
Ishige okamurae Attenuates Neuroinflammation and Cognitive Deficits in Mice Intracerebroventricularly Injected with LPS via Regulating TLR-4/MyD88-Dependent Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animal Experiments
2.3. Y-Maze Test
2.4. Morris Water Maze Test
2.5. Tissue Staining
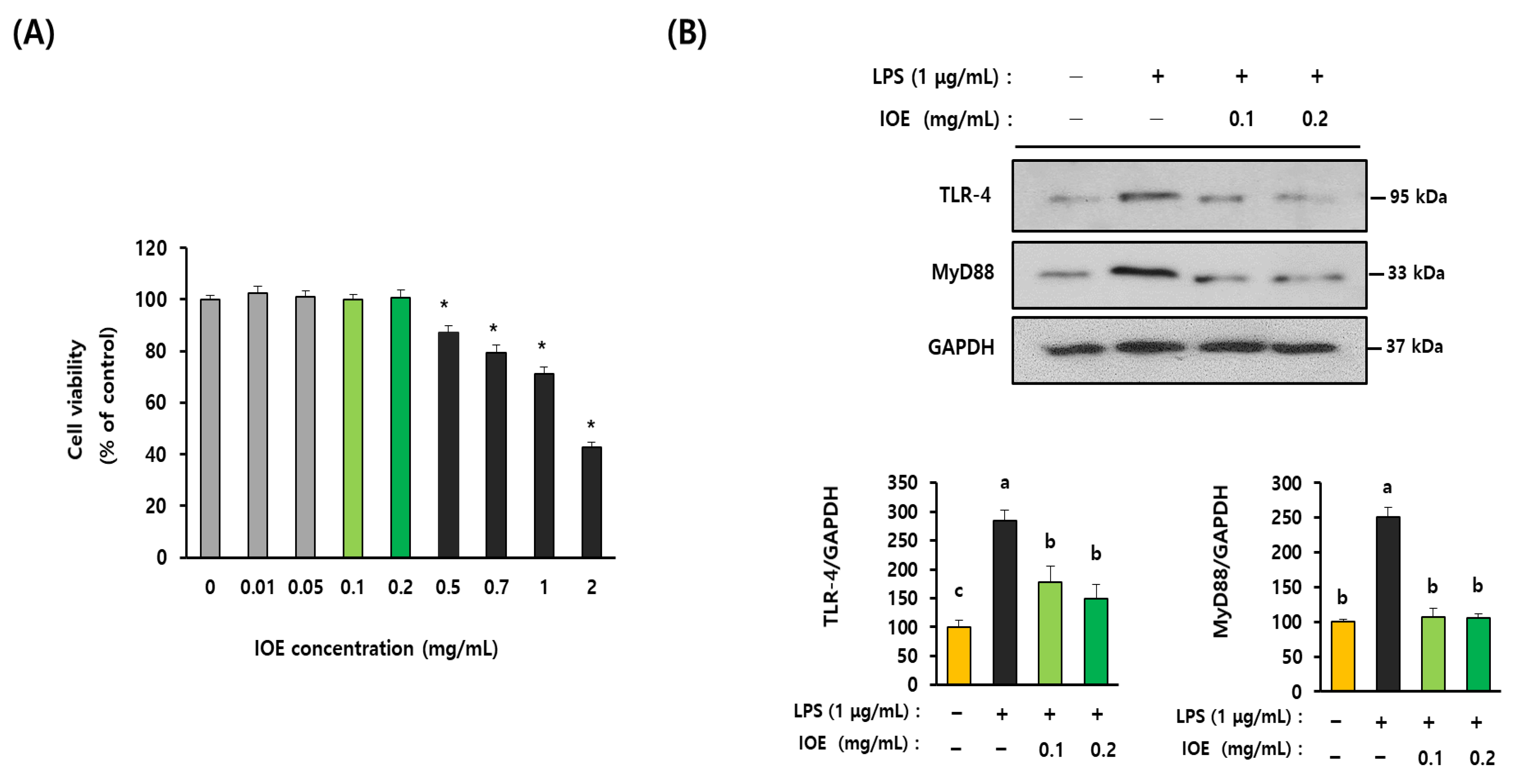
2.6. Cell Culture and Cell Viability Assay
2.7. Nitric Oxide Assay
2.8. Reactive Oxygen Species Measurements
2.9. Real-Time Polymerase Chain Reaction
2.10. Western Blot Analysis
2.11. Statistical Analysis
3. Results
3.1. Oral Administration of Ishige okamurae Extract (IOE) Attenuated LPS-Induced Cognitive Deficits
3.2. LPS-Induced Neuronal Loss and Neuroinflammation in Mouse Brains Were Attenuated by IOE Administration
3.3. IOE Administration Attenuated the LPS-Induced Aβ Plaque Formation in Mouse Brains
3.4. Activation of TLR-4 and its Downstream Signaling Pathway in LPS-Injected Mouse Brains Was Downregulated by Oral Administration of IOE
3.5. IOE Attenuated LPS-Induced TLR-4 Activation in C6 Glioma Cells
3.6. LPS-Mediated Expression of Inflammation Mediators Was Inhibited by IOE Treatment in C6 Glioma Cells
3.7. IOE Has Equivalent Efficacy with the MyD88 Inhibitor in the Inhibition of LPS-Mediated Inflammatory Responses
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, X.; Zhang, J.; Xia, X.; Li, H.; Qiu, C.; Liao, Y.; Chen, H.; He, Z.; Song, Z.; et al. Activated STAT3 signaling pathway by ligature-induced periodontitis could contribute to neuroinflammation and cognitive impairment in rats. J. Neuroinflammation 2021, 18, 80. [Google Scholar] [CrossRef]
- Joffre, J.; Hellman, J. Oxidative Stress and Endothelial Dysfunction in Sepsis and Acute Inflammation. Antioxid. Redox Signal 2021, 35, 1291–1307. [Google Scholar] [CrossRef]
- Qin, Z.; Zhou, C.; Xiao, X.; Guo, C. Metformin attenuates sepsis-induced neuronal injury and cognitive impairment. BMC Neurosci. 2021, 22, 78. [Google Scholar] [CrossRef]
- Jiang, C.; Li, G.; Huang, P.; Liu, Z.; Zhao, B. The Gut Microbiota and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 1–15. [Google Scholar] [CrossRef]
- Fulop, T.; Witkowski, J.M.; Bourgade, K.; Khalil, A.; Zerif, E.; Larbi, A.; Hirokawa, K.; Pawelec, G.; Bocti, C.; Lacombe, G.; et al. Can an Infection Hypothesis Explain the Beta Amyloid Hypothesis of Alzheimer’s Disease? Front. Aging Neurosci. 2018, 10, 224. [Google Scholar] [CrossRef]
