
Reduced Ribose-5-Phosphate Isomerase A-1 Expression in Specific Neurons and Time Points Promotes Longevity in Caenorhabditis elegans
 , ,
, ,
Abstract
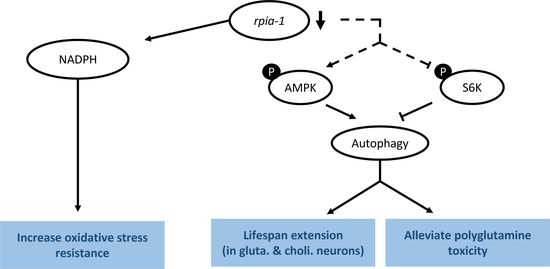
:
1. Introduction
2. Materials and Methods
2.1. Strains and Cultivation Conditions
2.2. RNA Interference
2.3. Oxidative Stress Assay
2.4. Quantification of NADP+ and NADPH Levels
2.5. Lifespan Assay
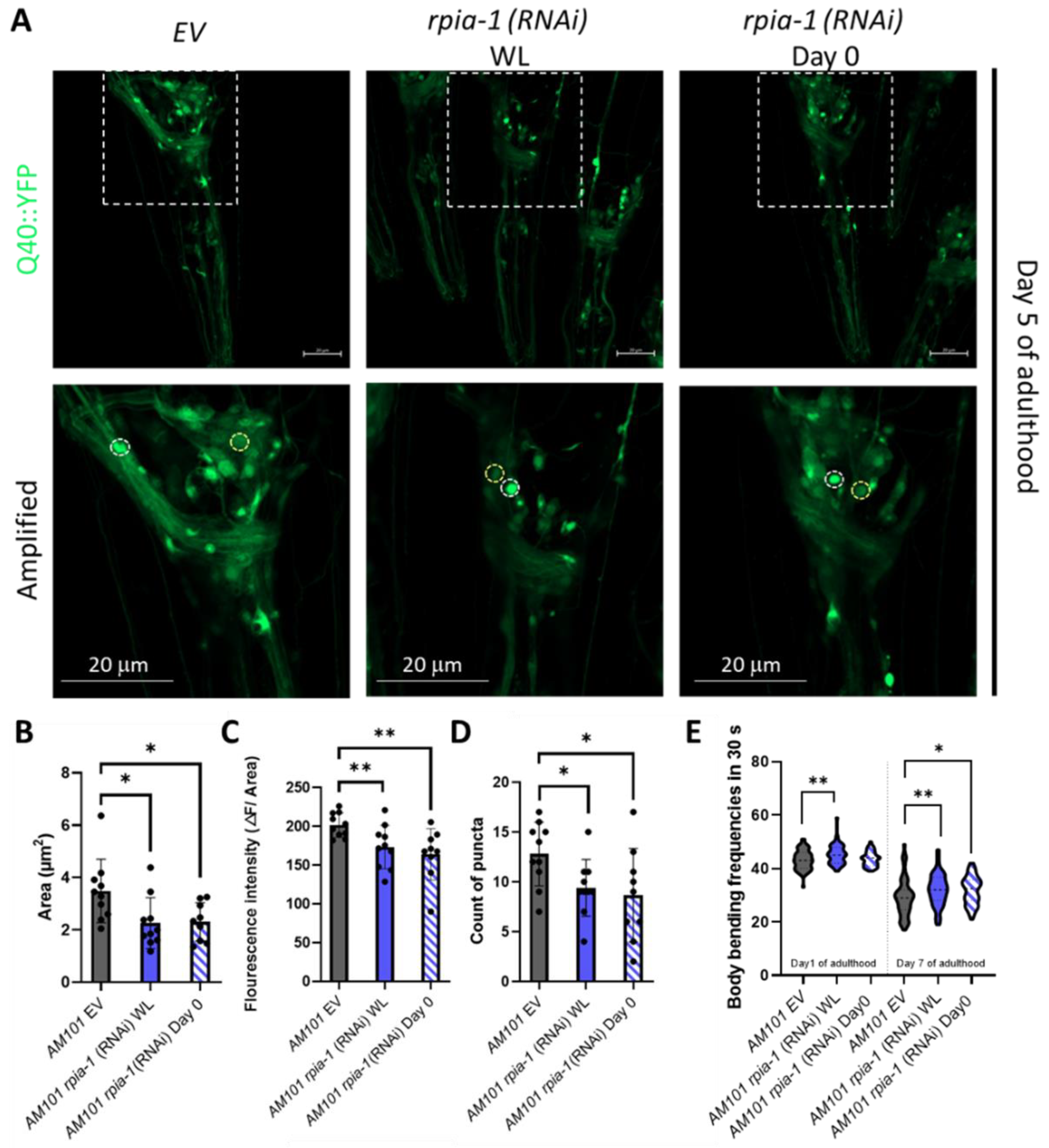
2.6. Polyglutamine Toxicity
2.7. RNA Isolation and Reverse Transcription Followed by Quantitative PCR (RT-qPCR)
2.8. Protein Extraction and Western Blotting
2.9. RNA-Seq Analysis
2.10. Developmental Delay Screening
2.11. Generation of rpia-1 Overexpression Construct
3. Results
3.1. Knockdown of rpia-1 Exhibits Increased Tolerance to Oxidative Stress, Elevated Levels of NADPH, and Attenuated Polyglutamine Toxicity in C. elegans
3.2. Knockdown of rpia-1 in Specific Time Points and Tissues Displays Extended Lifespan
3.3. Knockdown of rpia-1 Extends Lifespan by Activating Autophagy and AMPK Pathway and by Inhibiting TOR Pathway
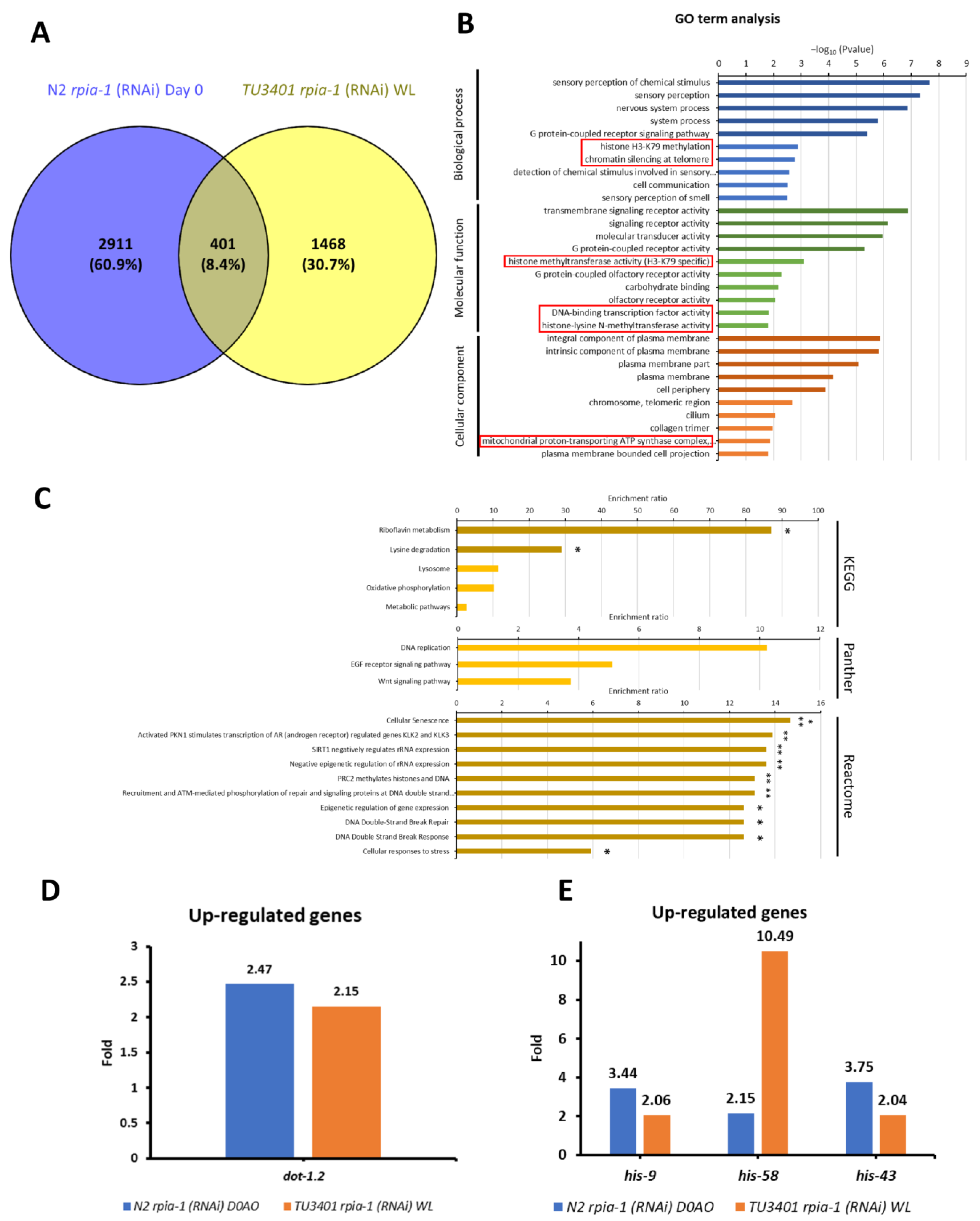
3.4. RNA Sequencing Analysis Reveals Potential Downstream Target Genes in rpia-1 Knockdown-Mediated Longevity Regulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hofmeister, F.; Baber, L.; Ferrari, U.; Hintze, S.; Jarmusch, S.; Krause, S.; Meinke, P.; Mehaffey, S.; Neuerburg, C.; Tangenelli, F.; et al. Late-onset neuromuscular disorders in the differential diagnosis of sarcopenia. BMC Neurol. 2021, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, J.N.; Kaeberlein, M. Why is aging conserved and what can we do about it? PLoS Biol. 2015, 13, e1002131. [Google Scholar] [CrossRef]
- Kapahi, P.; Kaeberlein, M.; Hansen, M. Dietary restriction and lifespan: Lessons from invertebrate models. Ageing Res. Rev. 2017, 39, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Kim, J.; Guan, K.L. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu. Rev. Pharm. Toxicol. 2012, 52, 381–400. [Google Scholar] [CrossRef] [PubMed]
- Trefts, E.; Shaw, R.J. AMPK: Restoring metabolic homeostasis over space and time. Mol. Cell 2021, 81, 3677–3690. [Google Scholar] [CrossRef] [PubMed]
- Dennis, P.B.; Jaeschke, A.; Saitoh, M.; Fowler, B.; Kozma, S.C.; Thomas, G. Mammalian TOR: A homeostatic ATP sensor. Science 2001, 294, 1102–1105. [Google Scholar] [CrossRef] [PubMed]
