
Specificity Protein 1-Mediated Promotion of CXCL12 Advances Endothelial Cell Metabolism and Proliferation in Pulmonary Hypertension
 , , and
, , and 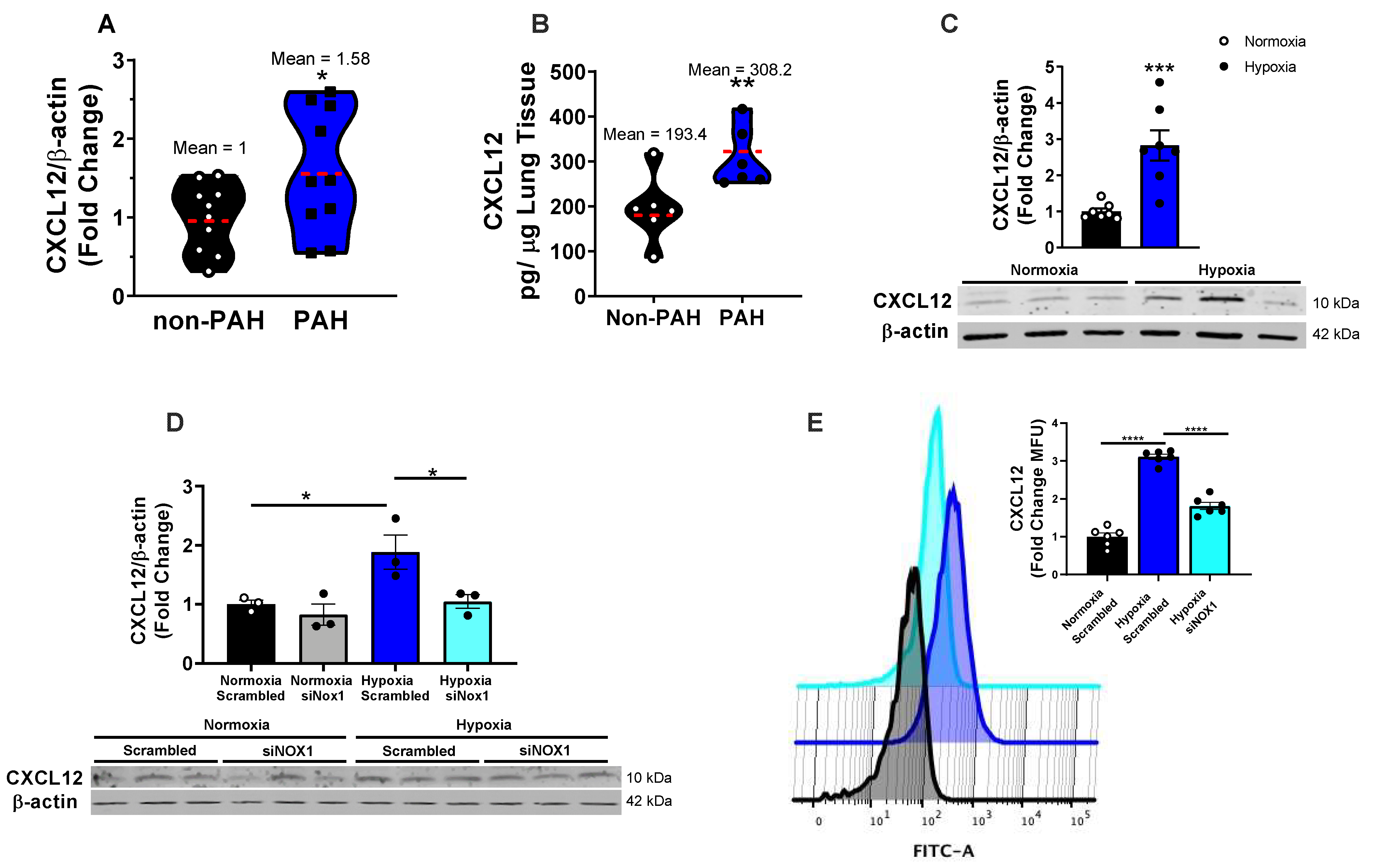{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Reagents
2.2. Human Samples
2.3. Cell Culture
2.4. In Vivo CXCL12 Infusion, mPAP and RV Hypertrophy
2.5. Protein Expression (Western Blot, ELISA, and Flow Cytometry)
2.6. ROS Measurement
2.7. TransAM Assay
2.8. Cell Proliferation
2.9. Luciferase Reporter Assay
2.10. Migration; Wound Healing Assay
2.11. Metabolic Enzyme Activity
2.12. Glucose Uptake
2.13. Statistical Analysis
3. Results
3.1. NOX1 Induces CXCL12 in Hypoxic hPAECs and Human PAH
3.2. NOX1 Mediates SP1-Induced CXCL12 in PAH-Associated Endothelial Signaling
3.3. CXCL12 Promotes hPAEC Proliferation/Migration
3.4. CXCL12 Activates Pro-Proliferative Metabolic Pathways
3.5. CXCL12 Activates the Master Metabolic Regulator mTOR in hPAECs
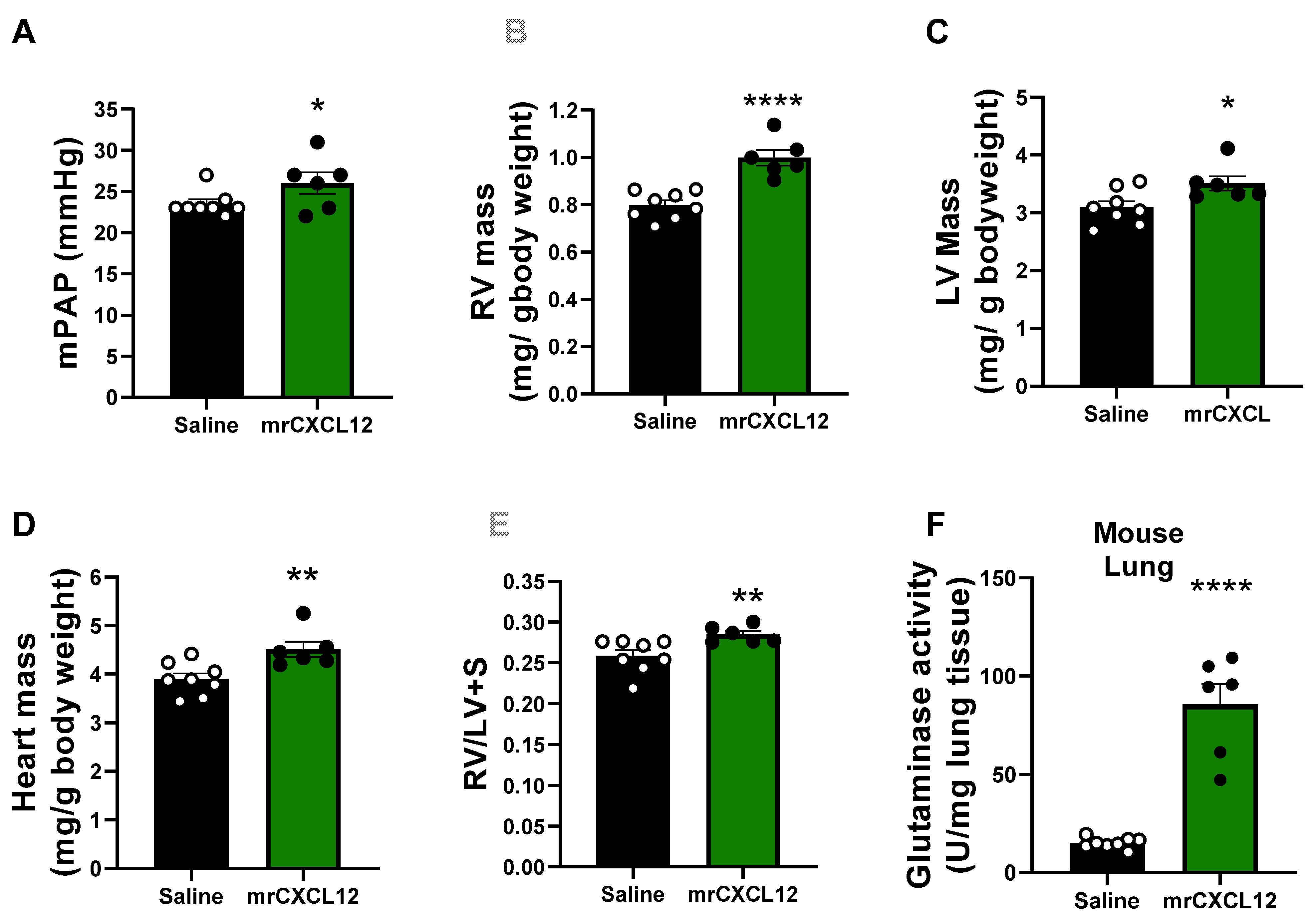
3.6. Infusion of rCXCL12 in Mice Causes an Increase in Pulmonary Artery Pressure, Cardiac Remodeling and Increased Glutaminase and PFK Activity in the Lung
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Farber, H.W.; Miller, D.P.; Poms, A.D.; Badesch, D.B.; Frost, A.E.; Rouzic, E.M.-L.; Romero, A.J.; Benton, W.W.; Elliott, C.G.; McGoon, M.D.; et al. Five-Year Outcomes of Patients Enrolled in the REVEAL Registry. Chest 2015, 148, 1043–1054. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Sikirica, M.; Iorga, S.R.; Bancroft, T.; Potash, J. The economic burden of pulmonary arterial hypertension (PAH) in the US on payers and patients. BMC Heal. Serv. Res. 2014, 14, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutendra G and Michelakis ED. The metabolic basis of pulmonary arterial hypertension. Cell metabolism. 2014, 19, 558–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benza, R.L.; Miller, D.P.; Barst, R.J.; Badesch, D.B.; Frost, A.E.; McGoon, M.D. An Evaluation of Long-term Survival from Time of Diagnosis in Pulmonary Arterial Hypertension from the REVEAL Registry. Chest 2012, 142, 448–456. [Google Scholar] [CrossRef]
- Sakao, S.; Tatsumi, K.; Voelkel, N.F. Endothelial cells and pulmonary arterial hypertension: Apoptosis, proliferation, interaction and transdifferentiation. Respir. Res. 2009, 10, 95. [Google Scholar] [CrossRef] [Green Version]
