
Antioxidant Therapy Significantly Attenuates Hepatotoxicity following Low Dose Exposure to Microcystin-LR in a Murine Model of Diet-Induced Non-Alcoholic Fatty Liver Disease

 ,
,  , ,
, ,  , and
, and 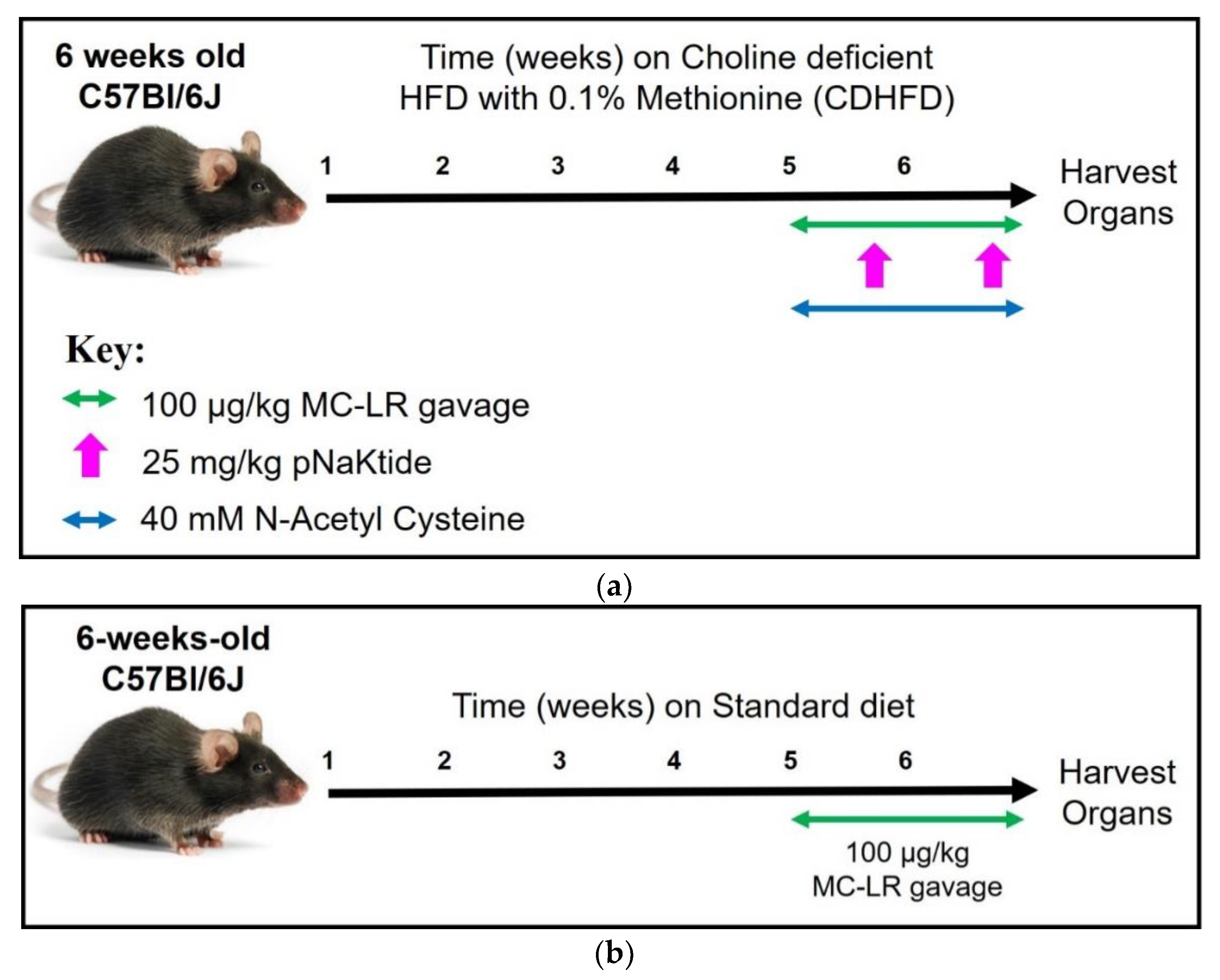{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Design
2.3. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.4. Histology
2.5. Oxidative Stress Analysis
2.5.1. Quantification of 8-Hydroxy 2 Deoxyguanosine (8-OHDG)
2.5.2. Protein Carbonylation Immunostaining
2.6. RNA Extraction and RealTime—PCR Analysis
2.7. Drug Metabolism Array
2.8. MC-LR and MC-LR Cysteine Determination in Urine
2.9. Glutathione-S-Transferase Activity Assay
2.10. Statistical Analysis
3. Results
3.1. Survival and Weights
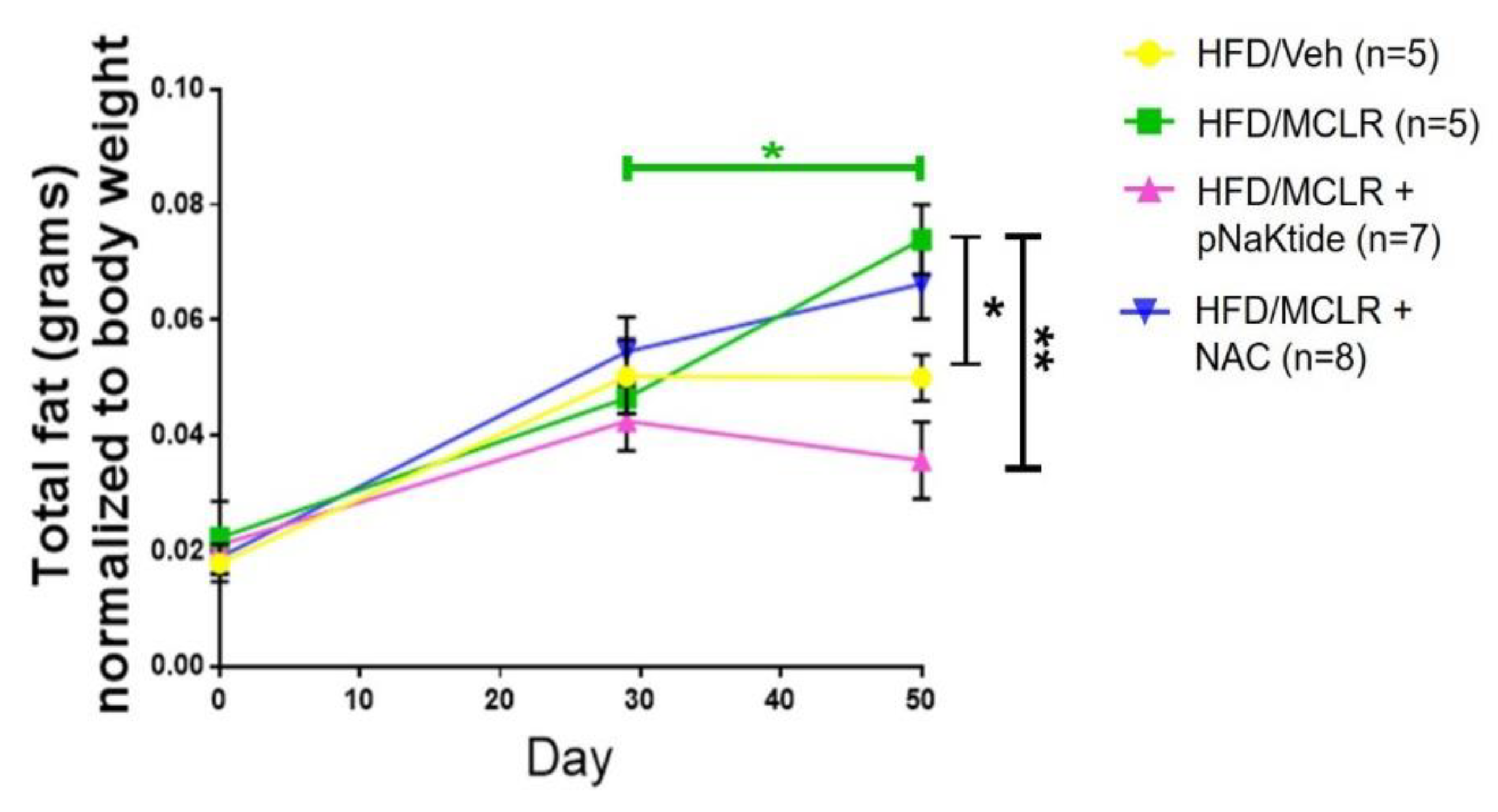
3.2. Assessing Lipid Content—Whole Body and Liver
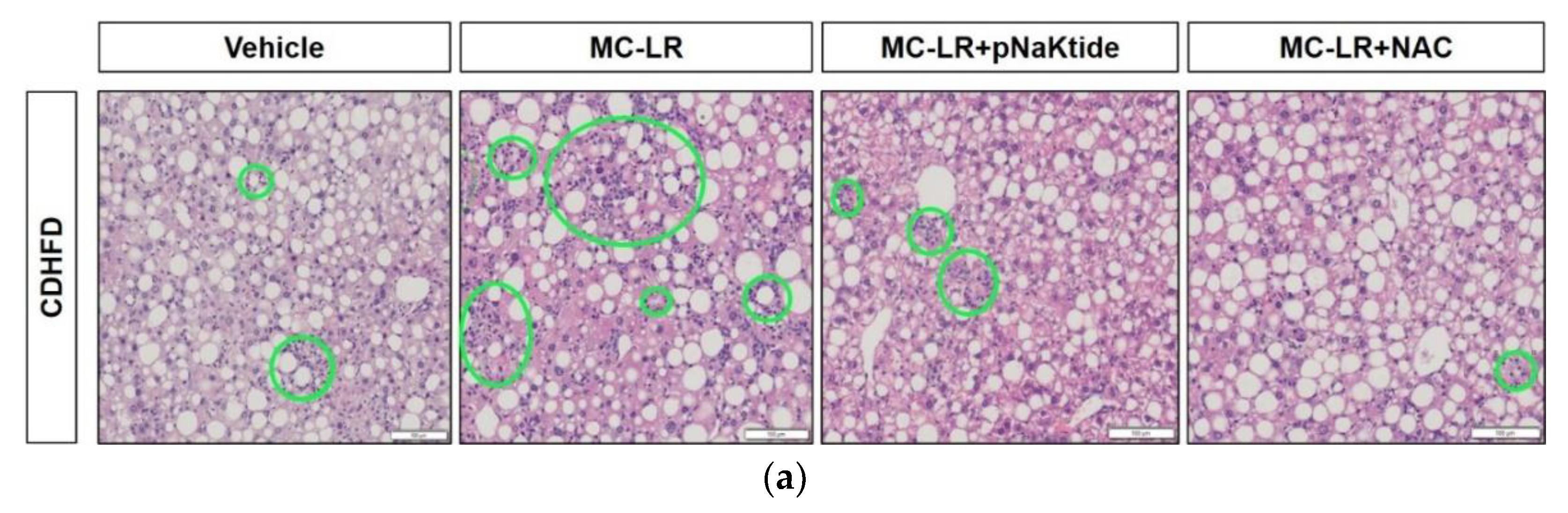
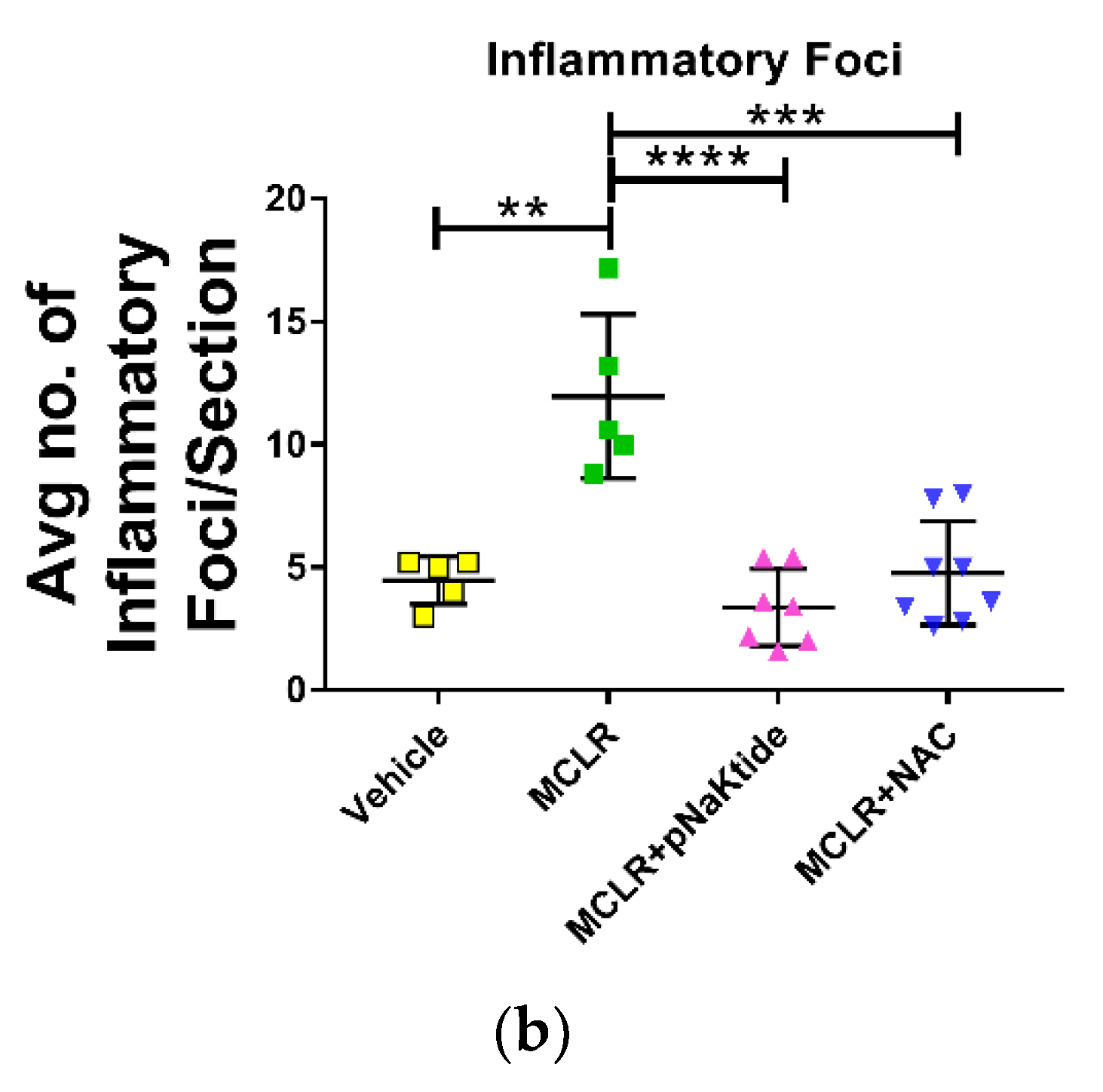
3.3. Liver Histology
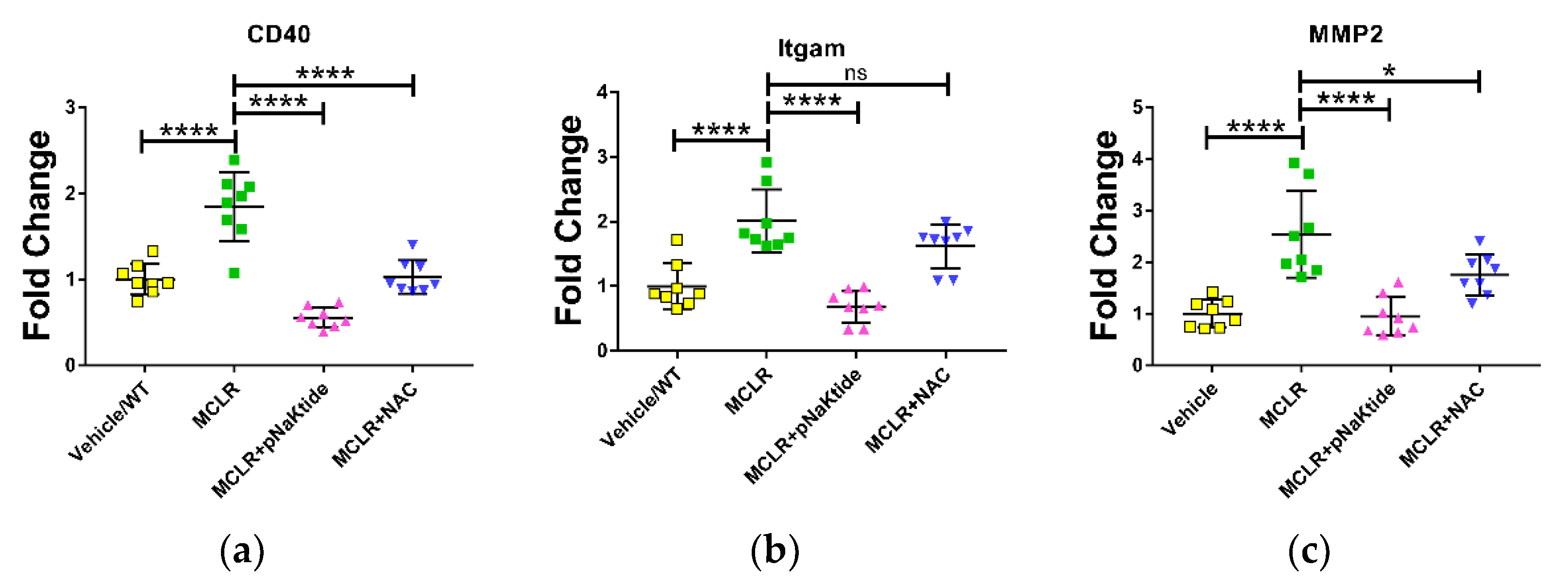
3.4. Gene Expression in Liver
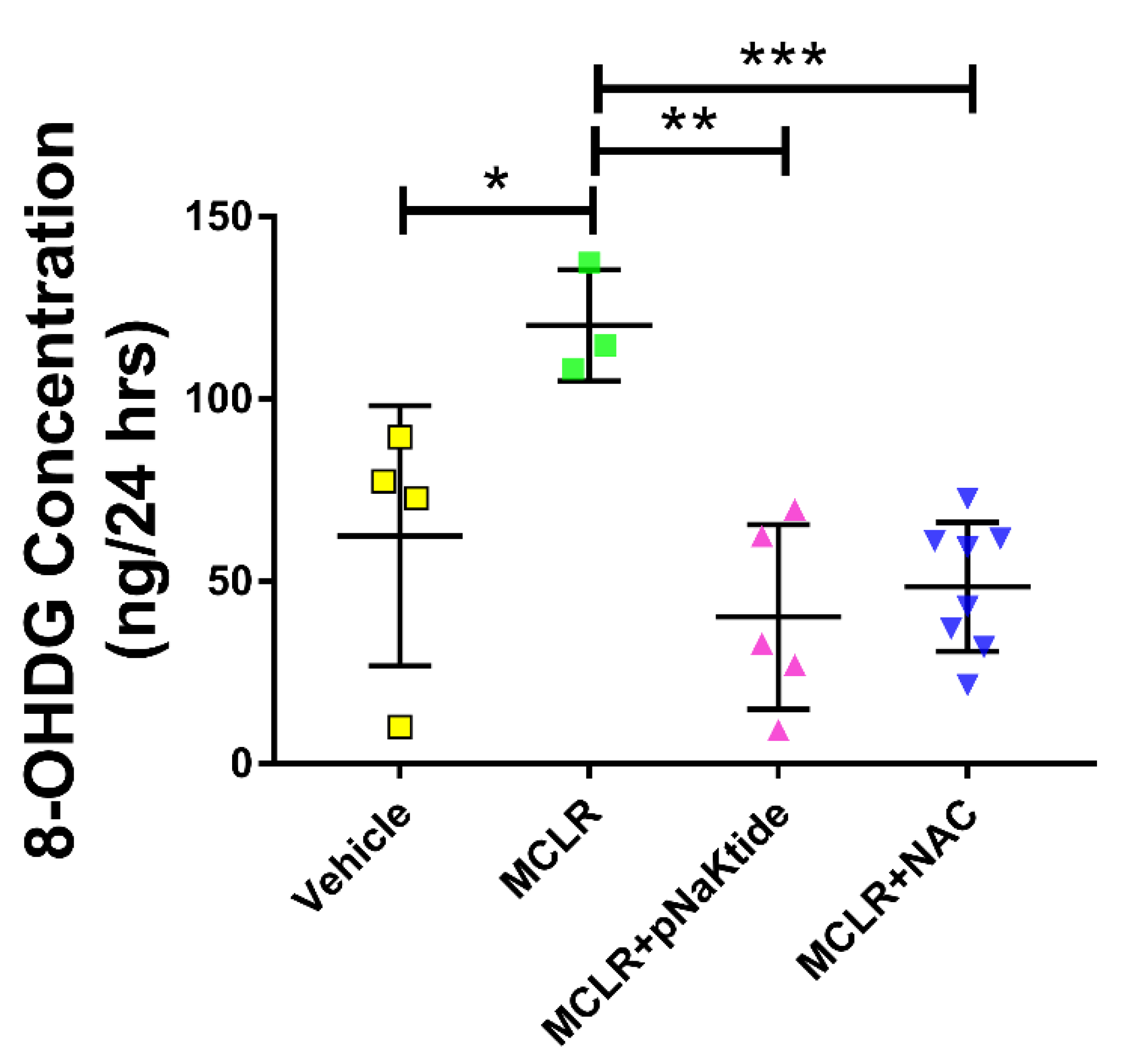
3.5. Assessment of Oxidative Stress Markers
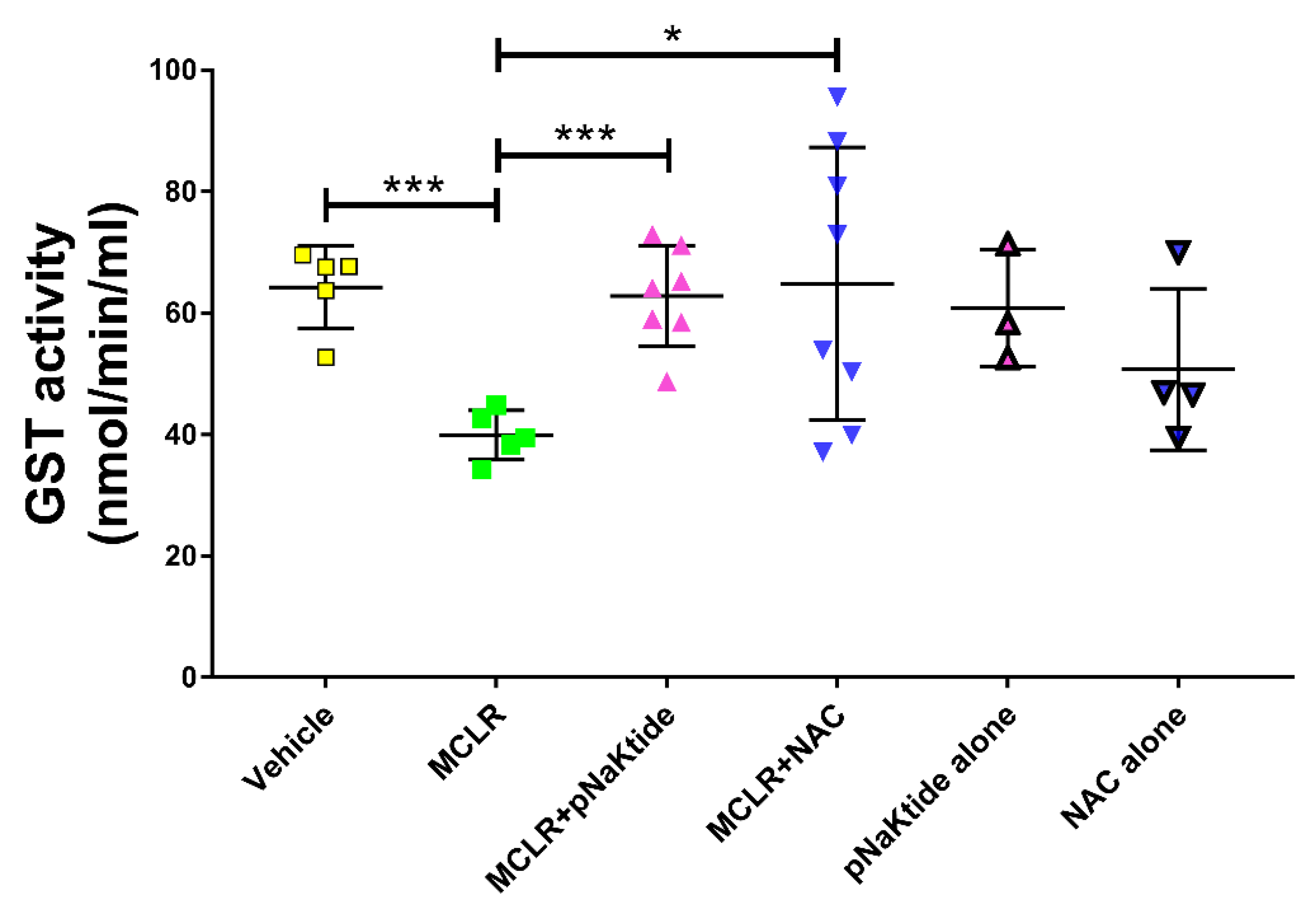
3.6. GST Activity in the Liver
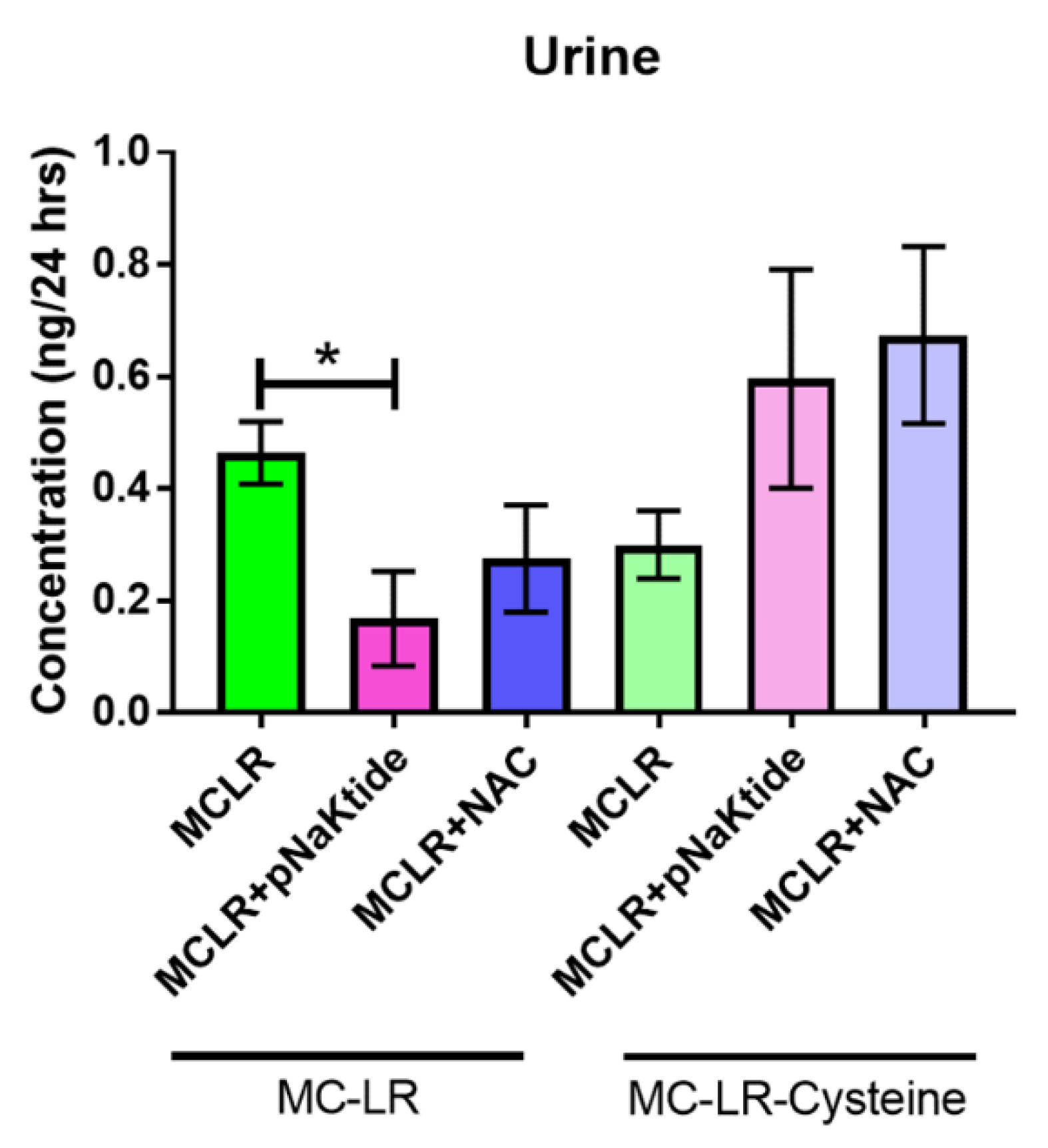
3.7. Mass Spectrometric Analysis for MC-LR and MC-LR-Cysteine in Plasma and Urine
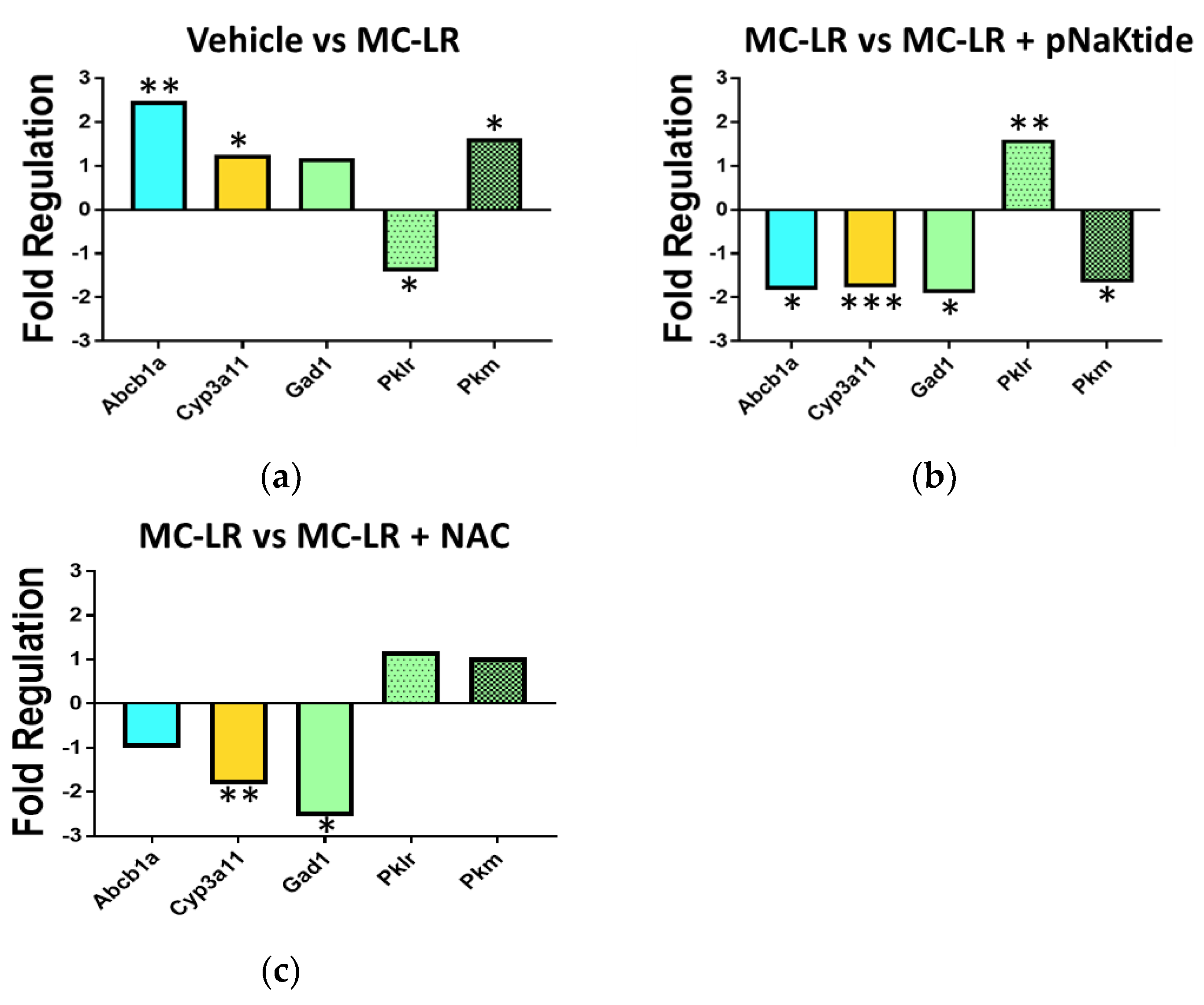
3.8. Genetic Analysis of Drug Metabolic Enzymes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Büdel, B. Cyanobacteria: Habitats and species. In Plant Desiccation Tolerance; Springer: Berlin/Heidelberg, Germany, 2011; pp. 11–21. [Google Scholar]
- Vasas, G.; Farkas, O.; Borics, G.; Felföldi, T.; Sramkó, G.; Batta, G.; Bácsi, I.; Gonda, S. Appearance of Planktothrix rubescens bloom with [D-Asp3, Mdha7] MC–RR in gravel pit pond of a shallow lake-dominated area. Toxins 2013, 5, 2434–2455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, Z.A. First report of toxic Cylindrospermopsis raciborskii and Raphidiopsis mediterranea (Cyanoprokaryota) in Egyptian fresh waters. FEMS Microbiol. Ecol. 2007, 59, 749–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantzouki, E.; Lürling, M.; Fastner, J.; de Senerpont Domis, L.; Wilk-Woźniak, E.; Koreivienė, J.; Seelen, L.; Teurlincx, S.; Verstijnen, Y.; Krztoń, W.; et al. Temperature effects explain continental scale distribution of cyanobacterial toxins. Toxins 2018, 10, 156. [Google Scholar] [CrossRef] [Green Version]
- Ueno, Y.; Nagat, S.; Suttajit, M.; Mebs, D.; Vasconcelos, V. Immunochemical survey of microcystins in environmental water in various countries. Mycotoxins Phycotoxins—Dev. Chem. Toxicol. Food Saf. P 1998, 449–453. [Google Scholar]
- Rinehart, K.L.; Harada, K.; Namikoshi, M.; Chen, C.; Harvis, C.A.; Munro, M.H.; Blunt, J.W.; Mulligan, P.E.; Beasley, V.R. Nodularin, microcystin, and the configuration of Adda. J. Am. Chem. Soc. 1988, 110, 8557–8558. [Google Scholar] [CrossRef]
- Chen, L.; Xie, P. Mechanisms of microcystin-induced cytotoxicity and apoptosis. Mini Rev. Med. Chem. 2016, 16, 1018–1031. [Google Scholar] [CrossRef]
