li, Y.; Sahil, A.; Lv, J.; Liu, Y.; Yang, Z.; Dong, R.; Xue, H.; et al. Melatonin alleviates cardiac fibrosis via inhibiting lncRNA MALAT1/miR-141-mediated NLRP3 inflammasome and TGF-β1/Smads signaling in diabetic cardiomyopathy. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 5282–5298. [Google Scholar] [CrossRef]
- Peng, Z.; Zhang, W.; Qiao, J.; He, B. Melatonin attenuates airway inflammation via SIRT1 dependent inhibition of NLRP3 inflammasome and IL-1β in rats with COPD. Int. Immunopharmacol. 2018, 62, 23–28. [Google Scholar] [CrossRef]
- Dubocovich, M.L. Pharmacology and function of melatonin receptors. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1988, 2, 2765–2773. [Google Scholar] [CrossRef]
- Ebisawa, T.; Karne, S.; Lerner, M.R.; Reppert, S.M. Expression cloning of a high-affinity melatonin receptor from Xenopus dermal melanophores. Proc. Natl. Acad. Sci. USA 1994, 91, 6133–6137. [Google Scholar] [CrossRef]
- Molis, T.M.; Spriggs, L.L.; Hill, S.M. Modulation of estrogen receptor mRNA expression by melatonin in MCF-7 human breast cancer cells. Mol. Endocrinol. 1994, 8, 1681–1690. [Google Scholar] [CrossRef]
- Acuña-Castroviejo, D.; Reiter, R.J.; Menéndez-Peláez, A.; Pablos, M.I.; Burgos, A. Characterization of high-affinity melatonin binding sites in purified cell nuclei of rat liver. J. Pineal Res. 1994, 16, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Dubocovich, M.L.; Delagrange, P.; Krause, D.N.; Sugden, D.; Cardinali, D.P.; Olcese, J. International Union of Basic and Clinical Pharmacology. LXXV. Nomenclature, classification, and pharmacology of G protein-coupled melatonin receptors. Pharmacol. Rev. 2010, 62, 343–380. [Google Scholar] [CrossRef] [PubMed]
- Steinhilber, D.; Brungs, M.; Werz, O.; Wiesenberg, I.; Danielsson, C.; Kahlen, J.P.; Nayeri, S.; Schräder, M.; Carlberg, C. The nuclear receptor for melatonin represses 5-lipoxygenase gene expression in human B lymphocytes. J. Biol. Chem. 1995, 270, 7037–7040. [Google Scholar] [CrossRef] [PubMed]
- Benítez-King, G.; Huerto-Delgadillo, L.; Antón-Tay, F. Melatonin modifies calmodulin cell levels in MDCK and N1E-115 cell lines and inhibits phosphodiesterase activity in vitro. Brain Res. 1991, 557, 289–292. [Google Scholar] [CrossRef]
- Benítez-King, G.; Huerto-Delgadillo, L.; Antón-Tay, F. Binding of 3H-melatonin to calmodulin. Life Sci. 1993, 53, 201–207. [Google Scholar] [CrossRef]
- Benítez-King, G.; Antón-Tay, F. Calmodulin mediates melatonin cytoskeletal effects. Experientia 1993, 49, 635–641. [Google Scholar] [CrossRef]
- Domínguez-Alonso, A.; Valdés-Tovar, M.; Solís-Chagoyán, H.; Benítez-King, G. Melatonin stimulates dendrite formation and complexity in the hilar zone of the rat hippocampus: Participation of the Ca++/Calmodulin complex. Int. J. Mol. Sci. 2015, 16, 1907–1927. [Google Scholar] [CrossRef]
- Turjanski, A.G.; Vaqué, J.P.; Gutkind, J.S. MAP kinases and the control of nuclear events. Oncogene 2007, 26, 3240–3253. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin: Signaling mechanisms of a pleiotropic agent. Biofactors 2009, 35, 183–192. [Google Scholar] [CrossRef]
- Dai, J.; Inscho, E.W.; Yuan, L.; Hill, S.M. Modulation of intracellular calcium and calmodulin by melatonin in MCF-7 human breast cancer cells. J. Pineal Res. 2002, 32, 112–119. [Google Scholar] [CrossRef]
- Skelding, K.A.; Rostas, J.A.P. Regulation of CaMKII in vivo: The importance of targeting and the intracellular microenvironment. Neurochem. Res. 2009, 34, 1792–1804. [Google Scholar] [CrossRef]
- Babu, Y.S.; Bugg, C.E.; Cook, W.J. Structure of calmodulin refined at 2.2 A resolution. J. Mol. Biol. 1988, 204, 191–204. [Google Scholar] [CrossRef]
- Colbran, R.J.; Brown, A.M. Calcium/calmodulin-dependent protein kinase II and synaptic plasticity. Curr. Opin. Neurobiol. 2004, 14, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Araki, S.; Osuka, K.; Takata, T.; Tsuchiya, Y.; Watanabe, Y. Coordination between Calcium/Calmodulin-Dependent Protein Kinase II and Neuronal Nitric Oxide Synthase in Neurons. Int. J. Mol. Sci. 2020, 21, 7997. [Google Scholar] [CrossRef] [PubMed]