- Escobar, K.A.; Cole, N.H.; Mermier, C.M.; VanDusseldorp, T.A. Autophagy and aging: Maintaining the proteome through exercise and caloric restriction. Aging Cell 2019, 18, e12876. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, M.C.; Grosso, R.A.; Fader, C.M. Hallmarks of Aging: An Autophagic Perspective. Front. Endocrinol. 2019, 9, 790. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and Aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Kumsta, C.; Chang, J.T.; Lee, R.; Tan, E.P.; Yang, Y.; Loureiro, R.; Choy, E.H.; Lim, S.H.Y.; Saez, I.; Springhorn, A.; et al. The autophagy receptor p62/SQST-1 promotes proteostasis and longevity in C. elegans by inducing autophagy. Nat. Commun. 2019, 10, 5648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.P. Redox theory of aging. Redox Biol. 2015, 5, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Go, Y.M.; Jones, D.P. Redox theory of aging: Implications for health and disease. Clin. Sci. 2017, 131, 1669–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohal, R.S.; Orr, W.C. The redox stress hypothesis of aging. Free Radic. Biol. Med. 2012, 52, 539–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, K.; Roychoudhury, A. Reactive oxygen species (ROS) and response of antioxidants as ROS-scavengers during environmental stress in plants. Front. Environ. Sci. 2014, 2, 53. [Google Scholar] [CrossRef] [Green Version]
- Omar, H. Mycotoxins-Induced Oxidative Stress and Disease. In Mycotoxin and Food Safety in Developing Countries; InTech: Rijeka, Croatia, 2013; pp. 63–92. [Google Scholar]
- Bradshaw, P.C. Cytoplasmic and Mitochondrial NADPH-Coupled Redox Systems in the Regulation of Aging. Nutrients 2019, 11, 504. [Google Scholar] [CrossRef] [Green Version]
- Farkas, R.; Daniš, P.; Medved'ová, L.; Mechler, B.M.; Knopp, J. Regulation of cytosolic malate dehydrogenase by juvenile hormone in Drosophila melanogaster. Cell Biochem. Biophys. 2002, 37, 37–52. [Google Scholar] [CrossRef]
- Legan, S.K.; Rebrin, I.; Mockett, R.J.; Radyuk, S.N.; Klichko, V.I.; Sohal, R.S.; Orr, W.C. Overexpression of Glucose-6-phosphate Dehydrogenase Extends the Life Span of Drosophila melanogaster. J. Biol. Chem. 2008, 283, 32492–32499. [Google Scholar] [CrossRef] [Green Version]