- Sakao, S.; Tatsumi, K.; Voelkel, N.F. Reversible or Irreversible Remodeling in Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2010, 43, 629–634. [Google Scholar] [CrossRef] [Green Version]
- de Jesus, D.S.; DeVallance, E.; Li, Y.; Falabella, M.; Guimaraes, D.; Shiva, S.; Kaufman, B.A.; Gladwin, M.T.; Pagano, P.J. Nox1/Ref-1-mediated activation of CREB promotes Gremlin1-driven endothelial cell proliferation and migration. Redox Biol. 2019, 22, 101138. [Google Scholar] [CrossRef]
- Budhiraja, R.; Tuder, R.M.; Hassoun, P.M. Endothelial Dysfunction in Pulmonary Hypertension. Circulation 2004, 109, 159–165. [Google Scholar] [CrossRef]
- Wong, C.-M.; Bansal, G.; Pavlickova, L.; Marcocci, L.; Suzuki, Y.J. Reactive Oxygen Species and Antioxidants in Pulmonary Hypertension. Antioxid. Redox Signal 2013, 18, 1789–1796. [Google Scholar] [CrossRef]
- Adesina, S.E.; Kang, B.-Y.; Bijli, K.M.; Ma, J.; Cheng, J.; Murphy, T.C.; Hart, C.M.; Sutliff, R.L. Targeting mitochondrial reactive oxygen species to modulate hypoxia-induced pulmonary hypertension. Free. Radic. Biol. Med. 2015, 87, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Fulton, D.J.; Li, X.; Bordan, Z.; Haigh, S.; Bentley, A.; Chen, F.; Barman, S.A. Reactive Oxygen and Nitrogen Species in the Development of Pulmonary Hypertension. Antioxidants 2017, 6, 54. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Al Ghouleh, I.; Sahoo, S.; Meijles, D.N.; Amaral, J.H.; de Jesus, D.S.; Sembrat, J.; Rojas, M.; Goncharov, D.A.; Goncharova, E.A.; Pagano, P.J. Endothelial Nox1 oxidase assembly in human pulmonary arterial hypertension; driver of Gremlin1-mediated proliferation. Clin. Sci. 2017, 131, 2019–2035. [Google Scholar] [CrossRef]
- Mccullagh, B.N.; Costello, C.M.; Li, L.; O’Connell, C.; Codd, M.; Lawrie, A.; Morton, A.; Kiely, D.G.; Condliffe, R.; Elliot, C.; et al. Elevated Plasma CXCL12α Is Associated with a Poorer Prognosis in Pulmonary Arterial Hypertension. PLoS ONE 2015, 10, e0123709. [Google Scholar] [CrossRef]
- Costello, C.M.; Mccullagh, B.; Howell, K.; Sands, M.; Belperio, J.A.; Keane, M.P.; Gaine, S.; McLoughlin, P. A role for the CXCL12 receptor, CXCR7, in the pathogenesis of human pulmonary vascular disease. Eur. Respir. J. 2011, 39, 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- Bordenave, J.; Thuillet, R.; Tu, L.; Phan, C.; Cumont, A.; Marsol, C.; Huertas, A.; Savale, L.; Hibert, M.; Galzi, J.-L.; et al. Neutralization of CXCL12 attenuates established pulmonary hypertension in rats. Cardiovasc. Res. 2019, 116, 686–697. [Google Scholar] [CrossRef]