- Yoshizawa, S.; Matsushima, R.; Watanabe, M.F.; Harada, K.I.; Ichihara, A.; Carmichael, W.W.; Fujiki, H. Inhibition of protein phosphatases by microcystis and nodularin associated with hepatotoxicity. J. Cancer Res. Clin. Oncol. 1990, 116, 609–614. [Google Scholar] [CrossRef]
- Ding, W.-X.; Shen, H.-M.; Zhu, H.-G.; Ong, C.-N. Studies on oxidative damage induced by cyanobacteria extract in primary cultured rat hepatocytes. Environ. Res. 1998, 78, 12–18. [Google Scholar] [CrossRef]
- Solter, P.F.; Wollenberg, G.K.; Huang, X.; Chu, F.S.; Runnegar, M.T. Prolonged sublethal exposure to the protein phosphatase inhibitor microcystin-LR results in multiple dose-dependent hepatotoxic effects. Toxicol. Sci. 1998, 44, 87–96. [Google Scholar] [CrossRef]
- Codd, G.A. Mechanisms of action and health effects associated with cyanobacterial toxins. Toxicol. Lett. 1996, 88, 21. [Google Scholar] [CrossRef]
- Milutinović, A.; Živin, M.; Zorc-Pleskovič, R.; Sedmak, B.; Šuput, D. Nephrotoxic effects of chronic administration of microcystins-LR and-YR. Toxicon 2003, 42, 281–288. [Google Scholar] [CrossRef]
- Pahan, K.; Sheikh, F.G.; Namboodiri, A.M.; Singh, I. Inhibitors of protein phosphatase 1 and 2A differentially regulate the expression of inducible nitric-oxide synthase in rat astrocytes and macrophages. J. Biol. Chem. 1998, 273, 12219–12226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzman, R.; Solter, P. Characterization of sublethal microcystin-LR exposure in mice. Vet. Pathol. 2002, 39, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Valério, E.; Vasconcelos, V.; Campos, A. New Insights on the Mode of Action of Microcystins in Animal Cells—A Review. Mini Rev. Med. Chem. 2016, 16, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.D.; Dzierlenga, A.; Arman, T.; Toth, E.; Li, H.; Lynch, K.D.; Tian, D.D.; Goedken, M.; Paine, M.F.; Cherrington, N. Nonalcoholic fatty liver disease alters microcystin-LR toxicokinetics and acute toxicity. Toxicon 2019, 162, 1–8. [Google Scholar] [CrossRef]
- Lone, Y.; Koiri, R.K.; Bhide, M. An overview of the toxic effect of potential human carcinogen Microcystin-LR on testis. Toxicol. Rep. 2015, 2, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, A.; Vasconcelos, V. Molecular mechanisms of microcystin toxicity in animal cells. Int. J. Mol. Sci. 2010, 11, 268–287. [Google Scholar] [CrossRef] [Green Version]
- Arman, T.; Clarke, J.D. Microcystin toxicokinetics, molecular toxicology, and pathophysiology in preclinical rodent models and humans. Toxins 2021, 13, 537. [Google Scholar] [CrossRef]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015, 149, 389–397.e10. [Google Scholar] [CrossRef] [Green Version]