- Luckinbill, L.S.; Riha, V.; Rhine, S.; Grudzien, T.A. The role of glucose-6-phosphate dehydrogenase in the evolution of longevity in Drosophila melanogaster. Heredity 1990, 65, 29–38. [Google Scholar] [CrossRef]
- Wang, C.T.; Chen, Y.C.; Wang, Y.Y.; Huang, M.H.; Yen, T.L.; Li, H.; Liang, C.J.; Sang, T.K.; Ciou, S.C.; Yuh, C.H.; et al. Reduced neuronal expression of ribose-5-phosphate isomerase enhances tolerance to oxidative stress, extends lifespan, and attenuates polyglutamine toxicity in Drosophila. Aging Cell 2012, 11, 93–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.D.; Kazemi-Esfarjani, P.; Benzer, S. Multiple-stress analysis for isolation of Drosophila longevity genes. Proc. Natl. Acad. Sci. USA 2004, 101, 12610–12615. [Google Scholar] [CrossRef] [Green Version]
- Heintze, J.; Costa, J.R.; Weber, M.; Ketteler, R. Ribose 5-phosphate isomerase inhibits LC3 processing and basal autophagy. Cell Signal 2016, 28, 1380–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieh, Y.C.; Chou, Y.T.; Chou, Y.T.; Wang, C.Y.; Lin, S.X.; Ciou, S.C.; Yuh, C.H.; Wang, H.D. Suppression of Ribose-5-Phosphate Isomerase a Induces ROS to Activate Autophagy, Apoptosis, and Cellular Senescence in Lung Cancer. Int. J. Mol. Sci. 2022, 23, 7883. [Google Scholar] [CrossRef] [PubMed]
- Stiernagle, T. Maintenance of C. elegans. In WormBook; Oxford University Press: Oxford, UK, 2006; pp. 1–11. [Google Scholar]
- Lin, Y.H.; Chen, Y.C.; Kao, T.Y.; Lin, Y.C.; Hsu, T.E.; Wu, Y.C.; Ja, W.W.; Brummel, T.J.; Kapahi, P.; Yuh, C.H.; et al. Diacylglycerol lipase regulates lifespan and oxidative stress response by inversely modulating TOR signaling in Drosophila and C. elegans. Aging Cell 2014, 13, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Conte, D., Jr.; MacNeil, L.T.; Walhout, A.J.M.; Mello, C.C. RNA Interference in Caenorhabditis elegans. Curr. Protoc. Mol. Biol. 2015, 109, 26.3.1–26.3.30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.L.; Lu, W.C.; Brummel, T.J.; Yuh, C.H.; Lin, P.T.; Kao, T.Y.; Li, F.Y.; Liao, P.C.; Benzer, S.; Wang, H.D. Reduced expression of alpha-1,2-mannosidase I extends lifespan in Drosophila melanogaster and Caenorhabditis elegans. Aging Cell 2009, 8, 370–379. [Google Scholar] [CrossRef] [Green Version]