- Guo, F.; Wang, Y.; Liu, J.; Mok, S.C.; Xue, F.; Zhang, W. CXCL12/CXCR4: A symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene 2016, 35, 816–826. [Google Scholar] [CrossRef]
- Braun, M.; Qorraj, M.; Büttner, M.; Klein, F.A.; Saul, D.; Aigner, M.; Huber, W.; Mackensen, A.; Jitschin, R.; Mougiakakos, D. CXCL12 promotes glycolytic reprogramming in acute myeloid leukemia cells via the CXCR4/mTOR axis. Leukemia 2016, 30, 1788–1792. [Google Scholar] [CrossRef] [PubMed]
- Scala, S. Molecular Pathways: Targeting the CXCR4–CXCL12 Axis—Untapped Potential in the Tumor Microenvironment. Clin. Cancer Res. 2015, 21, 4278–4285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucherat, O.; Vitry, G.; Trinh, I.; Paulin, R.; Provencher, S.; Bonnet, S. The cancer theory of pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Moruja, C.; Alonso-Lobo, J.M.; Rueda, P.; Torres, C.; González, N.; Bermejo, M.; Luque, F.; Arenzana-Seisdedos, F.; Alcamí, J.; Caruz, A. Functional Characterization of SDF-1 Proximal Promoter. J. Mol. Biol. 2005, 348, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Marković, J.; Uskoković, A.; Grdović, N.; Dinić, S.; Mihailović, M.; Jovanović, J.A.; Poznanović, G.; Vidaković, M. Identification of transcription factors involved in the transcriptional regulation of the CXCL12 gene in rat pancreatic insulinoma Rin-5F cell line. Biochem. Cell Biol. 2015, 93, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Koizume, S.; Miyagi, Y. Diverse Mechanisms of Sp1-Dependent Transcriptional Regulation Potentially Involved in the Adaptive Response of Cancer Cells to Oxygen-Deficient Conditions. Cancers 2016, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Sohl, M.; Lanner, F.; Farnebo, F. Sp1 mediate hypoxia induced ephrinB2 expression via a hypoxia-inducible factor independent mechanism. Biochem. Biophys. Res. Commun. 2010, 391, 24–27. [Google Scholar] [CrossRef]
- Ryu, H.; Lee, J.; Zaman, K.; Kubilis, J.; Ferrante, R.J.; Ross, B.D.; Neve, R.; Ratan, R.R. Sp1 and Sp3 Are Oxidative Stress-Inducible, Antideath Transcription Factors in Cortical Neurons. J. Neurosci. 2003, 23, 3597–3606. [Google Scholar] [CrossRef] [Green Version]
- Zielonka, J.; Joseph, J.; Sikora, A.; Kalyanaraman, B. Real-Time Monitoring of Reactive Oxygen and Nitrogen Species in a Multiwell Plate Using the Diagnostic Marker Products of Specific Probes. Methods Enzym. 2013, 526, 145–157. [Google Scholar] [CrossRef]
