
Nimbolide Inhibits SOD2 to Control Pancreatic Ductal Adenocarcinoma Growth and Metastasis
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Immunohistochemistry
2.3. Determining SOD2-Induced Alteration in Tumor Using In Silico Analysis
2.4. Survival Analysis
2.5. Immunofluorescence Analysis
2.6. Immunoblotting
2.7. Silencing SOD2 in Pancreatic Cell Line
2.8. Overexpression of SOD2 in Pancreatic Cell Line
2.9. SOD2 Zymography
2.10. Cell Proliferation Using MTS Assay
2.11. ROS Generation Assay
2.12. Transwell Migration and Matrigel Invasion Assay
2.13. Soft Agar Colony Formation Assay
2.14. Wound-Healing Assay
2.15. Apoptosis Analysis and Flow Cytometry
2.16. Xenograft Model
2.17. Statistical Analysis
3. Results
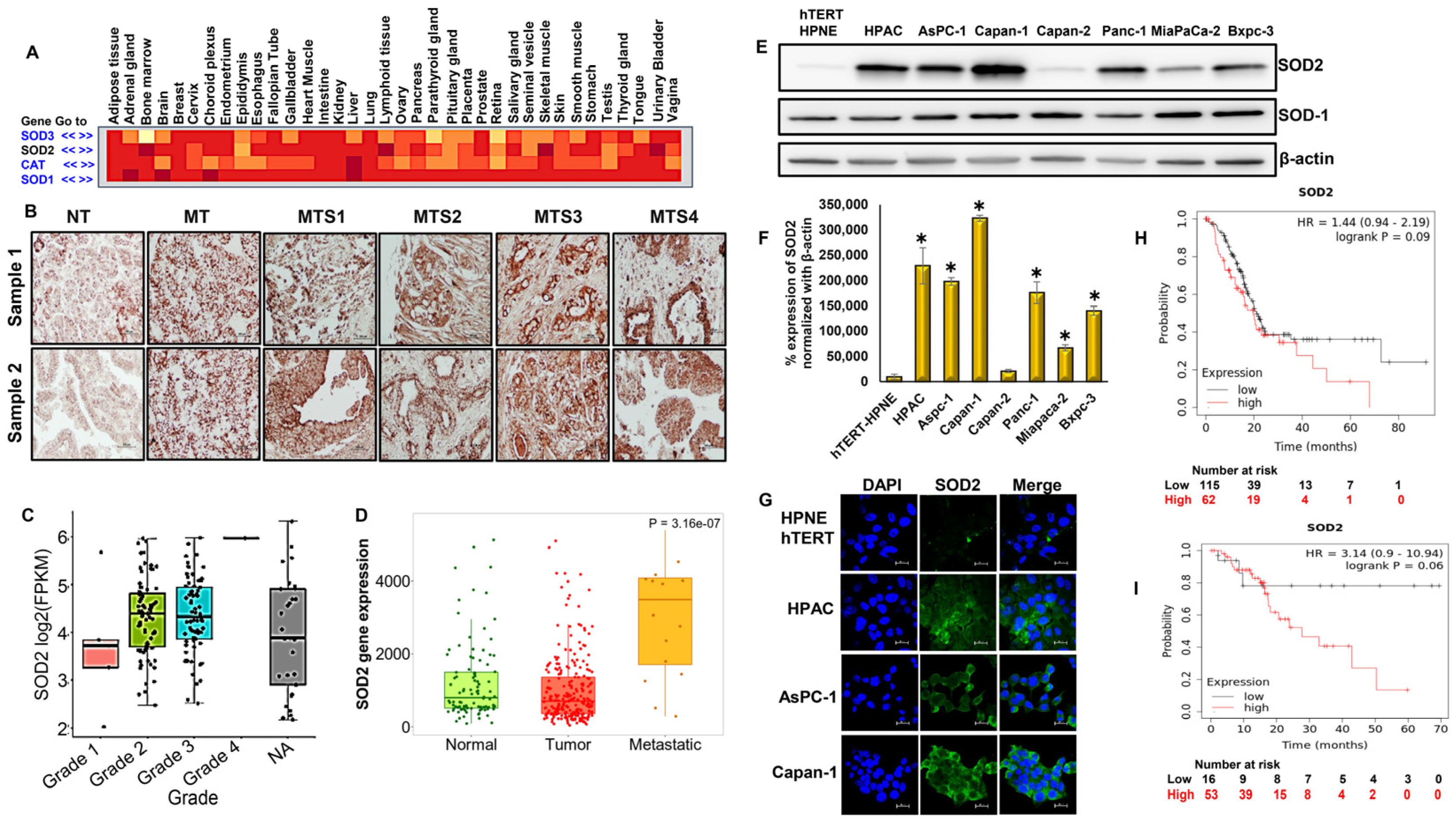
3.1. Abundant Expression of Superoxide Dismutase-2 (SOD2) in PDACs
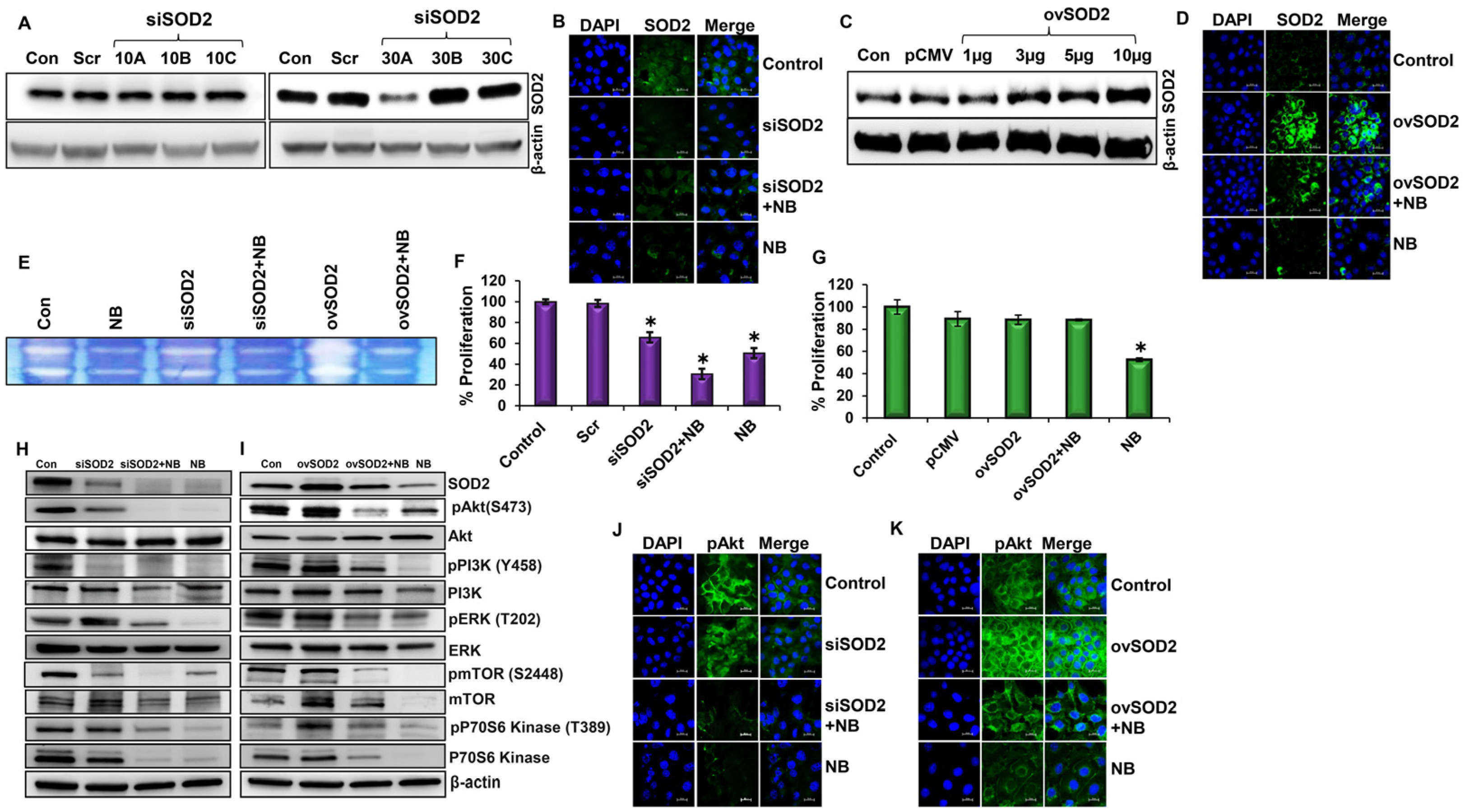
3.2. SOD2 Influences Cell Survival and Proliferation through PI3K/Akt/mTOR Pathway in Pancreatic Cancer
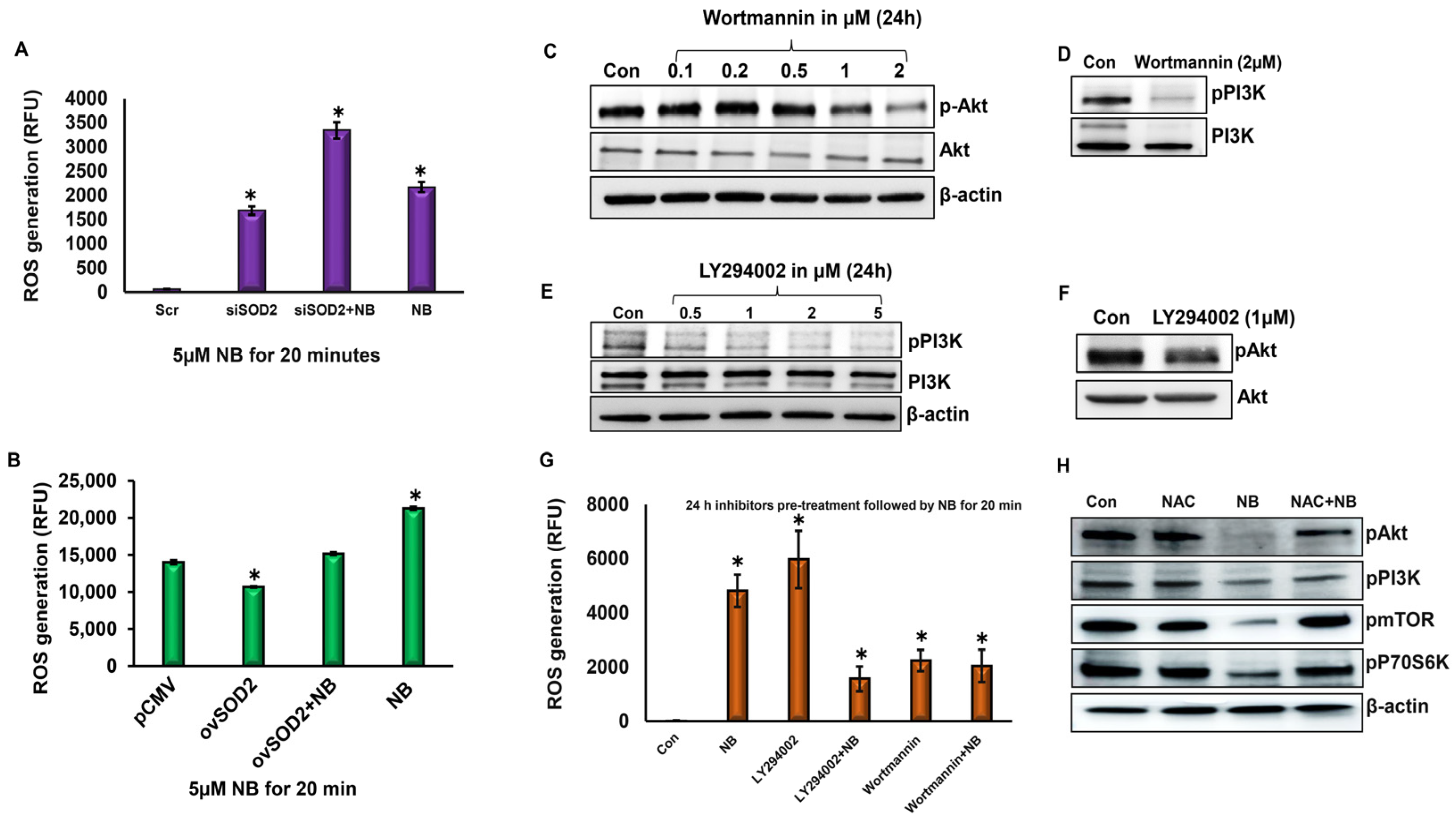
3.3. Inhibition of PI3K/Akt Signaling by NB Is Caused by the Induction of ROS via SOD2 Silencing
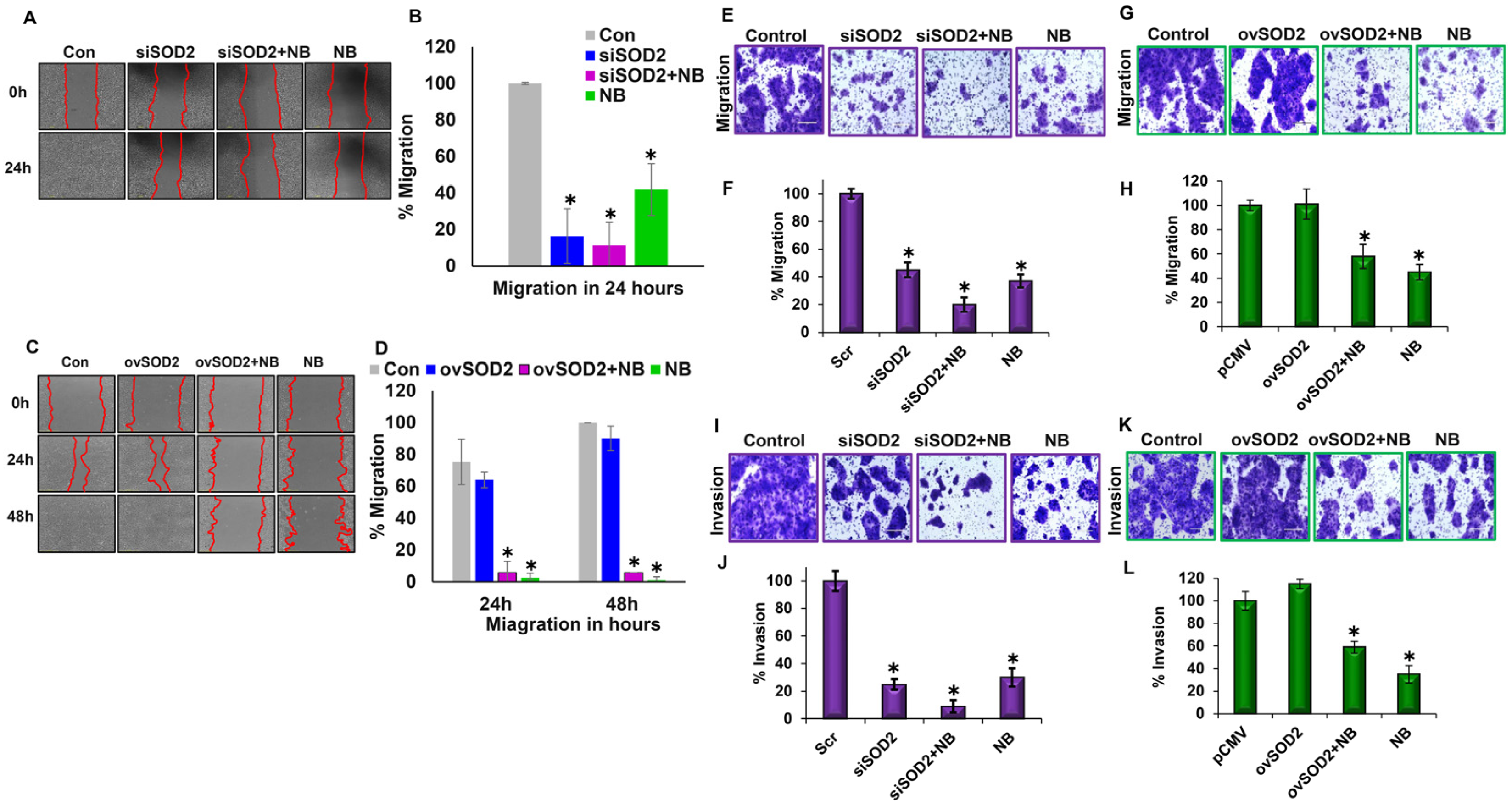
3.4. Differential SOD2 Expression Levels Influence Migration and Invasion Capabilities
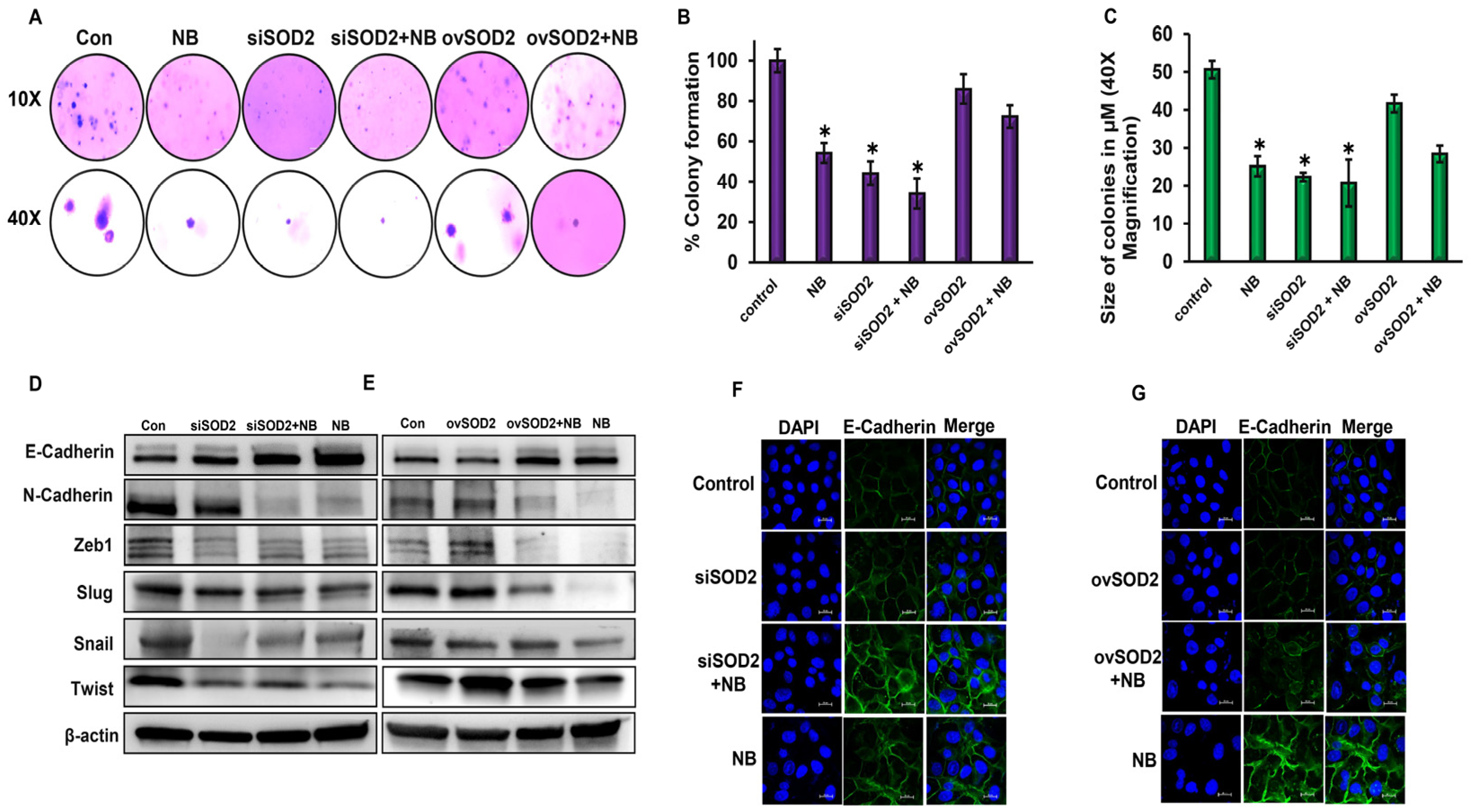
3.5. SOD2 Regulates Anchorage-Independent Growth of Pancreatic Cancer Cells
3.6. SOD2 Inhibits Epithelial–Mesenchymal Transition of PDAC Cells
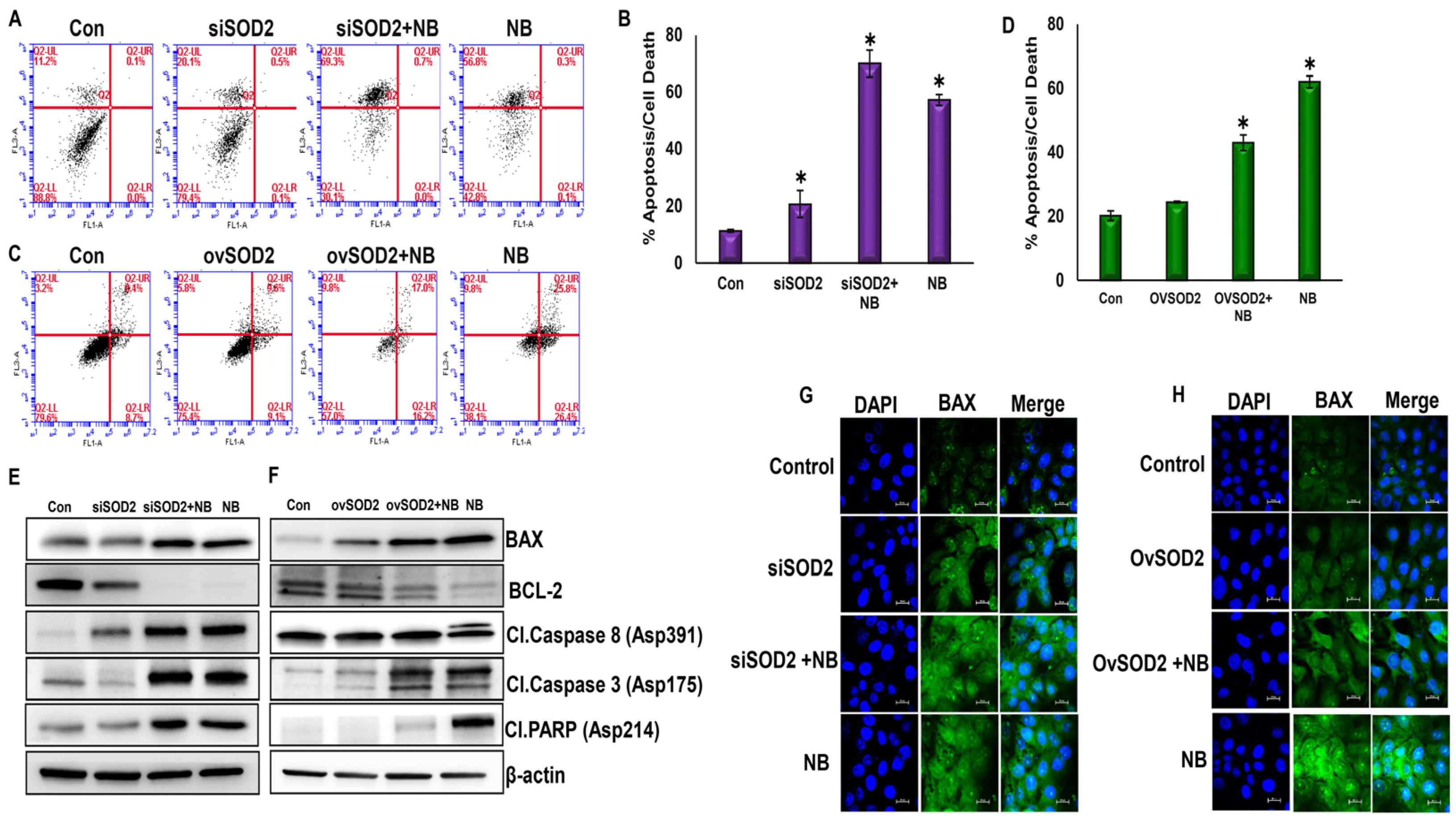
3.7. NB-Induced ROS Regulate Apoptosis through SOD2 in PDAC Cells