- Senchuk, M.M.; Dues, D.J.; Van Raamsdonk, J.M. Measuring Oxidative Stress in Caenorhabditis elegans: Paraquat and Juglone Sensitivity Assays. Bio Protoc. 2017, 7, e2086. [Google Scholar] [CrossRef] [Green Version]
- Govindan, J.A.; Jayamani, E.; Zhang, X.; Mylonakis, E.; Ruvkun, G. Dialogue between E. coli free radical pathways and the mitochondria of C. elegans. Proc. Natl. Acad. Sci. USA 2015, 112, 12456–12461. [Google Scholar] [CrossRef] [Green Version]
- Corpas, F.J.; Barroso, J.B. NADPH-generating dehydrogenases: Their role in the mechanism of protection against nitro-oxidative stress induced by adverse environmental conditions. Front. Environ. Sci. 2014, 2, 55. [Google Scholar] [CrossRef]
- Ju, H.-Q.; Lin, J.-F.; Tian, T.; Xie, D.; Xu, R.-H. NADPH homeostasis in cancer: Functions, mechanisms and therapeutic implications. Signal Transduct. Target. Ther. 2020, 5, 231. [Google Scholar] [CrossRef] [PubMed]
- Gkekas, I.; Gioran, A.; Boziki, M.K.; Grigoriadis, N.; Chondrogianni, N.; Petrakis, S. Oxidative Stress and Neurodegeneration: Interconnected Processes in PolyQ Diseases. Antioxidants 2021, 10, 1450. [Google Scholar] [CrossRef] [PubMed]
- Bertoni, A.; Giuliano, P.; Galgani, M.; Rotoli, D.; Ulianich, L.; Adornetto, A.; Santillo, M.R.; Porcellini, A.; Avvedimento, V.E. Early and late events induced by polyQ-expanded proteins: Identification of a common pathogenic property of polYQ-expanded proteins. J. Biol. Chem. 2011, 286, 4727–4741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajayi, A.; Yu, X.; Lindberg, S.; Langel, Ü.; Ström, A.-L. Expanded ataxin-7 cause toxicity by inducing ROS production from NADPH oxidase complexes in a stable inducible Spinocerebellar ataxia type 7 (SCA7) model. BMC Neurosci. 2012, 13, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brignull, H.R.; Moore, F.E.; Tang, S.J.; Morimoto, R.I. Polyglutamine Proteins at the Pathogenic Threshold Display Neuron-Specific Aggregation in a Pan-Neuronal Caenorhabditis elegans Model. J. Neurosci. 2006, 26, 7597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- C. elegans Deletion Mutant Consortium. Large-Scale Screening for Targeted Knockouts in the Caenorhabditis elegans Genome. G3 Genes|Genomes|Genet 2012, 2, 1415–1425. [CrossRef] [Green Version]
- Muschiol, D.; Schroeder, F.; Traunspurger, W. Life cycle and population growth rate of Caenorhabditis elegans studied by a new method. BMC Ecol. 2009, 9, 14. [Google Scholar] [CrossRef] [Green Version]