- Le, M.H.; Devaki, P.; Ha, N.B.; Jun, D.W.; Te, H.S.; Cheung, R.C.; Nguyen, M.H. Prevalence of non-alcoholic fatty liver disease and risk factors for advanced fibrosis and mortality in the United States. PLoS ONE 2017, 12, e0173499. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.T.; Kleiner, D.E. Histopathology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Metabolism 2016, 65, 1080–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazel, Y.; Koenig, A.B.; Sayiner, M.; Goodman, Z.D.; Younossi, Z.M. Epidemiology and natural history of non-alcoholic fatty liver disease. Metabolism 2016, 65, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedogni, G.; Miglioli, L.; Masutti, F.; Tiribelli, C.; Marchesini, G.; Bellentani, S. Prevalence of and risk factors for nonalcoholic fatty liver disease: The Dionysos nutrition and liver study. Hepatology 2005, 42, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Creeden, J.F.; Kipp, Z.A.; Xu, M.; Flight, R.M.; Moseley, H.N.B.; Martinez, G.J.; Lee, W.H.; Alganem, K.; Imami, A.S.; McMullen, M.R.; et al. Hepatic Kinome Atlas: An In-Depth Identification of Kinase Pathways in Liver Fibrosis of Humans and Rodents. Hepatology 2022. [Google Scholar] [CrossRef] [PubMed]
- Lad, A.; Su, R.C.; Breidenbach, J.D.; Stemmer, P.M.; Carruthers, N.J.; Sanchez, N.K.; Khalaf, F.K.; Zhang, S.; Kleinhenz, A.L.; Dube, P.; et al. Chronic Low Dose Oral Exposure to Microcystin-LR Exacerbates Hepatic Injury in a Murine Model of Non-Alcoholic Fatty Liver Disease. Toxins 2019, 11, 486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhang, Z.; Xie, J.X.; Li, X.; Tian, J.; Cai, T.; Cui, H.; Ding, H.; Shapiro, J.I.; Xie, Z. Na/K-ATPase mimetic pNaKtide peptide inhibits the growth of human cancer cells. J. Biol. Chem. 2011, 286, 32394–32403. [Google Scholar] [CrossRef] [Green Version]
- Udoh, U.-A.S.; Banerjee, M.; Rajan, P.K.; Sanabria, J.D.; Smith, G.; Schade, M.; Sanabria, J.A.; Nakafuku, Y.; Sodhi, K.; Pierre, S.V.; et al. Tumor-Suppressor Role of the α1-Na/K-ATPase Signalosome in NASH Related Hepatocellular Carcinoma. Int. J. Mol. Sci. 2022, 23, 7359. [Google Scholar] [CrossRef]
- Clark, J.; Clore, E.L.; Zheng, K.; Adame, A.; Masliah, E.; Simon, D.K. Oral N-acetyl-cysteine attenuates loss of dopaminergic terminals in α-synuclein overexpressing mice. PLoS ONE 2010, 5, e12333. [Google Scholar] [CrossRef] [Green Version]
- Fawell, J.; Mitchell, R.; Everett, D.; Hill, R. The toxicity of cyanobacterial toxins in the mouse: I microcystin-LR. Hum. Exp. Toxicol. 1999, 18, 162–167. [Google Scholar] [CrossRef]
- Itoh, M.; Kato, H.; Suganami, T.; Konuma, K.; Marumoto, Y.; Terai, S.; Sakugawa, H.; Kanai, S.; Hamaguchi, M.; Fukaishi, T.; et al. Hepatic crown-like structure: A unique histological feature in non-alcoholic steatohepatitis in mice and humans. PLoS ONE 2013, 8, e82163. [Google Scholar] [CrossRef] [Green Version]