3.8. Impact of SOD2 Expression on Tumor Growth, Proliferation, Epithelial-to-Mesenchymal Transition, and Apoptosis in Pancreatic Cancer Xenograft Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kikuyama, M.; Kamisawa, T.; Kuruma, S.; Chiba, K.; Kawaguchi, S.; Terada, S.; Satoh, T. Early Diagnosis to Improve the Poor Prognosis of Pancreatic Cancer. Cancers 2018, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Takesue, S.; Ohuchida, K.; Shinkawa, T.; Otsubo, Y.; Matsumoto, S.; Sagara, A.; Yonenaga, A.; Ando, Y.; Kibe, S.; Nakayama, H.; et al. Neutrophil extracellular traps promote liver micrometastasis in pancreatic ductal adenocarcinoma via the activation of cancer-associated fibroblasts. Int. J. Oncol. 2020, 56, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Caldas, C.; Kern, S.E. K-ras mutation and pancreatic adenocarcinoma. Int. J. Pancreatol. 1995, 18, 1–6. [Google Scholar] [CrossRef]
- Wilkes, J.G.; Alexander, M.S.; Cullen, J.J. Superoxide Dismutases in Pancreatic Cancer. Antioxidants 2017, 6, 66. [Google Scholar] [CrossRef]
- Liou, G.Y.; Doppler, H.; DelGiorno, K.E.; Zhang, L.; Leitges, M.; Crawford, H.C.; Murphy, M.P.; Storz, P. Mutant KRas-Induced Mitochondrial Oxidative Stress in Acinar Cells Upregulates EGFR Signaling to Drive Formation of Pancreatic Precancerous Lesions. Cell Rep. 2016, 14, 2325–2336. [Google Scholar] [CrossRef]