- Uno, M.; Tani, Y.; Nono, M.; Okabe, E.; Kishimoto, S.; Takahashi, C.; Abe, R.; Kurihara, T.; Nishida, E. Neuronal DAF-16-to-intestinal DAF-16 communication underlies organismal lifespan extension in C. elegans. iScience 2021, 24, 102706. [Google Scholar] [CrossRef]
- Schmeisser, S.; Li, S.; Bouchard, B.; Ruiz, M.; Des Rosiers, C.; Roy, R. Muscle-Specific Lipid Hydrolysis Prolongs Lifespan through Global Lipidomic Remodeling. Cell Rep. 2019, 29, 4540–4552. [Google Scholar] [CrossRef] [Green Version]
- Filer, D.; Thompson, M.; Takhaveev, V.; Dobson, A.; Kotronaki, I.; Green, J.; Heinemann, M.; Tullet, J.; Alic, N. RNA polymerase III limits longevity downstream of TORC1. Nature 2017, 552, 263–267. [Google Scholar] [CrossRef]
- Mallick, A.; Ranawade, A.; van den Berg, W.; Gupta, B.P. Axin-Mediated Regulation of Lifespan and Muscle Health in C. elegans Requires AMPK-FOXO Signaling. iScience 2020, 23, 101843. [Google Scholar] [CrossRef] [PubMed]
- Firnhaber, C.; Hammarlund, M. Neuron-specific feeding RNAi in C. elegans and its use in a screen for essential genes required for GABA neuron function. PLoS Genet. 2013, 9, e1003921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H. Genetic and Transcriptomic Analysis of Axenic Longevity in Caenorhabditis elegans. Ph.D. Thesis, Ghent University, Ghent, Belgium, 2016. [Google Scholar]
- Ewald, C.Y.; Hourihan, J.M.; Bland, M.S.; Obieglo, C.; Katic, I.; Moronetti Mazzeo, L.E.; Alcedo, J.; Blackwell, T.K.; Hynes, N.E. NADPH oxidase-mediated redox signaling promotes oxidative stress resistance and longevity through memo-1 in C. elegans. eLife 2017, 6, e19493. [Google Scholar] [CrossRef] [PubMed]
- Spaans, S.; Weusthuis, R.; Van Der Oost, J.; Kengen, S. NADPH-generating systems in bacteria and archaea. Front. Microbiol. 2015, 6, 742. [Google Scholar] [CrossRef]
- Wong, S.Q.; Kumar, A.V.; Mills, J.; Lapierre, L.R. Autophagy in aging and longevity. Hum. Genet. 2020, 139, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Kumsta, C.; Chang, J.T.; Schmalz, J.; Hansen, M. Hormetic heat stress and HSF-1 induce autophagy to improve survival and proteostasis in C. elegans. Nat. Commun. 2017, 8, 14337. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [Green Version]