- Palagama, D.S.; Baliu-Rodriguez, D.; Lad, A.; Levison, B.S.; Kennedy, D.J.; Haller, S.T.; Westrick, J.; Hensley, K.; Isailovic, D. Development and applications of solid-phase extraction and liquid chromatography-mass spectrometry methods for quantification of microcystins in urine, plasma, and serum. J. Chromatogr. A 2018, 1573, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Baliu-Rodriguez, D.; Kucheriavaia, D.; Palagama, D.S.; Lad, A.; O’Neill, G.M.; Birbeck, J.A.; Kennedy, D.J.; Haller, S.T.; Westrick, J.A.; Isailovic, D. Development and Application of Extraction Methods for LC-MS Quantification of Microcystins in Liver Tissue. Toxins 2020, 12, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutz, T.A.; Woods, S.C. Overview of animal models of obesity. Curr. Protoc. Pharmacol. 2012, 58, 5–61. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, M.; Hada, N.; Sakamaki, Y.; Uno, A.; Shiga, T.; Tanaka, C.; Ito, T.; Katsume, A.; Sudoh, M. An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int. J. Exp. Pathol. 2013, 94, 93–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anstee, Q.M.; Goldin, R.D. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int. J. Exp. Pathol. 2006, 87, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Charlat, O.; Tartaglia, L.A.; Woolf, E.A.; Weng, X.; Ellis, S.J.; Lakey, N.D.; Culpepper, J.; More, K.J.; Breitbart, R.E.; et al. Evidence that the diabetes gene encodes the leptin receptor: Identification of a mutation in the leptin receptor gene in db/db mice. Cell 1996, 84, 491–495. [Google Scholar] [CrossRef] [Green Version]
- Conn, P.M. Animal Models for the Study of Human Disease; Academic Press: Cambridge, MA, USA, 2017. [Google Scholar]
- Kirsch, R.; Clarkson, V.; Shephard, E.G.; Marais, D.A.; Jaffer, M.A.; Woodburne, V.E.; Kirsch, R.E.; Hall, P.D.L.M. Rodent nutritional model of non-alcoholic steatohepatitis: Species, strain and sex difference studies. J. Gastroenterol. Hepatol. 2003, 18, 1272–1282. [Google Scholar] [CrossRef]
- Rangnekar, A.S.; Lammert, F.; Igolnikov, A.; Green, R.M. Quantitative trait loci analysis of mice administered the methionine–choline deficient dietary model of experimental steatohepatitis. Liver Int. 2006, 26, 1000–1005. [Google Scholar] [CrossRef]
- Radhakrishnan, S.; Yeung, S.F.; Ke, J.-Y.; Antunes, M.M.; Pellizzon, M.A. Considerations When Choosing High-Fat, High-Fructose, and High-Cholesterol Diets to Induce Experimental Nonalcoholic Fatty Liver Disease in Laboratory Animal Models. Curr. Dev. Nutr. 2021, 5, nzab138. [Google Scholar] [CrossRef]
- Ames, P.R.; Bucci, T.; Merashli, M.; Amaral, M.; Arcaro, A.; Gentile, F.; Nourooz-Zadeh, J.; DelgadoAlves, J. Oxidative/nitrative stress in the pathogenesis of systemic sclerosis: Are antioxidants beneficial? Free Radic. Res. 2018, 52, 1063–1082. [Google Scholar] [CrossRef]
- Sahin, S.; Alatas, O. The protective effects of n-acetylcysteine against acute hepatotoxicity. Indian J. Gastroenterol. 2013, 32, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.H.H.; Messiha, B.A.S.; Abdel-Latif, H.A.-T. Protective effect of ursodeoxycholic acid, resveratrol, and N-acetylcysteine on nonalcoholic fatty liver disease in rats. Pharm. Biol. 2016, 54, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Tian, F.; Zhai, Q.; Yu, R.; Zhang, H.; Gu, Z.; Chen, W. Protective effects of a cocktail of lactic acid bacteria on microcystin-LR-induced hepatotoxicity and oxidative damage in BALB/c mice. RSC Adv. 2017, 7, 20480–20487. [Google Scholar] [CrossRef] [Green Version]