- Fink, C.C.; Meyer, T. Molecular mechanisms of CaMKII activation in neuronal plasticity. Curr. Opin. Neurobiol. 2002, 12, 293–299. [Google Scholar] [CrossRef]
- Tao-Cheng, J.H.; Dosemeci, A.; Winters, C.A.; Reese, T.S. Changes in the distribution of calcium calmodulin-dependent protein kinase II at the presynaptic bouton after depolarization. Brain Cell Biol. 2006, 35, 117–124. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, B.; Ge, Q.; Wang, Z.W. Presynaptic Ca2+/calmodulin-dependent protein kinase II modulates neurotransmitter release by activating BK channels at Caenorhabditis elegans neuromuscular junction. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 10404–10413. [Google Scholar] [CrossRef] [PubMed]
- Strack, S.; Choi, S.; Lovinger, D.M.; Colbran, R.J. Translocation of autophosphorylated calcium/calmodulin-dependent protein kinase II to the postsynaptic density. J. Biol. Chem. 1997, 272, 13467–13470. [Google Scholar] [CrossRef]
- Fong, D.K.; Rao, A.; Crump, F.T.; Craig, A.M. Rapid synaptic remodeling by protein kinase C: Reciprocal translocation of NMDA receptors and calcium/calmodulin-dependent kinase II. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 2153–2164. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R. Antioxidative protection by melatonin: Multiplicity of mechanisms from radical detoxification to radical avoidance. Endocrine 2005, 27, 119–130. [Google Scholar] [CrossRef]
- Reiter, R.J.; Paredes, S.D.; Manchester, L.C.; Tan, D.X. Reducing oxidative/nitrosative stress: A newly-discovered genre for melatonin. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 175–200. [Google Scholar] [CrossRef]
- Camp, O.G.; Bai, D.; Awonuga, A.; Goud, P.T.; Abu-Soud, H.M. Hypochlorous acid facilitates inducible nitric oxide synthase subunit dissociation: The link between heme destruction, disturbance of the zinc-tetrathiolate center, and the prevention by melatonin. Nitric Oxide Biol. Chem. 2022, 124, 32–38. [Google Scholar] [CrossRef]
- Galijasevic, S.; Abdulhamid, I.; Abu-Soud, H.M. Melatonin is a potent inhibitor for myeloperoxidase. Biochemistry 2008, 47, 2668–2677. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Galijasevic, S.; Abdulhamid, I.; Abu-Soud, H.M. Analysis of the mechanism by which melatonin inhibits human eosinophil peroxidase. Br. J. Pharmacol. 2008, 154, 1308–1317. [Google Scholar] [CrossRef] [PubMed]
- Gilad, E.; Cuzzocrea, S.; Zingarelli, B.; Salzman, A.L.; Szabó, C. Melatonin is a scavenger of peroxynitrite. Life Sci. 1997, 60, PL169–PL174. [Google Scholar] [CrossRef] [PubMed]
- Gulcin, I.; Buyukokuroglu, M.E.; Kufrevioglu, O.I. Metal chelating and hydrogen peroxide scavenging effects of melatonin. J. Pineal Res. 2003, 34, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Xie, Q.W. Regulation of biosynthesis of nitric oxide. J. Biol. Chem. 1994, 269, 13725–13728. [Google Scholar] [CrossRef]
- Stuehr, D.J.; Haque, M.M. Nitric oxide synthase enzymology in the 20 years after the Nobel Prize. Br. J. Pharmacol. 2019, 176, 177–188. [Google Scholar] [CrossRef]
- Abu-Soud, H.M.; Loftus, M.; Stuehr, D.J. Subunit dissociation and unfolding of macrophage NO synthase: Relationship between enzyme structure, prosthetic group binding, and catalytic function. Biochemistry 1995, 34, 11167–11175. [Google Scholar] [CrossRef]
- Li, H.; Raman, C.S.; Glaser, C.B.; Blasko, E.; Young, T.A.; Parkinson, J.F.; Whitlow, M.; Poulos, T.L. Crystal structures of zinc-free and -bound heme domain of human inducible nitric-oxide synthase. Implications for dimer stability and comparison with endothelial nitric-oxide synthase. J. Biol. Chem. 1999, 274, 21276–21284. [Google Scholar] [CrossRef]