- Zielonka, J.; Sikora, A.; Hardy, M.; Joseph, J.; Dranka, B.P.; Kalyanaraman, B. Boronate Probes as Diagnostic Tools for Real Time Monitoring of Peroxynitrite and Hydroperoxides. Chem. Res. Toxicol. 2012, 25, 1793–1799. [Google Scholar] [CrossRef] [Green Version]
- DeVallance, E.; Branyan, K.W.; Lemaster, K.C.; Anderson, R.; Marshall, K.L.; Olfert, I.M.; Smith, D.M.; Kelley, E.E.; Bryner, R.W.; Frisbee, J.C.; et al. Exercise training prevents the perivascular adipose tissue-induced aortic dysfunction with metabolic syndrome. Redox Biol. 2019, 26, 101285. [Google Scholar] [CrossRef]
- Li, Y.; Cifuentes-Pagano, E.; DeVallance, E.R.; de Jesus, D.; Sahoo, S.; Meijles, D.; Koes, D.; Camacho, C.J.; Ross, M.; St Croix, C.; et al. NADPH oxidase 2 inhibitors CPP11G and CPP11H attenuate endothelial cell inflammation & vessel dysfunction and restore mouse hind-limb flow. Redox Biol. 2019, 22, 101143. [Google Scholar] [CrossRef]
- Novelli, E.M.; Little-Ihrig, L.; Knupp, H.E.; Rogers, N.M.; Yao, M.; Baust, J.J.; Meijles, D.; Croix, C.M.S.; Ross, M.A.; Pagano, P.J.; et al. Vascular TSP1-CD47 signaling promotes sickle cell-associated arterial vasculopathy and pulmonary hypertension in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L1150–L1164. [Google Scholar] [CrossRef]
- D’Addario, M.; Arora, P.D.; Ellen, R.P.; McCulloch, C.A. Interaction of p38 and Sp1 in a mechanical force-induced, beta 1 integrin-mediated transcriptional circuit that regulates the actin-binding protein filamin-A. J. Biol. Chem. 2002, 277, 47541–47550. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.Y.; Rubin, L.J. Metabolic dysfunction in pulmonary hypertension: From basic science to clinical practice. Eur. Respir Rev. 2017, 26, 170094. [Google Scholar] [CrossRef] [Green Version]
- Leopold, J.A.; Walker, J.; Scribner, A.W.; Voetsch, B.; Zhang, Y.-Y.; Loscalzo, A.J.; Stanton, R.C.; Loscalzo, J. Glucose-6-phosphate Dehydrogenase Modulates Vascular Endothelial Growth Factor-mediated Angiogenesis. J. Biol. Chem. 2003, 278, 32100–32106. [Google Scholar] [CrossRef] [Green Version]
- Wellen, K.E.; Lu, C.; Mancuso, A.; Lemons, J.M.; Ryczko, M.; Dennis, J.W.; Rabinowitz, J.D.; Coller, H.A.; Thompson, C.B. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev. 2010, 24, 2784–2799. [Google Scholar] [CrossRef] [Green Version]