- Khoshbaten, M.; Aliasgarzadeh, A.; Masnadi, K.; Tarzamani, M.K.; Farhang, S.; Babaei, H.; Kiani, J.; Zaare, M.; Najafipoor, F. N-acetylcysteine improves liver function in patients with non-alcoholic Fatty liver disease. Hepat. Mon. 2010, 10, 12. [Google Scholar] [PubMed]
- Lakhani, H.V.; Sharma, D.; Dodrill, M.W.; Nawab, A.; Sharma, N.; Cottrill, C.L.; Shapiro, J.I.; Sodhi, K. Phenotypic alteration of hepatocytes in non-alcoholic fatty liver disease. Int. J. Med. Sci. 2018, 15, 1591. [Google Scholar] [CrossRef] [Green Version]
- Sedan, D.; Laguens, M.; Copparoni, G.; Aranda, J.O.; Giannuzzi, L.; Marra, C.A.; Andrinolo, D. Hepatic and intestine alterations in mice after prolonged exposure to low oral doses of Microcystin-LR. Toxicon 2015, 104, 26–33. [Google Scholar] [CrossRef]
- He, J.; Li, G.; Chen, J.; Lin, J.; Zeng, C.; Chen, J.; Deng, J.; Xie, P. Prolonged exposure to low-dose microcystin induces nonalcoholic steatohepatitis in mice: A systems toxicology study. Arch. Toxicol. 2017, 91, 465–480. [Google Scholar] [CrossRef]
- Guzman, R.E.; Solter, P.F. Hepatic oxidative stress following prolonged sublethal microcystin LR exposure. Toxicol. Pathol. 1999, 27, 582–588. [Google Scholar] [CrossRef]
- Yoshida, T.; Takeda, M.; Tsutsumi, T.; Nagata, S.; Yoshida, F.; Maita, K.; Harada, T.; Ueno, Y. Tumor necrosis factor-α expression and Kupffer cell activation in hepatotoxicity caused by microcystin-LR in mice. J. Toxicol. Pathol. 2001, 14, 259. [Google Scholar] [CrossRef]
- Danese, S.; Sans, M.; Fiocchi, C. The CD40/CD40L costimulatory pathway in inflammatory bowel disease. Gut 2004, 53, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.A.; Farrah, T.; Goodwin, R.G. The TNF receptor superfamily of cellular and viral proteins: Activation, costimulation, and death. Cell 1994, 76, 959–962. [Google Scholar] [CrossRef]
- Shiraki, K.; Sugimoto, K.; Okano, H.; Wagayama, H.; Fujikawa, K.; Yamanaka, T.; Ito, T.; Ohmori, S.; Sakai, T.; Takase, K.; et al. CD40 expression in HCV-associated chronic liver diseases. Int. J. Mol. Med. 2006, 18, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbi, A.; Kishimoto, T.; Miller, L.; Springer, T. The human leukocyte adhesion glycoprotein Mac-1 (complement receptor type 3, CD11b) alpha subunit. Cloning, primary structure, and relation to the integrins, von Willebrand factor and factor B. J. Biol. Chem. 1988, 263, 12403–12411. [Google Scholar] [CrossRef]
- Sevastianova, K.; Sutinen, J.; Kannisto, K.; Hamsten, A.; Ristola, M.; Yki-Jarvinen, H. Adipose tissue inflammation and liver fat in patients with highly active antiretroviral therapy-associated lipodystrophy. Am. J. Physiol.-Endocrinol. Metab. 2008, 295, E85–E91. [Google Scholar] [CrossRef] [PubMed]