- González, A.; Hall, M.N.; Lin, S.C.; Hardie, D.G. AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef]
- Kim, W.; Kim, R.; Park, G.; Park, J.-W.; Kim, J.-E. Deficiency of H3K79 histone methyltransferase Dot1-like protein (DOT1L) inhibits cell proliferation. J. Biol. Chem. 2012, 287, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Karnewar, S.; Neeli, P.K.; Panuganti, D.; Kotagiri, S.; Mallappa, S.; Jain, N.; Jerald, M.K.; Kotamraju, S. Metformin regulates mitochondrial biogenesis and senescence through AMPK mediated H3K79 methylation: Relevance in age-associated vascular dysfunction. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2018, 1864(Pt. A), 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Heesbeen, H.J.; Oerthel, L.; Vries, P.M.; Wagemans, M.R.J.; Smidt, M.P. Neuronal Dot1l is a broad mitochondrial gene-repressor associated with human brain aging via H3K79 hypermethylation. bioRxiv 2021. [Google Scholar]
- Feser, J.; Truong, D.; Das, C.; Carson, J.J.; Kieft, J.; Harkness, T.; Tyler, J.K. Elevated histone expression promotes life span extension. Mol. Cell 2010, 39, 724–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sural, S.; Liang, C.Y.; Wang, F.Y.; Ching, T.T.; Hsu, A.L. HSB-1/HSF-1 pathway modulates histone H4 in mitochondria to control mtDNA transcription and longevity. Sci. Adv. 2020, 6, eaaz4452. [Google Scholar] [CrossRef]
- Silva, I.; Lopes, C.; Liz, M. Transthyretin interacts with actin regulators in a Drosophila model of familial amyloid polyneuropathy. Sci. Rep. 2020, 10, 13596. [Google Scholar] [CrossRef]
- Chou, Y.T.; Jiang, J.K.; Yang, M.H.; Lu, J.W.; Lin, H.K.; Wang, H.D.; Yuh, C.H. Identification of a noncanonical function for ribose-5-phosphate isomerase A promotes colorectal cancer formation by stabilizing and activating beta-catenin via a novel C-terminal domain. PLoS Biol. 2018, 16, e2003714. [Google Scholar] [CrossRef]
- Chou, Y.T.; Chen, L.Y.; Tsai, S.L.; Tu, H.C.; Lu, J.W.; Ciou, S.C.; Wang, H.D.; Yuh, C.H. Ribose-5-phosphate isomerase A overexpression promotes liver cancer development in transgenic zebrafish via activation of ERK and beta-catenin pathways. Carcinogenesis 2019, 40, 461–473. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Andriotis, V.M.E.; Smith, A.M. The plastidial pentose phosphate pathway is essential for postglobular embryo development in Arabidopsis. Proc. Natl. Acad. Sci. USA 2019, 116, 15297–15306. [Google Scholar] [CrossRef] [Green Version]