- Pan, T.; Gao, L.; Wu, G.; Shen, G.; Xie, S.; Wen, H.; Yang, J.; Zhou, Y.; Tu, Z.; Qian, W. Elevated expression of glutaminase confers glucose utilization via glutaminolysis in prostate cancer. Biochem. Biophys. Res. Commun. 2015, 456, 452–458. [Google Scholar] [CrossRef]
- Wang, J.-B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.; Dias, S.M.; Dang, C.V.; et al. Targeting Mitochondrial Glutaminase Activity Inhibits Oncogenic Transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Magaway, C.; Kim, E.; Jacinto, E. Targeting mTOR and Metabolism in Cancer: Lessons and Innovations. Cells 2019, 8, 1584. [Google Scholar] [CrossRef] [Green Version]
- Csibi, A.; Fendt, S.-M.; Li, C.; Poulogiannis, G.; Choo, A.Y.; Chapski, D.J.; Jeong, S.M.; Dempsey, J.M.; Parkhitko, A.; Morrison, T.; et al. TThe mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 2013, 153, 840–854. [Google Scholar] [CrossRef]
- Csibi, A.; Lee, G.; Yoon, S.O.; Tong, H.; Ilter, D.; Elia, I.; Fendt, S.M.; Roberts, T.M.; Blenis, J. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr. Biol. 2014, 24, 2274–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekulić, A.; Hudson, C.C.; Homme, J.L.; Yin, P.; Otterness, D.M.; Karnitz, L.M.; Abraham, R.T. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res. 2000, 60, 3504–3513. [Google Scholar] [PubMed]
- Copp, J.; Manning, G.; Hunter, T. TORC-Specific Phosphorylation of Mammalian Target of Rapamycin (mTOR): Phospho-Ser2481 Is a Marker for Intact mTOR Signaling Complex 2. Cancer Res. 2009, 69, 1821–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranayhossaini, D.J.; Rodriguez, A.I.; Sahoo, S.; Chen, B.B.; Mallampalli, R.K.; Kelley, E.E.; Csanyi, G.; Gladwin, M.T.; Romero, G.; Pagano, P.J. Selective Recapitulation of Conserved and Nonconserved Regions of Putative NOXA1 Protein Activation Domain Confers Isoform-specific Inhibition of Nox1 Oxidase and Attenuation of Endothelial Cell Migration. J. Biol. Chem. 2013, 288, 36437–36450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milanini-Mongiat, J.; Pouysségur, J.; Pagès, G. Identification of two Sp1 phosphorylation sites for p42/p44 mitogen-activated protein kinases: Their implication in vascular endothelial growth factor gene transcription. J. Biol. Chem. 2002, 277, 20631–20639. [Google Scholar] [CrossRef] [Green Version]
- D’Addario, M.; Arora, P.D.; McCulloch, C. Role of p38 in stress activation of Sp1. Gene 2006, 379, 51–61. [Google Scholar] [CrossRef]