- Knowles, R.G.; Moncada, S. Nitric oxide synthases in mammals. Biochem. J. 1994, 298 Pt 2, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Stuehr, D.J. Structure-function aspects in the nitric oxide synthases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 339–359. [Google Scholar] [CrossRef] [PubMed]
- Garthwaite, J. NO as a multimodal transmitter in the brain: Discovery and current status. Br. J. Pharmacol. 2019, 176, 197–211. [Google Scholar] [CrossRef]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357 Pt 3, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflug. Arch. 2010, 460, 525–542. [Google Scholar] [CrossRef] [PubMed]
- Kashfi, K.; Kannikal, J.; Nath, N. Macrophage Reprogramming and Cancer Therapeutics: Role of iNOS-Derived NO. Cells 2021, 10, 3194. [Google Scholar] [CrossRef]
- Murphy, M.P. Nitric oxide and cell death. Biochim. Biophys. Acta 1999, 1411, 401–414. [Google Scholar] [CrossRef]
- Shi, Y.; Liu, H.; Liu, H.; Yu, Y.; Zhang, J.; Li, Y.; Luo, G.; Zhang, X.; Xu, N. Increased expression levels of inflammatory cytokines and adhesion molecules in lipopolysaccharide-induced acute inflammatory apoM-/- mice. Mol. Med. Rep. 2020, 22, 3117–3126. [Google Scholar] [CrossRef]
- Blacher, E.; Tsai, C.; Litichevskiy, L.; Shipony, Z.; Iweka, C.A.; Schneider, K.M.; Chuluun, B.; Heller, H.C.; Menon, V.; Thaiss, C.A.; et al. Aging disrupts circadian gene regulation and function in macrophages. Nat. Immunol. 2022, 23, 229–236. [Google Scholar] [CrossRef]
- Rahman, S.A.; Castanon-Cervantes, O.; Scheer, F.A.J.L.; Shea, S.A.; Czeisler, C.A.; Davidson, A.J.; Lockley, S.W. Endogenous circadian regulation of pro-inflammatory cytokines and chemokines in the presence of bacterial lipopolysaccharide in humans. Brain. Behav. Immun. 2015, 47, 4–13. [Google Scholar] [CrossRef]
- Schmidt, H.H.; Walter, U. NO at work. Cell 1994, 78, 919–925. [Google Scholar] [CrossRef]
- Geiger, S.S.; Curtis, A.M.; O’Neill, L.A.J.; Siegel, R.M. Daily variation in macrophage phagocytosis is clock-independent and dispensable for cytokine production. Immunology 2019, 157, 122–136. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef]
- Benoit, M.; Desnues, B.; Mege, J.L. Macrophage polarization in bacterial infections. J. Immunol. 2008, 181, 3733–3739. [Google Scholar] [CrossRef]
- Harry, G.J. Microglia during development and aging. Pharmacol. Ther. 2013, 139, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Rodríguez, G.; Ortíz-López, L.; Domínguez-Alonso, A.; Benítez-King, G.A.; Kempermann, G. Chronic treatment with melatonin stimulates dendrite maturation and complexity in adult hippocampal neurogenesis of mice. J. Pineal Res. 2011, 50, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Rodríguez, G.; Klempin, F.; Babu, H.; Benítez-King, G.; Kempermann, G. Melatonin modulates cell survival of new neurons in the hippocampus of adult mice. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2009, 34, 2180–2191. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Haba, M.G.; Garcia-Mauriño, S.; Calvo, J.R.; Goberna, R.; Guerrero, J.M. High-affinity binding of melatonin by human circulating T lymphocytes (CD4+). FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1995, 9, 1331–1335. [Google Scholar] [CrossRef]
- Maestroni, G.J. T-helper-2 lymphocytes as a peripheral target of melatonin. J. Pineal Res. 1995, 18, 84–89. [Google Scholar] [CrossRef]
- Cuzzocrea, S.; Zingarelli, B.; Gilad, E.; Hake, P.; Salzman, A.L.; Szabó, C. Protective effect of melatonin in carrageenan-induced models of local inflammation: Relationship to its inhibitory effect on nitric oxide production and its peroxynitrite scavenging activity. J. Pineal Res. 1997, 23, 106–116. [Google Scholar] [CrossRef]
- Sonoli, S.S.; Shivprasad, S.; Prasad, C.V.B.; Patil, A.B.; Desai, P.B.; Somannavar, M.S. Visfatin—A review. Eur. Rev. Med. Pharmacol. Sci. 2011, 15, 9–14. [Google Scholar] [PubMed]