- Indo, H.P.; Hawkins, C.L.; Nakanishi, I.; Matsumoto, K.I.; Matsui, H.; Suenaga, S.; Davies, M.J.; St Clair, D.K.; Ozawa, T.; Majima, H.J. Role of Mitochondrial Reactive Oxygen Species in the Activation of Cellular Signals, Molecules, and Function. Handb. Exp. Pharmacol. 2017, 240, 439–456. [Google Scholar] [CrossRef]
- Marengo, B.; Nitti, M.; Furfaro, A.L.; Colla, R.; Ciucis, C.D.; Marinari, U.M.; Pronzato, M.A.; Traverso, N.; Domenicotti, C. Redox Homeostasis and Cellular Antioxidant Systems: Crucial Players in Cancer Growth and Therapy. Oxid. Med. Cell Longev. 2016, 2016, 6235641. [Google Scholar] [CrossRef]
- Mittler, R. ROS Are Good. Trends Plant. Sci. 2017, 22, 11–19. [Google Scholar] [CrossRef]
- Veeraraghavan, J.; Natarajan, M.; Lagisetty, P.; Awasthi, V.; Herman, T.S.; Aravindan, N. Impact of curcumin, raspberry extract, and neem leaf extract on rel protein-regulated cell death/radiosensitization in pancreatic cancer cells. Pancreas 2011, 40, 1107–1119. [Google Scholar] [CrossRef]
- Eleutherio, E.C.A.; Silva Magalhaes, R.S.; de Araujo Brasil, A.; Monteiro Neto, J.R.; de Holanda Paranhos, L. SOD1, more than just an antioxidant. Arch. Biochem. Biophys. 2021, 697, 108701. [Google Scholar] [CrossRef]
- Natu, J.; Nagaraju, G.P. Gemcitabine effects on tumor microenvironment of pancreatic ductal adenocarcinoma: Special focus on resistance mechanisms and metronomic therapies. Cancer Lett. 2023, 573, 216382. [Google Scholar] [CrossRef]
- Maity, P.; Biswas, K.; Chattopadhyay, I.; Banerjee, R.K.; Bandyopadhyay, U. The use of neem for controlling gastric hyperacidity and ulcer. Phytother. Res. 2009, 23, 747–755. [Google Scholar] [CrossRef]