- Brown, G.C. The endotoxin hypothesis of neurodegeneration. J. Neuroinflammation 2019, 16, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Miller, R.G.; Gascon, R.; Champion, S.; Katz, J.; Lancero, M.; Narvaez, A.; Honrada, R.; Ruvalcaba, D.; McGrath, M.S. Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis (sALS). J. Neuroimmunol. 2009, 206, 121–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Joo, B.; Nam, J.H.; Nam, H.Y.; Lee, W.; Nam, Y.; Seo, Y.; Kang, H.J.; Cho, H.J.; Jang, Y.P.; et al. An Aqueous Extract of Herbal Medicine ALWPs Enhances Cognitive Performance and Inhibits LPS-Induced Neuroinflammation via FAK/NF-kappaB Signaling Pathways. Front. Aging Neurosci. 2018, 10, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H.; et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 2019, 9, 5790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitner, G.R.; Wenzel, T.J.; Marshall, N.; Gates, E.J.; Klegeris, A. Targeting toll-like receptor 4 to modulate neuroinflammation in central nervous system disorders. Expert Opin. Ther. Targets 2019, 23, 865–882. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhang, J.G.; Yang, W.; Xu, P.; Xiao, Y.L.; Zhang, H.T. 6-Gingerol attenuates LPS-induced neuroinflammation and cognitive impairment partially via suppressing astrocyte overactivation. Biomed. Pharmacother. 2018, 107, 1523–1529. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. TLR signaling pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef]
- Park, M.H.; Jeon, Y.J.; Kim, H.J.; Han, J.S. Effect of diphlorethohydroxycarmalol isolated from Ishige okamurae on apoptosis in 3 T3-L1 preadipocytes. Phytother. Res. 2013, 27, 931–936. [Google Scholar] [CrossRef]
- Yang, H.W.; Fernando, K.H.N.; Oh, J.Y.; Li, X.; Jeon, Y.J.; Ryu, B. Anti-Obesity and Anti-Diabetic Effects of Ishige okamurae. Mar. Drugs 2019, 17, 202. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.M.; Rajapakse, N.; Kim, S.K. Anti-inflammatory effect of Ishige okamurae ethanolic extract via inhibition of NF-kappaB transcription factor in RAW 264.7 cells. Phytother. Res. 2009, 23, 628–634. [Google Scholar] [CrossRef]
- Min, K.H.; Kim, H.J.; Jeon, Y.J.; Han, J.S. Ishige okamurae ameliorates hyperglycemia and insulin resistance in C57BL/KsJ-db/db mice. Diabetes Res. Clin. Pract. 2011, 93, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Cho, C.; Lee, C.; Ryu, B.; Kim, S.; Hur, J.; Lee, S.H. Ishige okamurae Ameliorates Methylglyoxal-Induced Nephrotoxicity via Reducing Oxidative Stress, RAGE Protein Expression, and Modulating MAPK, Nrf2/ARE Signaling Pathway in Mouse Glomerular Mesangial Cells. Foods 2021, 10, 2000. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.Y.; Lee, S.H. Ameliorating Activity of Ishige okamurae on the Amyloid Beta-Induced Cognitive Deficits and Neurotoxicity through Regulating ERK, p38 MAPK, and JNK Signaling in Alzheimer’s Disease-Like Mice Model. Mol. Nutr. Food Res. 2020, 64, e19012202020-64. [Google Scholar] [CrossRef]