- Luk, T.; Malam, Z.; Marshall, J.C. Pre-B cell colony-enhancing factor (PBEF)/visfatin: A novel mediator of innate immunity. J. Leukoc. Biol. 2008, 83, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.R.; Gerner, R.R.; Tilg, H. Pre-B cell colony enhancing factor/NAMPT/visfatin in inflammation and obesity-related disorders. Curr. Pharm. Des. 2010, 16, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Ok, C.Y.; Park, S.; Jang, H.O.; Takata, T.; Bae, M.K.; Kim, Y.D.; Ryu, M.H.; Bae, S.K. Visfatin Induces Senescence of Human Dental Pulp Cells. Cells 2020, 9, 193. [Google Scholar] [CrossRef]
- Anderson, G.; Reiter, R.J. Melatonin: Roles in influenza, COVID-19, and other viral infections. Rev. Med. Virol. 2020, 30, e2109. [Google Scholar] [CrossRef]
- Bald, E.M.; Nance, C.S.; Schultz, J.L. Melatonin may slow disease progression in amyotrophic lateral sclerosis: Findings from the Pooled Resource Open-Access ALS Clinic Trials database. Muscle Nerve 2021, 63, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A. Antioxidants and Neuron-Astrocyte Interplay in Brain Physiology: Melatonin, a Neighbor to Rely on. Neurochem. Res. 2021, 46, 34–50. [Google Scholar] [CrossRef]
- Guo, Y.L.; Wei, X.J.; Zhang, T.; Sun, T. Molecular mechanisms of melatonin-induced alleviation of synaptic dysfunction and neuroinflammation in Parkinson’s disease: A review. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 5070–5082. [Google Scholar] [CrossRef]
- Hardeland, R.; Cardinali, D.P.; Brown, G.M.; Pandi-Perumal, S.R. Melatonin and brain inflammaging. Prog. Neurobiol. 2015, 127–128, 46–63. [Google Scholar] [CrossRef]
- Mauriz, J.L.; Collado, P.S.; Veneroso, C.; Reiter, R.J.; González-Gallego, J. A review of the molecular aspects of melatonin’s anti-inflammatory actions: Recent insights and new perspectives. J. Pineal Res. 2013, 54, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Naseem, M.; Parvez, S. Role of melatonin in traumatic brain injury and spinal cord injury. ScientificWorldJournal 2014, 2014, 586270. [Google Scholar] [CrossRef]
- Paprocka, J.; Kijonka, M.; Rzepka, B.; Sokół, M. Melatonin in Hypoxic-Ischemic Brain Injury in Term and Preterm Babies. Int. J. Endocrinol. 2019, 2019, 9626715. [Google Scholar] [CrossRef]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.R.; Robitaille, R.; Volterra, A. Gliotransmitters travel in time and space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Dallérac, G.; Rouach, N. Astrocytes as new targets to improve cognitive functions. Prog. Neurobiol. 2016, 144, 48–67. [Google Scholar] [CrossRef] [PubMed]
- Simard, M.; Nedergaard, M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience 2004, 129, 877–896. [Google Scholar] [CrossRef]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef]
- Geyer, S.; Jacobs, M.; Hsu, N.J. Immunity Against Bacterial Infection of the Central Nervous System: An Astrocyte Perspective. Front. Mol. Neurosci. 2019, 12, 57. [Google Scholar] [CrossRef]
- Klegeris, A. Targeting neuroprotective functions of astrocytes in neuroimmune diseases. Expert Opin. Ther. Targets 2021, 25, 237–241. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Patabendige, A.; Singh, A.; Jenkins, S.; Sen, J.; Chen, R. Astrocyte Activation in Neurovascular Damage and Repair Following Ischaemic Stroke. Int. J. Mol. Sci. 2021, 22, 4280. [Google Scholar] [CrossRef]
- Guttenplan, K.A.; Weigel, M.K.; Prakash, P.; Wijewardhane, P.R.; Hasel, P.; Rufen-Blanchette, U.; Münch, A.E.; Blum, J.A.; Fine, J.; Neal, M.C.; et al. Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature 2021, 599, 102–107. [Google Scholar] [CrossRef]
- Huang, J.; Li, C.; Shang, H. Astrocytes in Neurodegeneration: Inspiration From Genetics. Front. Neurosci. 2022, 16, 882316. [Google Scholar] [CrossRef] [PubMed]