- Mahomoodally, M.F.; Protab, K.; Aumeeruddy, M.Z. Medicinal plants brought by Indian indentured immigrants: A comparative review of ethnopharmacological uses between Mauritius and India. J. Ethnopharmacol. 2019, 234, 245–289. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Wang, R.; Bian, P.; Wu, Q.; Seshadri, V.D.D.; Liu, L. Evaluation of antiarthritic activity of nimbolide against Freund’s adjuvant induced arthritis in rats. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3391–3398. [Google Scholar] [CrossRef]
- Wang, L.; Phan, D.D.; Zhang, J.; Ong, P.S.; Thuya, W.L.; Soo, R.; Wong, A.L.; Yong, W.P.; Lee, S.C.; Ho, P.C.; et al. Anticancer properties of nimbolide and pharmacokinetic considerations to accelerate its development. Oncotarget 2016, 7, 44790–44802. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, V.D.; Singh, A.; El-Kafraway, S.A.; Alandijany, T.A.; Faizo, A.A.; Bajrai, L.H.; Kamal, M.A.; Azhar, E.I. Mechanistic insights into the Japanese encephalitis virus RNA dependent RNA polymerase protein inhibition by bioflavonoids from Azadirachta indica. Sci. Rep. 2021, 11, 18125. [Google Scholar] [CrossRef]
- Dallaqua, B.; Saito, F.H.; Rodrigues, T.; Calderon, I.M.; Rudge, M.V.; Herrera, E.; Damasceno, D.C. Treatment with Azadirachta indica in diabetic pregnant rats: Negative effects on maternal outcome. J. Ethnopharmacol. 2012, 143, 805–811. [Google Scholar] [CrossRef]