- Dienel, G.A. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef]
- Zullo, J.M.; Drake, D.; Aron, L.; O’Hern, P.; Dhamne, S.C.; Davidsohn, N.; Mao, C.-A.; Klein, W.H.; Rotenberg, A.; Bennett, D.A.; et al. Regulation of lifespan by neural excitation and REST. Nature 2019, 574, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Maiuri, M.C.; Markaki, M.; Megalou, E.; Pasparaki, A.; Palikaras, K.; Criollo, A.; Galluzzi, L.; Malik, S.A.; Vitale, I.; et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010, 1, e10. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, K.; Iwadate, D.; Kato, H.; Nakai, Y.; Tateishi, K.; Fujishiro, M. Targeting autophagy as a therapeutic strategy against pancreatic cancer. J. Gastroenterol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Elf, S.; Shan, C.; Kang, H.B.; Ji, Q.; Zhou, L.; Hitosugi, T.; Zhang, L.; Zhang, S.; Seo, J.H.; et al. 6-Phosphogluconate dehydrogenase links oxidative PPP, lipogenesis and tumour growth by inhibiting LKB1-AMPK signalling. Nat. Cell Biol. 2015, 17, 1484–1496. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Zhao, L.; Liu, S.; Li, Y.; Xia, S.; Chen, D.; Wang, M.; Wu, S.; Dai, Q.; Vu, H.; et al. γ-6-Phosphogluconolactone, a Byproduct of the Oxidative Pentose Phosphate Pathway, Contributes to AMPK Activation through Inhibition of PP2A. Mol. Cell 2019, 76, 857–871. [Google Scholar]
- Dai, C.; Zhang, X.; Xie, D.; Tang, P.; Li, C.; Zuo, Y.; Jiang, B.; Xue, C. Targeting PP2A activates AMPK signaling to inhibit colorectal cancer cells. Oncotarget 2017, 8, 95810–95823. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Roeth, A.; Zhang, Y.; Yang, A.; Mashadova, O.; Asara, J.; Wang, X.; Bronson, R.; Lyssiotis, C.; Ying, H.; et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Stenesen, D.; Suh, J.M.; Seo, J.; Yu, K.; Lee, K.S.; Kim, J.S.; Min, K.J.; Graff, J.M. Adenosine nucleotide biosynthesis and AMPK regulate adult life span and mediate the longevity benefit of caloric restriction in flies. Cell Metab. 2013, 17, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Asby, D.J.; Cuda, F.; Beyaert, M.; Houghton, F.D.; Cagampang, F.R.; Tavassoli, A. AMPK Activation via Modulation of De Novo Purine Biosynthesis with an Inhibitor of ATIC Homodimerization. Chem. Biol. 2015, 22, 838–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain Name | Genotype and Description | Regulated Genes/Longevity Effects | Reference |