- Arman, T.; Lynch, K.D.; Montonye, M.L.; Goedken, M.; Clarke, J.D. Sub-chronic microcystin-LR liver toxicity in preexisting diet-induced nonalcoholic steatohepatitis in rats. Toxins 2019, 11, 398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, C. 8-hydroxy-2′-deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci. Health Part C 2009, 27, 120–139. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.L.; Chiou, C.-C.; Chang, P.-Y.; Wu, J.T. Urinary 8-OHdG: A marker of oxidative stress to DNA and a risk factor for cancer, atherosclerosis and diabetics. Clin. Chim. Acta 2004, 339, 1–9. [Google Scholar] [CrossRef]
- Creeden, J.F.; Gordon, D.M.; Stec, D.E.; Hinds, T.D., Jr. Bilirubin as a metabolic hormone: The physiological relevance of low levels. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E191–E207. [Google Scholar] [CrossRef]
- Stec, D.E.; Hinds, T.D., Jr. Natural Product Heme Oxygenase Inducers as Treatment for Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 9493. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Creeden, J.F.; Gordon, D.M.; Stec, D.F.; Donald, M.C.; Stec, D.E. Bilirubin Nanoparticles Reduce Diet-Induced Hepatic Steatosis, Improve Fat Utilization, and Increase Plasma beta-Hydroxybutyrate. Front. Pharmacol. 2020, 11, 594574. [Google Scholar] [CrossRef]
- Gordon, D.M.; Neifer, K.L.; Hamoud, A.A.; Hawk, C.F.; Nestor-Kalinoski, A.L.; Miruzzi, S.A.; Morran, M.P.; Adeosun, S.O.; Sarver, J.G.; Erhardt, P.W.; et al. Bilirubin remodels murine white adipose tissue by reshaping mitochondrial activity and the coregulator profile of peroxisome proliferator-activated receptor alpha. J. Biol. Chem. 2020, 295, 9804–9822. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Aldini, G.; Carini, M.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation, cellular dysfunction, and disease progression. J. Cell. Mol. Med. 2006, 10, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, M.; Bollineni, R.C.; Hoffmann, R. Protein carbonylation as a major hallmark of oxidative damage: Update of analytical strategies. Mass Spectrom. Rev. 2014, 33, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Su, R.C.; Meyers, C.M.; Warner, E.A.; Garcia, J.A.; Refsnider, J.M.; Lad, A.; Breidenbach, J.D.; Modyanov, N.; Malhotra, D.; Haller, S.T.; et al. Harmful Algal Bloom Toxicity in Lithobates catesbeiana Tadpoles. Toxins 2020, 12, 378. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.; Kim, H.-C.; Shin, E.-J. Repeated exposure to microcystin-leucine-arginine potentiates excitotoxicity induced by a low dose of kainate. Toxicology 2021, 460, 152887. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.R.; Wilhelm, S.W.; Boyer, G.L. The fate of microcystins in the environment and challenges for monitoring. Toxins 2014, 6, 3354–3387. [Google Scholar] [CrossRef] [Green Version]