- Suresh, G.; Gopalakrishnan, G.; Wesley, S.D.; Pradeep Singh, N.D.; Malathi, R.; Rajan, S.S. Insect antifeedant activity of tetranortriterpenoids from the Rutales. A perusal of structural relations. J. Agric. Food Chem. 2002, 50, 4484–4490. [Google Scholar] [CrossRef] [PubMed]
- Rochanakij, S.; Thebtaranonth, Y.; Yenjai, C.; Yuthavong, Y. Nimbolide, a constituent of Azadirachta indica, inhibits Plasmodium falciparum in culture. Southeast Asian J. Trop. Med. Public Health 1985, 16, 66–72. [Google Scholar] [PubMed]
- Tasanarong, T.; Patntirapong, S.; Aupaphong, V. The inhibitory effect of a novel neem paste against cariogenic bacteria. J. Clin. Exp. Dent. 2021, 13, e1083–e1088. [Google Scholar] [CrossRef]
- Udeinya, I.J.; Mbah, A.U.; Chijioke, C.P.; Shu, E.N. An antimalarial extract from neem leaves is antiretroviral. Trans. R. Soc. Trop. Med. Hyg. 2004, 98, 435–437. [Google Scholar] [CrossRef]
- Cohen, E.; Quistad, G.B.; Casida, J.E. Cytotoxicity of nimbolide, epoxyazadiradione and other limonoids from neem insecticide. Life Sci. 1996, 58, 1075–1081. [Google Scholar] [CrossRef]
- Gupta, S.C.; Prasad, S.; Tyagi, A.K.; Kunnumakkara, A.B.; Aggarwal, B.B. Neem (Azadirachta indica): An indian traditional panacea with modern molecular basis. Phytomedicine 2017, 34, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Bodduluru, L.N.; Kasala, E.R.; Thota, N.; Barua, C.C.; Sistla, R. Chemopreventive and therapeutic effects of nimbolide in cancer: The underlying mechanisms. Toxicol. Vitr. 2014, 28, 1026–1035. [Google Scholar] [CrossRef]