|---|---|---|---|
| TU3401 | uIs69 [pCFJ90 (myo-2p::mCherry) + unc-119p::sid-1]. Neuron-hypersensitive knockdown line. | daf-2↓/lifespan↑; daf-16↓/lifespan↓ | [40] |
| WM118 | neIs9 [myo-3::HA::RDE-1 + rol-6(su1006)]. Muscle-specific knockdown line. | kin-1↓/lifespan↑; kin-2↓/lifespan↓ | [41] |
| VP303 | kbIs7 [nhx-2p::rde-1 + rol-6(su1006)]. Intestine-specific knockdown line. | rpc-1↓/lifespan↑ | [42] |
| NR222 | kzIs9 [(pKK1260) lin-26p::NLS::GFP + (pKK1253) lin-26p::rde-1 + rol-6(su1006)]. Hypodermis-specific knockdown line. | pry-1↓/lifespan↓ | [43] |
| Strain Name | Genotype and Description | Regulated Genes/Longevity Effects | Reference |
|---|---|---|---|
| XE1582 | wpSi11 [eat-4p::rde-1::SL2::sid-1 + Cbr-unc-119(+)] II. Glutamatergic neuron-specific knockdown line. | cbp-1↓/lifespan↓ | [45] |
| XE1581 | wpSi10 [unc-17p::rde-1::SL2::sid-1 + Cbr-unc-119(+)] II. Cholinergic neuron-specific knockdown line. | ||
| XE1375 | wpIs36 [unc-47p::mCherry] I. wpSi1 [unc-47p::rde-1::SL2::sid-1 + Cbr-unc-119(+)] II. GABAergic neuron-specific knockdown line. | ||
| XE1474 | wpSi6 [dat-1p::rde-1::SL2::sid-1 + Cbr-unc-119(+)] II. Dopaminergic neuron-specific knockdown line. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, W.-C.; Yuh, C.-H.; Lu, Y.-T.; Lin, Y.-H.; Ching, T.-T.; Wang, C.-Y.; Wang, H.-D. Reduced Ribose-5-Phosphate Isomerase A-1 Expression in Specific Neurons and Time Points Promotes Longevity in Caenorhabditis elegans. Antioxidants 2023, 12, 124. https://doi.org/10.3390/antiox12010124
Shen W-C, Yuh C-H, Lu Y-T, Lin Y-H, Ching T-T, Wang C-Y, Wang H-D. Reduced Ribose-5-Phosphate Isomerase A-1 Expression in Specific Neurons and Time Points Promotes Longevity in Caenorhabditis elegans. Antioxidants. 2023; 12(1):124. https://doi.org/10.3390/antiox12010124
Chicago/Turabian StyleShen, Wen-Chi, Chiou-Hwa Yuh, Yu-Ting Lu, Yen-Hung Lin, Tsui-Ting Ching, Chao-Yung Wang, and Horng-Dar Wang. 2023. "Reduced Ribose-5-Phosphate Isomerase A-1 Expression in Specific Neurons and Time Points Promotes Longevity in Caenorhabditis elegans" Antioxidants 12, no. 1: 124. https://doi.org/10.3390/antiox12010124