- Lad, A.; Breidenbach, J.D.; Su, R.C.; Murray, J.; Kuang, R.; Mascarenhas, A.; Najjar, J.; Patel, S.; Hegde, P.; Youssef, M.; et al. As We Drink and Breathe: Adverse Health Effects of Microcystins and Other Harmful Algal Bloom Toxins in the Liver, Gut, Lungs and Beyond. Life 2022, 12, 418. [Google Scholar] [CrossRef]
- Gehringer, M.M.; Shephard, E.G.; Downing, T.G.; Wiegand, C.; Neilan, B.A. An investigation into the detoxification of microcystin-LR by the glutathione pathway in Balb/c mice. Int. J. Biochem. Cell Biol. 2004, 36, 931–941. [Google Scholar] [CrossRef]
- Li, S.; Chen, J.; Xie, P.; Guo, X.; Fan, H.; Yu, D.; Zeng, C.; Chen, L. The role of glutathione detoxification pathway in MCLR-induced hepatotoxicity in SD rats. Environ. Toxicol. 2015, 30, 1470–1480. [Google Scholar] [CrossRef]
- Liu, W.; Baker, S.S.; Baker, R.D.; Zhu, L. Antioxidant mechanisms in nonalcoholic fatty liver disease. Curr. Drug Targets 2015, 16, 1301–1314. [Google Scholar] [CrossRef]
- Seif El-Din, S.H.; Sabra, A.N.A.; Hammam, O.A.; Ebeid, F.A.; El-Lakkany, N.M. Pharmacological and antioxidant actions of garlic and/or onion in non-alcoholic fatty liver disease (NAFLD) in rats. J. Egypt. Soc. Parasitol. 2014, 44, 295–308. [Google Scholar]
- Jayaraj, R.; Anand, T.; Rao, P.L. Activity and gene expression profile of certain antioxidant enzymes to microcystin-LR induced oxidative stress in mice. Toxicology 2006, 220, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Peña-Llopis, S.; Ferrando, M.D.; Peña, J.B. Fish tolerance to organophosphate-induced oxidative stress is dependent on the glutathione metabolism and enhanced by N-acetylcysteine. Aquat. Toxicol. 2003, 65, 337–360. [Google Scholar] [CrossRef]
- Schaffner, F. Hepatic drug metabolism and adverse hepatic drug reactions. Vet. Pathol. 1975, 12, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Sundararaghavan, V.L.; Sindhwani, P.; Hinds, T.D., Jr. Glucuronidation and UGT isozymes in bladder: New targets for the treatment of uroepithelial carcinomas? Oncotarget 2017, 8, 3640–3648. [Google Scholar] [CrossRef] [Green Version]
- Cobbina, E.; Akhlaghi, F. Non-alcoholic fatty liver disease (NAFLD)–pathogenesis, classification, and effect on drug metabolizing enzymes and transporters. Drug Metab. Rev. 2017, 49, 197–211. [Google Scholar] [CrossRef]
- Aitken, A.E.; Richardson, T.A.; Morgan, E.T. Regulation of drug-metabolizing enzymes and transporters in inflammation. Annu. Rev. Pharm. Toxicol. 2006, 46, 123–149. [Google Scholar] [CrossRef]
- Morgan, E.T. Impact of infectious and inflammatory disease on cytochrome P450–mediated drug metabolism and pharmacokinetics. Clin. Pharmacol. Ther. 2009, 85, 434–438. [Google Scholar] [CrossRef]
- Hamoud, A.R.; Weaver, L.; Stec, D.E.; Hinds, T.D., Jr. Bilirubin in the Liver-Gut Signaling Axis. Trends Endocrinol. Metab. 2018, 29, 140–150. [Google Scholar] [CrossRef]
- Weaver, L.; Hamoud, A.R.; Stec, D.E.; Hinds, T.D., Jr. Biliverdin reductase and bilirubin in hepatic disease. Am. J. Physiol. Gastrointest Liver Physiol. 2018, 314, G668–G676. [Google Scholar] [CrossRef]