- Kashif, M.; Hwang, Y.; Hong, G.; Kim, G. In vitro comparative cytotoxic effect of Nimbolide: A limonoid from Azadirachta indica (Neem tree) on cancer cell lines and normal cell lines through MTT assay. Pak. J. Pharm. Sci. 2017, 30, 967–973. [Google Scholar] [PubMed]
- Subramani, R.; Gonzalez, E.; Arumugam, A.; Nandy, S.; Gonzalez, V.; Medel, J.; Camacho, F.; Ortega, A.; Bonkoungou, S.; Narayan, M.; et al. Nimbolide inhibits pancreatic cancer growth and metastasis through ROS-mediated apoptosis and inhibition of epithelial-to-mesenchymal transition. Sci. Rep. 2016, 6, 19819. [Google Scholar] [CrossRef]
- Subramani, R.; Camacho, F.A.; Levin, C.I.; Flores, K.; Clift, A.; Galvez, A.; Terres, M.; Rivera, S.; Kolli, S.N.; Dodderer, J.; et al. FOXC1 plays a crucial role in the growth of pancreatic cancer. Oncogenesis 2018, 7, 52. [Google Scholar] [CrossRef]
- Subramani, R.; Gonzalez, E.; Nandy, S.B.; Arumugam, A.; Camacho, F.; Medel, J.; Alabi, D.; Lakshmanaswamy, R. Gedunin inhibits pancreatic cancer by altering sonic hedgehog signaling pathway. Oncotarget 2017, 8, 10891–10904. [Google Scholar] [CrossRef]
- Subramani, R.; Medel, J.; Flores, K.; Perry, C.; Galvez, A.; Sandoval, M.; Rivera, S.; Pedroza, D.A.; Penner, E.; Chitti, M.; et al. Hepatocyte nuclear factor 1 alpha influences pancreatic cancer growth and metastasis. Sci. Rep. 2020, 10, 20225. [Google Scholar] [CrossRef]
- Subramani, R.; Lopez-Valdez, R.; Arumugam, A.; Nandy, S.; Boopalan, T.; Lakshmanaswamy, R. Targeting insulin-like growth factor 1 receptor inhibits pancreatic cancer growth and metastasis. PLoS ONE 2014, 9, e97016. [Google Scholar] [CrossRef] [PubMed]