- Qian, K.; Jiang, X.; Liu, Z.Q.; Zhang, J.; Fu, P.; Su, Y.; Brazhe, N.A.; Liu, D.; Zhu, L.Q. Revisiting the critical roles of reactive astrocytes in neurodegeneration. Mol. Psychiatry 2023. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.Y.; Huo, J. A1/A2 astrocytes in central nervous system injuries and diseases: Angels or devils? Neurochem. Int. 2021, 148, 105080. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Gliem, M.; Krammes, K.; Liaw, L.; van Rooijen, N.; Hartung, H.P.; Jander, S. Macrophage-derived osteopontin induces reactive astrocyte polarization and promotes re-establishment of the blood brain barrier after ischemic stroke. Glia 2015, 63, 2198–2207. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, G.R.; Ding, S. Reactive astrocytes and therapeutic potential in focal ischemic stroke. Neurobiol. Dis. 2016, 85, 234–244. [Google Scholar] [CrossRef]
- Li, H.; Zhang, N.; Lin, H.Y.; Yu, Y.; Cai, Q.Y.; Ma, L.; Ding, S. Histological, cellular and behavioral assessments of stroke outcomes after photothrombosis-induced ischemia in adult mice. BMC Neurosci. 2014, 15, 58. [Google Scholar] [CrossRef]
- Li, D.; He, T.; Zhang, Y.; Liu, J.; Zhao, H.; Wang, D.; Wang, Q.; Yuan, Y.; Zhang, S. Melatonin regulates microglial polarization and protects against ischemic stroke-induced brain injury in mice. Exp. Neurol. 2023, 367, 114464. [Google Scholar] [CrossRef] [PubMed]
- Yawoot, N.; Sengking, J.; Wicha, P.; Govitrapong, P.; Tocharus, C.; Tocharus, J. Melatonin attenuates reactive astrogliosis and glial scar formation following cerebral ischemia and reperfusion injury mediated by GSK-3β and RIP1K. J. Cell. Physiol. 2022, 237, 1818–1832. [Google Scholar] [CrossRef]
- Li, H.; Wang, Y.; Feng, D.; Liu, Y.; Xu, M.; Gao, A.; Tian, F.; Zhang, L.; Cui, Y.; Wang, Z.; et al. Alterations in the time course of expression of the Nox family in the brain in a rat experimental cerebral ischemia and reperfusion model: Effects of melatonin. J. Pineal Res. 2014, 57, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Ballester, C.; Robel, S. Astrocyte-mediated mechanisms contribute to traumatic brain injury pathology. WIREs Mech. Dis. 2023, 15, e1622. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.L.; Li, S.S.; Fan, Y.J.; Qi, M.M.; Li, Z.Z. Melatonin alleviates traumatic brain injury-induced anxiety-like behaviors in rats: Roles of the protein kinase A/cAMP-response element binding signaling pathway. Exp. Ther. Med. 2022, 23, 248. [Google Scholar] [CrossRef]
- Babaee, A.; Eftekhar-Vaghefi, S.H.; Asadi-Shekaari, M.; Shahrokhi, N.; Soltani, S.D.; Malekpour-Afshar, R.; Basiri, M. Melatonin treatment reduces astrogliosis and apoptosis in rats with traumatic brain injury. Iran. J. Basic Med. Sci. 2015, 18, 867–872. [Google Scholar]
- Yang, Z.; Bao, Y.; Chen, W.; He, Y. Melatonin exerts neuroprotective effects by attenuating astro- and microgliosis and suppressing inflammatory response following spinal cord injury. Neuropeptides 2020, 79, 102002. [Google Scholar] [CrossRef]
- Phatnani, H.; Maniatis, T. Astrocytes in neurodegenerative disease. Cold Spring Harb. Perspect. Biol. 2015, 7, a020628. [Google Scholar] [CrossRef]
- Li, Q.; Haney, M.S. The role of glia in protein aggregation. Neurobiol. Dis. 2020, 143, 105015. [Google Scholar] [CrossRef]
- Pathak, D.; Sriram, K. Neuron-astrocyte omnidirectional signaling in neurological health and disease. Front. Mol. Neurosci. 2023, 16, 1169320. [Google Scholar] [CrossRef] [PubMed]
- Knight, E.; Geetha, T.; Broderick, T.L.; Babu, J.R. The Role of Dietary Antioxidants and Their Potential Mechanisms in Alzheimer’s Disease Treatment. Metabolites 2023, 13, 438. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Wan, J.; Liu, A.; Sun, J. Melatonin regulates Aβ production/clearance balance and Aβ neurotoxicity: A potential therapeutic molecule for Alzheimer’s disease. Biomed. Pharmacother. 2020, 132, 110887. [Google Scholar] [CrossRef]