- Rohlenova, K.; Veys, K.; Miranda-Santos, I.; De Bock, K.; Carmeliet, P. Endothelial Cell Metabolism in Health and Disease. Trends Cell Biol. 2018, 28, 224–236. [Google Scholar] [CrossRef]
- Chen, G.; Chen, S.M.; Wang, X.; Ding, X.F.; Ding, J.; Meng, L.H. Inhibition of chemokine (CXC motif) ligand 12/chemokine (CXC motif) receptor 4 axis (CXCL12/CXCR4)-mediated cell migration by targeting mammalian target of rapamycin (mTOR) pathway in human gastric carcinoma cells. J. Biol. Chem. 2012, 287, 12132–12141. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, M.E.; Hatch, M.M.S.; Wu, N.; Muawad, S.A.; Hughes, C.C.W. mTORC2 mediates CXCL12-induced angiogenesis. Angiogenesis 2016, 19, 359–371. [Google Scholar] [CrossRef] [Green Version]
- Yakulov, T.A.; Todkar, A.P.; Slanchev, K.; Wiegel, J.; Bona, A.; Groß, M.; Scholz, A.; Hess, I.; Wurditsch, A.; Grahammer, F.; et al. CXCL12 and MYC control energy metabolism to support adaptive responses after kidney injury. Nat. Commun. 2018, 9, 3660. [Google Scholar] [CrossRef]
- Dai, Z.; Li, M.; Wharton, J.; Zhu, M.M.; Zhao, Y.-Y. Prolyl-4 Hydroxylase 2 (PHD2) Deficiency in Endothelial Cells and Hematopoietic Cells Induces Obliterative Vascular Remodeling and Severe Pulmonary Arterial Hypertension in Mice and Humans Through Hypoxia-Inducible Factor-2α. Circulation 2016, 133, 2447–2458. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
DeVallance, E.R.; Dustin, C.M.; de Jesus, D.S.; Ghouleh, I.A.; Sembrat, J.C.; Cifuentes-Pagano, E.; Pagano, P.J. Specificity Protein 1-Mediated Promotion of CXCL12 Advances Endothelial Cell Metabolism and Proliferation in Pulmonary Hypertension. Antioxidants 2023, 12, 71. https://doi.org/10.3390/antiox12010071
DeVallance ER, Dustin CM, de Jesus DS, Ghouleh IA, Sembrat JC, Cifuentes-Pagano E, Pagano PJ. Specificity Protein 1-Mediated Promotion of CXCL12 Advances Endothelial Cell Metabolism and Proliferation in Pulmonary Hypertension. Antioxidants. 2023; 12(1):71. https://doi.org/10.3390/antiox12010071
Chicago/Turabian StyleDeVallance, Evan R., Christopher M. Dustin, Daniel Simoes de Jesus, Imad Al Ghouleh, John C. Sembrat, Eugenia Cifuentes-Pagano, and Patrick J. Pagano. 2023. "Specificity Protein 1-Mediated Promotion of CXCL12 Advances Endothelial Cell Metabolism and Proliferation in Pulmonary Hypertension" Antioxidants 12, no. 1: 71. https://doi.org/10.3390/antiox12010071