- Kwon, O.Y.; Lee, S.H. Ishige okamurae Suppresses Trimethyltin-Induced Neurodegeneration and Glutamate-Mediated Excitotoxicity by Regulating MAPKs/Nrf2/HO-1 Antioxidant Pathways. Antioxidants 2021, 10, 440. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, D.K.; Chung, B.R.; Kim, H.V.; Kim, Y. Intracerebroventricular Injection of Amyloid-beta Peptides in Normal Mice to Acutely Induce Alzheimer-like Cognitive Deficits. J. Vis. Exp. 2016, 109, e533082016-109. [Google Scholar] [CrossRef] [Green Version]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [Green Version]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef] [Green Version]
- Sochocka, M.; Zwolinska, K.; Leszek, J. The Infectious Etiology of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 996–1009. [Google Scholar] [CrossRef] [Green Version]
- Giau, V.V.; Wu, S.Y.; Jamerlan, A.; An, S.S.A.; Kim, S.Y.; Hulme, J. Gut Microbiota and Their Neuroinflammatory Implications in Alzheimer’s Disease. Nutrients 2018, 10, 1765. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, A.B.; Crean, S.; Olsen, I.; Singhrao, S.K. Periodontitis, Microbiomes and their Role in Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 336. [Google Scholar] [CrossRef]
- Walter, S.; Letiembre, M.; Liu, Y.; Heine, H.; Penke, B.; Hao, W.; Bode, B.; Manietta, N.; Walter, J.; Schulz-Schuffer, W.; et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol. Biochem. 2007, 20, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Balducci, C.; Frasca, A.; Zotti, M.; La Vitola, P.; Mhillaj, E.; Grigoli, E.; Iacobellis, M.; Grandi, F.; Messa, M.; Colombo, L.; et al. Toll-like receptor 4-dependent glial cell activation mediates the impairment in memory establishment induced by beta-amyloid oligomers in an acute mouse model of Alzheimer’s disease. Brain Behav. Immun. 2017, 60, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, R.; Fusco, R.; Cuzzocrea, S. Astrocytes: Role and Functions in Brain Pathologies. Front. Pharmacol. 2019, 10, 1114. [Google Scholar] [CrossRef] [Green Version]
- Gorina, R.; Font-Nieves, M.; Marquez-Kisinousky, L.; Santalucia, T.; Planas, A.M. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFkappaB signaling, MAPK, and Jak1/Stat1 pathways. Glia 2011, 59, 242–255. [Google Scholar] [CrossRef]
- Garwood, C.J.; Ratcliffe, L.E.; Simpson, J.E.; Heath, P.R.; Ince, P.G.; Wharton, S.B. Review: Astrocytes in Alzheimer’s disease and other age-associated dementias: A supporting player with a central role. Neuropathol. Appl. Neurobiol. 2017, 43, 281–298. [Google Scholar] [CrossRef]