- Mack, J.M.; Schamne, M.G.; Sampaio, T.B.; Pértile, R.A.N.; Fernandes, P.A.C.M.; Markus, R.P.; Prediger, R.D. Melatoninergic System in Parkinson’s Disease: From Neuroprotection to the Management of Motor and Nonmotor Symptoms. Oxid. Med. Cell. Longev. 2016, 2016, 3472032. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Jurado, A.; Escribano, B.M.; Caballero-Villarraso, J.; Galván, A.; Agüera, E.; Santamaría, A.; Túnez, I. Melatonin and multiple sclerosis: Antioxidant, anti-inflammatory and immunomodulator mechanism of action. Inflammopharmacology 2022, 30, 1569–1596. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G. Amyotrophic Lateral Sclerosis Pathoetiology and Pathophysiology: Roles of Astrocytes, Gut Microbiome, and Muscle Interactions via the Mitochondrial Melatonergic Pathway, with Disruption by Glyphosate-Based Herbicides. Int. J. Mol. Sci. 2022, 24, 587. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, P.; Ren, L.; Hu, C.; Bi, J. Protective effect of melatonin on soluble Aβ1-42-induced memory impairment, astrogliosis, and synaptic dysfunction via the Musashi1/Notch1/Hes1 signaling pathway in the rat hippocampus. Alzheimers Res. Ther. 2016, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Andrade, M.K.; Souza, L.C.; Azevedo, E.M.; Bail, E.L.; Zanata, S.M.; Andreatini, R.; Vital, M.A.B.F. Melatonin reduces β-amyloid accumulation and improves short-term memory in streptozotocin-induced sporadic Alzheimer’s disease model. IBRO Neurosci. Rep. 2023, 14, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Labunets, I.; Rodnichenko, A.; Savosko, S.; Pivneva, T. Reaction of different cell types of the brain on neurotoxin cuprizone and hormone melatonin treatment in young and aging mice. Front. Cell. Neurosci. 2023, 17, 1131130. [Google Scholar] [CrossRef]
- Xi, Y.; Liu, M.; Xu, S.; Hong, H.; Chen, M.; Tian, L.; Xie, J.; Deng, P.; Zhou, C.; Zhang, L.; et al. Inhibition of SERPINA3N-dependent neuroinflammation is essential for melatonin to ameliorate trimethyltin chloride-induced neurotoxicity. J. Pineal Res. 2019, 67, e12596. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Liu, X.; Wu, J.; Yan, B.; Zhang, Y.; Lu, Y.; Wu, Y.; Liu, C.; Guo, J.; Nanberg, E.; et al. Cognitive deficits and anxiety induced by diisononyl phthalate in mice and the neuroprotective effects of melatonin. Sci. Rep. 2015, 5, 14676. [Google Scholar] [CrossRef]
- Lwin, T.; Yang, J.L.; Ngampramuan, S.; Viwatpinyo, K.; Chancharoen, P.; Veschsanit, N.; Pinyomahakul, J.; Govitrapong, P.; Mukda, S. Melatonin ameliorates methamphetamine-induced cognitive impairments by inhibiting neuroinflammation via suppression of the TLR4/MyD88/NFκB signaling pathway in the mouse hippocampus. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 111, 110109. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef] [PubMed]
- Favero, G.; Franceschetti, L.; Bonomini, F.; Rodella, L.F.; Rezzani, R. Melatonin as an Anti-Inflammatory Agent Modulating Inflammasome Activation. Int. J. Endocrinol. 2017, 2017, 1835195. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R. Aging, Melatonin, and the Pro- and Anti-Inflammatory Networks. Int. J. Mol. Sci. 2019, 20, 1223. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef]
- Williams, L.; Bradley, L.; Smith, A.; Foxwell, B. Signal transducer and activator of transcription 3 is the dominant mediator of the anti-inflammatory effects of IL-10 in human macrophages. J. Immunol. 2004, 172, 567–576. [Google Scholar] [CrossRef]
- Azedi, F.; Mehrpour, M.; Talebi, S.; Zendedel, A.; Kazemnejad, S.; Mousavizadeh, K.; Beyer, C.; Zarnani, A.H.; Joghataei, M.T. Melatonin regulates neuroinflammation ischemic stroke damage through interactions with microglia in reperfusion phase. Brain Res. 2019, 1723, 146401. [Google Scholar] [CrossRef]