- Subramani, R.; Lopez-Valdez, R.; Salcido, A.; Boopalan, T.; Arumugam, A.; Nandy, S.; Lakshmanaswamy, R. Growth hormone receptor inhibition decreases the growth and metastasis of pancreatic ductal adenocarcinoma. Exp. Mol. Med. 2014, 46, e117. [Google Scholar] [CrossRef]
- Pan, J.S.; Hong, M.Z.; Ren, J.L. Reactive oxygen species: A double-edged sword in oncogenesis. World J. Gastroenterol. 2009, 15, 1702–1707. [Google Scholar] [CrossRef]
- Mateen, S.; Moin, S.; Khan, A.Q.; Zafar, A.; Fatima, N. Increased Reactive Oxygen Species Formation and Oxidative Stress in Rheumatoid Arthritis. PLoS ONE 2016, 11, e0152925. [Google Scholar] [CrossRef]
- Kim, Y.S.; Gupta Vallur, P.; Phaeton, R.; Mythreye, K.; Hempel, N. Insights into the Dichotomous Regulation of SOD2 in Cancer. Antioxidants 2017, 6, 86. [Google Scholar] [CrossRef] [PubMed]
- Chien, S.Y.; Hsu, C.H.; Lin, C.C.; Chuang, Y.C.; Lo, Y.S.; Hsi, Y.T.; Hsieh, M.J.; Chen, M.K. Nimbolide induces apoptosis in human nasopharyngeal cancer cells. Env. Toxicol. 2017, 32, 2085–2092. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef]
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; Gonzalez-Baron, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, H.; Yang, Y.; Liao, J.; Yang, G.Y. Knockdown or inhibition of aldo-keto reductase 1B10 inhibits pancreatic carcinoma growth via modulating Kras-E-cadherin pathway. Cancer Lett. 2014, 355, 273–280. [Google Scholar] [CrossRef] [PubMed]