- Tao, F.; Zhu, J.; Duan, L.; Wu, J.; Zhang, J.; Yao, K.; Bo, J.; Zu, H. Anti-inflammatory effects of doxepin hydrochloride against LPS-induced C6-glioma cell inflammatory reaction by PI3K-mediated Akt signaling. J. Biochem. Mol. Toxicol. 2020, 34, e224242020-34. [Google Scholar] [CrossRef]
- Galland, F.; Seady, M.; Taday, J.; Smaili, S.S.; Goncalves, C.A.; Leite, M.C. Astrocyte culture models: Molecular and function characterization of primary culture, immortalized astrocytes and C6 glioma cells. Neurochem. Int. 2019, 131, 104538. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, C.S. Sigma-1 receptor activation ameliorates LPS-induced NO production and ROS formation through the Nrf2/HO-1 signaling pathway in cultured astrocytes. Neurosci. Lett. 2019, 711, 134387. [Google Scholar] [CrossRef]
- Wu, H.; Wang, Y.; Zhang, Y.; Xu, F.; Chen, J.; Duan, L.; Zhang, T.; Wang, J.; Zhang, F. Breaking the vicious loop between inflammation, oxidative stress and coagulation, a novel anti-thrombus insight of nattokinase by inhibiting LPS-induced inflammation and oxidative stress. Redox Biol. 2020, 32, 101500. [Google Scholar] [CrossRef]
- Ye, S.; Zheng, Q.; Zhou, Y.; Bai, B.; Yang, D.; Zhao, Z. Chlojaponilactone B Attenuates Lipopolysaccharide-Induced Inflammatory Responses by Suppressing TLR4-Mediated ROS Generation and NF-kappaB Signaling Pathway. Molecules 2019, 24, 3731. [Google Scholar] [CrossRef] [Green Version]
- Ju Woo, H.; Jun, D.Y.; Lee, J.Y.; Park, H.S.; Woo, M.H.; Park, S.J.; Kim, S.C.; Yang, C.H.; Kim, Y.H. Anti-inflammatory action of 2-carbomethoxy-2,3-epoxy-3-prenyl-1,4-naphthoquinone (CMEP-NQ) suppresses both the MyD88-dependent and TRIF-dependent pathways of TLR4 signaling in LPS-stimulated RAW264.7 cells. J. Ethnopharmacol. 2017, 205, 103–115. [Google Scholar] [CrossRef]
- Ren, J.; Su, D.; Li, L.; Cai, H.; Zhang, M.; Zhai, J.; Li, M.; Wu, X.; Hu, K. Anti-inflammatory effects of Aureusidin in LPS-stimulated RAW264.7 macrophages via suppressing NF-kappaB and activating ROS- and MAPKs-dependent Nrf2/HO-1 signaling pathways. Toxicol. Appl. Pharmacol. 2020, 387, 114846. [Google Scholar] [CrossRef]
- Kawai, T.; Adachi, O.; Ogawa, T.; Takeda, K.; Akira, S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 1999, 11, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, O.; Takeda, K.; Hoshino, K.; Adachi, O.; Ogawa, T.; Akira, S. Cellular responses to bacterial cell wall components are mediated through MyD88-dependent signaling cascades. Int. Immunol. 2000, 12, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Hacker, H.; Vabulas, R.M.; Takeuchi, O.; Hoshino, K.; Akira, S.; Wagner, H. Immune cell activation by bacterial CpG-DNA through myeloid differentiation marker 88 and tumor necrosis factor receptor-associated factor (TRAF)6. J. Exp. Med. 2000, 192, 595–600. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, F.; Smith, K.D.; Ozinsky, A.; Hawn, T.R.; Yi, E.C.; Goodlett, D.R.; Eng, J.K.; Akira, S.; Underhill, D.M.; Aderem, A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 2001, 410, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Luo, W.; Wu, G.; Wu, L.; Huang, S.; Li, J.; Wang, J.; Hu, X.; Huang, W.; Liang, G. A novel MyD88 inhibitor LM9 prevents atherosclerosis by regulating inflammatory responses and oxidative stress in macrophages. Toxicol. Appl. Pharmacol. 2019, 370, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Shiratori, E.; Itoh, M.; Tohda, S. MYD88 Inhibitor ST2825 Suppresses the Growth of Lymphoma and Leukaemia Cells. Anticancer Res. 2017, 37, 6203–6209. [Google Scholar] [CrossRef]
- Wang, L.; Hu, D.; Xie, B.; Xie, L. Blockade of Myd88 signaling by a novel MyD88 inhibitor prevents colitis-associated colorectal cancer development by impairing myeloid-derived suppressor cells. Investig. New Drugs 2022, 40, 506–518. [Google Scholar] [CrossRef]
- Heo, S.J.; Hwang, J.Y.; Choi, J.I.; Lee, S.H.; Park, P.J.; Kang, D.H.; Oh, C.; Kim, D.W.; Han, J.S.; Jeon, Y.J.; et al. Protective effect of diphlorethohydroxycarmalol isolated from Ishige okamurae against high glucose-induced-oxidative stress in human umbilical vein endothelial cells. Food Chem. Toxicol. 2010, 48, 1448–1454. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Ahn, G.; Kim, H.S.; Je, J.G.; Kim, K.N.; Jeon, Y.J. Diphlorethohydroxycarmalol (DPHC) Isolated from the Brown Alga Ishige okamurae Acts on Inflammatory Myopathy as an Inhibitory Agent of TNF-alpha. Mar. Drugs 2020, 18, 529. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, H.W.; Jiang, Y.; Oh, J.Y.; Jeon, Y.J.; Ryu, B. Ishophloroglucin A Isolated from Ishige okamurae Suppresses Melanogenesis Induced by alpha-MSH: In Vitro and In Vivo. Mar. Drugs 2020, 18, 470. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, O.-Y.; Lee, S.-H. Ishige okamurae Attenuates Neuroinflammation and Cognitive Deficits in Mice Intracerebroventricularly Injected with LPS via Regulating TLR-4/MyD88-Dependent Pathways. Antioxidants 2023, 12, 78. https://doi.org/10.3390/antiox12010078
Kwon O-Y, Lee S-H. Ishige okamurae Attenuates Neuroinflammation and Cognitive Deficits in Mice Intracerebroventricularly Injected with LPS via Regulating TLR-4/MyD88-Dependent Pathways. Antioxidants. 2023; 12(1):78. https://doi.org/10.3390/antiox12010078
Chicago/Turabian StyleKwon, Oh-Yun, and Seung-Ho Lee. 2023. "Ishige okamurae Attenuates Neuroinflammation and Cognitive Deficits in Mice Intracerebroventricularly Injected with LPS via Regulating TLR-4/MyD88-Dependent Pathways" Antioxidants 12, no. 1: 78. https://doi.org/10.3390/antiox12010078