- Chumboatong, W.; Khamchai, S.; Tocharus, C.; Govitrapong, P.; Tocharus, J. Agomelatine Exerts an Anti-inflammatory Effect by Inhibiting Microglial Activation Through TLR4/NLRP3 Pathway in pMCAO Rats. Neurotox. Res. 2022, 40, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Suofu, Y.; Jauhari, A.; Nirmala, E.S.; Mullins, W.A.; Wang, X.; Li, F.; Carlisle, D.L.; Friedlander, R.M. Neuronal melatonin type 1 receptor overexpression promotes M2 microglia polarization in cerebral ischemia/reperfusion-induced injury. Neurosci. Lett. 2023, 795, 137043. [Google Scholar] [CrossRef]
- Ding, K.; Wang, H.; Xu, J.; Lu, X.; Zhang, L.; Zhu, L. Melatonin reduced microglial activation and alleviated neuroinflammation induced neuron degeneration in experimental traumatic brain injury: Possible involvement of mTOR pathway. Neurochem. Int. 2014, 76, 23–31. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, C.; Zhang, K.; Lan, X.; Chen, X.; Zang, W.; Wang, Z.; Guan, F.; Zhu, C.; Yang, X.; et al. Melatonin receptor activation provides cerebral protection after traumatic brain injury by mitigating oxidative stress and inflammation via the Nrf2 signaling pathway. Free Radic. Biol. Med. 2019, 131, 345–355. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Z.; Zhang, W.; Wu, Q.; Zhang, Y.; Liu, Y.; Guan, Y.; Chen, X. Melatonin improves functional recovery in female rats after acute spinal cord injury by modulating polarization of spinal microglial/macrophages. J. Neurosci. Res. 2019, 97, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Song, G.J.; Suk, K. Pharmacological Modulation of Functional Phenotypes of Microglia in Neurodegenerative Diseases. Front. Aging Neurosci. 2017, 9, 139. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, H.; Wang, X.; Xia, Y.; Huang, J.; Wang, T.; Lin, Z.; Xiong, N. Melatonin ameliorates Parkinson’s disease via regulating microglia polarization in a RORα-dependent pathway. NPJ Park. Dis. 2022, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Meng, J.; Zhu, Y.; Ding, M.; Zhang, Y.; Zhou, J. Melatonin enhances SIRT1 to ameliorate mitochondrial membrane damage by activating PDK1/Akt in granulosa cells of PCOS. J. Ovarian Res. 2021, 14, 152. [Google Scholar] [CrossRef]
- Caruso, G.I.; Spampinato, S.F.; Costantino, G.; Merlo, S.; Sortino, M.A. SIRT1-Dependent Upregulation of BDNF in Human Microglia Challenged with Aβ: An Early but Transient Response Rescued by Melatonin. Biomedicines 2021, 9, 466. [Google Scholar] [CrossRef]
- Das, R.; Balmik, A.A.; Chinnathambi, S. Melatonin Reduces GSK3β-Mediated Tau Phosphorylation, Enhances Nrf2 Nuclear Translocation and Anti-Inflammation. ASN Neuro 2020, 12, 1759091420981204. [Google Scholar] [CrossRef]
- Campbell, B.C.V.; De Silva, D.A.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic stroke. Nat. Rev. Dis. Primer 2019, 5, 70. [Google Scholar] [CrossRef]
- Acciarresi, M.; Bogousslavsky, J.; Paciaroni, M. Post-stroke fatigue: Epidemiology, clinical characteristics and treatment. Eur. Neurol. 2014, 72, 255–261. [Google Scholar] [CrossRef]
- Cohen, D.L.; Roffe, C.; Beavan, J.; Blackett, B.; Fairfield, C.A.; Hamdy, S.; Havard, D.; McFarlane, M.; McLauglin, C.; Randall, M.; et al. Post-stroke dysphagia: A review and design considerations for future trials. Int. J. Stroke Off. J. Int. Stroke Soc. 2016, 11, 399–411. [Google Scholar] [CrossRef]
- Guo, J.; Wang, J.; Sun, W.; Liu, X. The advances of post-stroke depression: 2021 update. J. Neurol. 2022, 269, 1236–1249. [Google Scholar] [CrossRef]
- Kalaria, R.N.; Akinyemi, R.; Ihara, M. Stroke injury, cognitive impairment and vascular dementia. Biochim. Biophys. Acta 2016, 1862, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Ihara, M. Post-stroke epilepsy. Neurochem. Int. 2017, 107, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Tater, P.; Pandey, S. Post-stroke Movement Disorders: Clinical Spectrum, Pathogenesis, and Management. Neurol. India 2021, 69, 272–283. [Google Scholar] [CrossRef]