- San Martin, A.; Griendling, K.K. Redox control of vascular smooth muscle migration. Antioxid. Redox Signal. 2010, 12, 625–640. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Feng, J.; Li, Y.; Guan, D.; Chen, H.; Zhai, X.; Zhang, L.; Li, C.; Li, C. The vicious cycle between ferritinophagy and ROS production triggered EMT inhibition of gastric cancer cells was through p53/AKT/mTor pathway. Chem. Biol. Interact. 2020, 328, 109196. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehmetoglu-Gurbuz, T.; Lakshmanaswamy, R.; Perez, K.; Sandoval, M.; Jimenez, C.A.; Rocha, J.; Goldfarb, R.M.; Perry, C.; Bencomo, A.; Neela, N.; et al. Nimbolide Inhibits SOD2 to Control Pancreatic Ductal Adenocarcinoma Growth and Metastasis. Antioxidants 2023, 12, 1791. https://doi.org/10.3390/antiox12101791
Mehmetoglu-Gurbuz T, Lakshmanaswamy R, Perez K, Sandoval M, Jimenez CA, Rocha J, Goldfarb RM, Perry C, Bencomo A, Neela N, et al. Nimbolide Inhibits SOD2 to Control Pancreatic Ductal Adenocarcinoma Growth and Metastasis. Antioxidants. 2023; 12(10):1791. https://doi.org/10.3390/antiox12101791
Chicago/Turabian StyleMehmetoglu-Gurbuz, Tugba, Rajkumar Lakshmanaswamy, Karla Perez, Mayra Sandoval, Casandra A. Jimenez, Jackelyn Rocha, Rachel Madeline Goldfarb, Courtney Perry, Alejandra Bencomo, Nishkala Neela, and et al. 2023. "Nimbolide Inhibits SOD2 to Control Pancreatic Ductal Adenocarcinoma Growth and Metastasis" Antioxidants 12, no. 10: 1791. https://doi.org/10.3390/antiox12101791