- Jaiswal, A.K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic. Biol. Med. 2004, 36, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Nakata, K.; Tanaka, Y.; Nakano, T.; Adachi, T.; Tanaka, H.; Kaminuma, T. and Ishikawa, T. Nuclear receptor-mediated transcriptional regulation in Phase I, II, and III xenobiotic metabolizing systems. Drug Metab. Pharmacokinet. 2006, 21, 437–457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab. Rev. 2006, 38, 769–789. [Google Scholar] [CrossRef] [PubMed]
- Canet, M.J.; Hardwick, R.N.; Lake, A.D.; Dzierlenga, A.L.; Clarke, J.D.; Cherrington, N.J. Modeling human nonalcoholic steatohepatitis-associated changes in drug transporter expression using experimental rodent models. Drug Metab. Dispos. 2014, 42, 586–595. [Google Scholar] [CrossRef] [Green Version]
- Canet, M.J.; Merrell, M.D.; Hardwick, R.N.; Bataille, A.M.; Campion, S.N.; Ferreira, D.W.; Xanthakos, S.A.; Manautou, J.E.; A-Kader, H.H.; Erickson, R.P.; et al. Altered regulation of hepatic efflux transporters disrupts acetaminophen disposition in pediatric nonalcoholic steatohepatitis. Drug Metab. Dispos. 2015, 43, 829–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liu, Y.; Li, X. Alteration in the expression of cytochrome P450s (CYP1A1, CYP2E1, and CYP3A11) in the liver of mouse induced by microcystin-LR. Toxins 2015, 7, 1102–1115. [Google Scholar] [CrossRef] [Green Version]
- Buechler, C.; Sweiss, T. Does hepatic steatosis affect drug metabolizing enzymes in the liver? Curr. Drug Metab. 2011, 12, 24–34. [Google Scholar] [CrossRef]
- Naik, A.; Belič, A.; Zanger, U.; Rozman, D. Molecular Interactions between NAFLD and Xenobiotic Metabolism. Front. Genet. 2013, 4, 2. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lad, A.; Hunyadi, J.; Connolly, J.; Breidenbach, J.D.; Khalaf, F.K.; Dube, P.; Zhang, S.; Kleinhenz, A.L.; Baliu-Rodriguez, D.; Isailovic, D.; et al. Antioxidant Therapy Significantly Attenuates Hepatotoxicity following Low Dose Exposure to Microcystin-LR in a Murine Model of Diet-Induced Non-Alcoholic Fatty Liver Disease. Antioxidants 2022, 11, 1625. https://doi.org/10.3390/antiox11081625
Lad A, Hunyadi J, Connolly J, Breidenbach JD, Khalaf FK, Dube P, Zhang S, Kleinhenz AL, Baliu-Rodriguez D, Isailovic D, et al. Antioxidant Therapy Significantly Attenuates Hepatotoxicity following Low Dose Exposure to Microcystin-LR in a Murine Model of Diet-Induced Non-Alcoholic Fatty Liver Disease. Antioxidants. 2022; 11(8):1625. https://doi.org/10.3390/antiox11081625
Chicago/Turabian StyleLad, Apurva, Jonathan Hunyadi, Jacob Connolly, Joshua D. Breidenbach, Fatimah K. Khalaf, Prabhatchandra Dube, Shungang Zhang, Andrew L. Kleinhenz, David Baliu-Rodriguez, Dragan Isailovic, and et al. 2022. "Antioxidant Therapy Significantly Attenuates Hepatotoxicity following Low Dose Exposure to Microcystin-LR in a Murine Model of Diet-Induced Non-Alcoholic Fatty Liver Disease" Antioxidants 11, no. 8: 1625. https://doi.org/10.3390/antiox11081625