- Klit, H.; Finnerup, N.B.; Jensen, T.S. Central post-stroke pain: Clinical characteristics, pathophysiology, and management. Lancet Neurol. 2009, 8, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Kaur, T.; Shih, H.C.; Huang, A.C.W.; Shyu, B.C. Modulation of melatonin to the thalamic lesion-induced pain and comorbid sleep disturbance in the animal model of the central post-stroke hemorrhage. Mol. Pain 2022, 18, 17448069221127180. [Google Scholar] [CrossRef]
- Rancan, L.; Paredes, S.D.; García, C.; González, P.; Rodríguez-Bobada, C.; Calvo-Soto, M.; Hyacinthe, B.; Vara, E.; Tresguerres, J.A.F. Comparison of the Effect of Melatonin Treatment before and after Brain Ischemic Injury in the Inflammatory and Apoptotic Response in Aged Rats. Int. J. Mol. Sci. 2018, 19, 2097. [Google Scholar] [CrossRef]
- Nair, S.M.; Rahman, R.M.A.; Clarkson, A.N.; Sutherland, B.A.; Taurin, S.; Sammut, I.A.; Appleton, I. Melatonin treatment following stroke induction modulates L-arginine metabolism. J. Pineal Res. 2011, 51, 313–323. [Google Scholar] [CrossRef]
- Bseiso, E.A.; AbdEl-Aal, S.A.; Nasr, M.; Sammour, O.A.; El Gawad, N.A.A. Nose to brain delivery of melatonin lipidic nanocapsules as a promising post-ischemic neuroprotective therapeutic modality. Drug Deliv. 2022, 29, 2469–2480. [Google Scholar] [CrossRef]
- Molina-Carballo, A.; Palacios-López, R.; Jerez-Calero, A.; Augustín-Morales, M.C.; Agil, A.; Muñoz-Hoyos, A.; Muñoz-Gallego, A. Protective Effect of Melatonin Administration against SARS-CoV-2 Infection: A Systematic Review. Curr. Issues Mol. Biol. 2021, 44, 31–45. [Google Scholar] [CrossRef]
- Ramos, E.; López-Muñoz, F.; Gil-Martín, E.; Egea, J.; Álvarez-Merz, I.; Painuli, S.; Semwal, P.; Martins, N.; Hernández-Guijo, J.M.; Romero, A. The Coronavirus Disease 2019 (COVID-19): Key Emphasis on Melatonin Safety and Therapeutic Efficacy. Antioxidants 2021, 10, 1152. [Google Scholar] [CrossRef] [PubMed]
- Renn, T.Y.; Yang, C.P.; Wu, U.I.; Chen, L.Y.; Mai, F.D.; Tikhonova, M.A.; Amstislavskaya, T.G.; Liao, W.C.; Lin, C.T.; Liu, Y.C.; et al. Water composed of reduced hydrogen bonds activated by localized surface plasmon resonance effectively enhances anti-viral and anti-oxidative activities of melatonin. Chem. Eng. J. 2022, 427, 131626. [Google Scholar] [CrossRef]
- Tan, D.X.; Hardeland, R. Potential utility of melatonin in deadly infectious diseases related to the overreaction of innate immune response and destructive inflammation: Focus on COVID-19. Melatonin Res. 2020, 3, 120–143. [Google Scholar] [CrossRef]
- Wongchitrat, P.; Yasawong, M.; Jumpathong, W.; Chanmanee, T.; Samutpong, A.; Dangsakul, W.; Govitrapong, P.; Reiter, R.J.; Puthavathana, P. Melatonin inhibits Zika virus replication in Vero epithelial cells and SK-N-SH human neuroblastoma cells. Melatonin Res. 2022, 5, 171–185. [Google Scholar] [CrossRef]
- Tan, D.X.; Korkmaz, A.; Reiter, R.J.; Manchester, L.C. Ebola virus disease: Potential use of melatonin as a treatment. J. Pineal Res. 2014, 57, 381–384. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carretero, V.J.; Ramos, E.; Segura-Chama, P.; Hernández, A.; Baraibar, A.M.; Álvarez-Merz, I.; Muñoz, F.L.; Egea, J.; Solís, J.M.; Romero, A.; et al. Non-Excitatory Amino Acids, Melatonin, and Free Radicals: Examining the Role in Stroke and Aging. Antioxidants 2023, 12, 1844. https://doi.org/10.3390/antiox12101844
Carretero VJ, Ramos E, Segura-Chama P, Hernández A, Baraibar AM, Álvarez-Merz I, Muñoz FL, Egea J, Solís JM, Romero A, et al. Non-Excitatory Amino Acids, Melatonin, and Free Radicals: Examining the Role in Stroke and Aging. Antioxidants. 2023; 12(10):1844. https://doi.org/10.3390/antiox12101844
Chicago/Turabian StyleCarretero, Victoria Jiménez, Eva Ramos, Pedro Segura-Chama, Adan Hernández, Andrés M Baraibar, Iris Álvarez-Merz, Francisco López Muñoz, Javier Egea, José M. Solís, Alejandro Romero, and et al. 2023. "Non-Excitatory Amino Acids, Melatonin, and Free Radicals: Examining the Role in Stroke and Aging" Antioxidants 12, no. 10: 1844. https://doi.org/10.3390/antiox12101844