
mTOR Signalling Pathway: A Potential Therapeutic Target for Ocular Neurodegenerative Diseases


Abstract
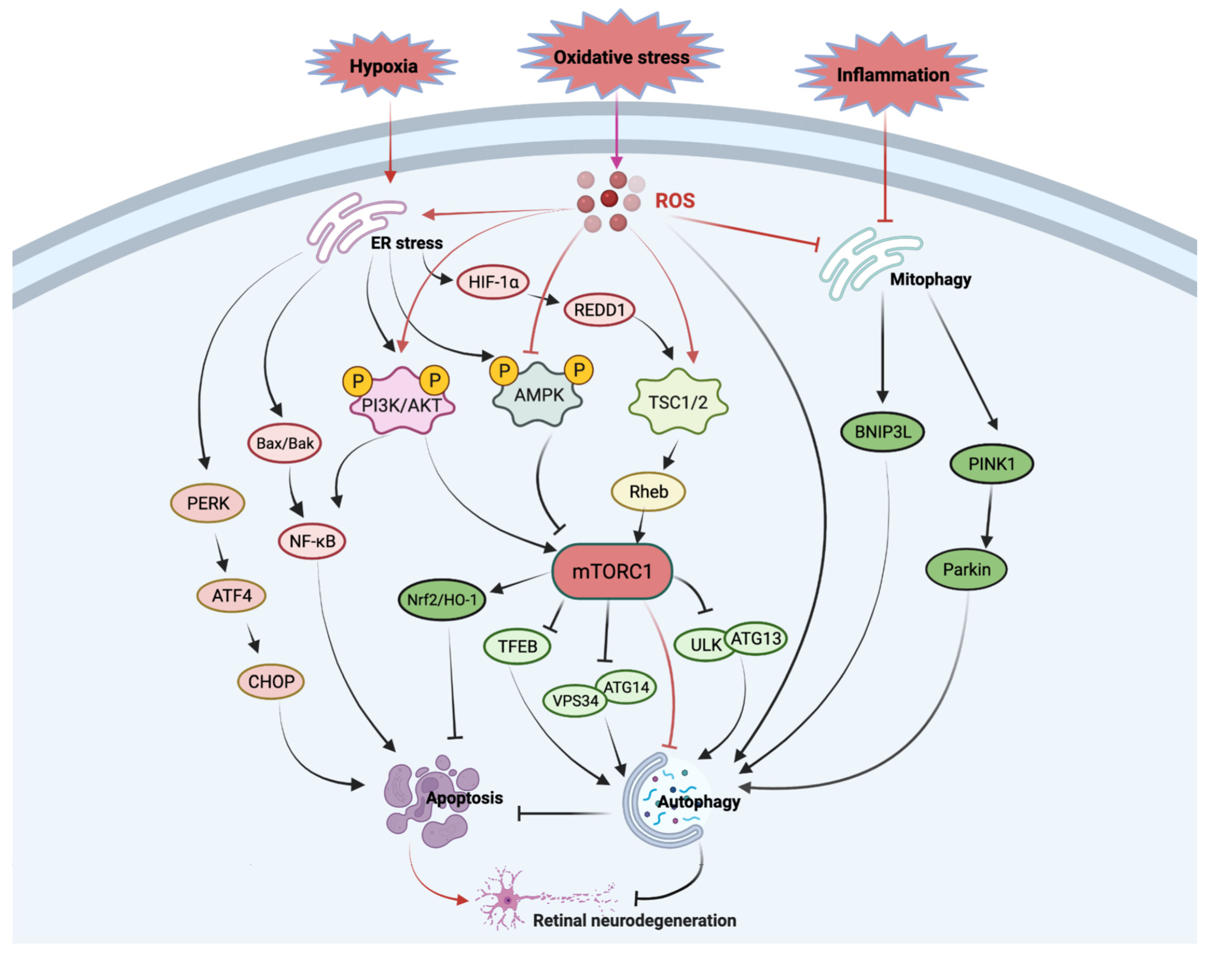
:1. Introduction
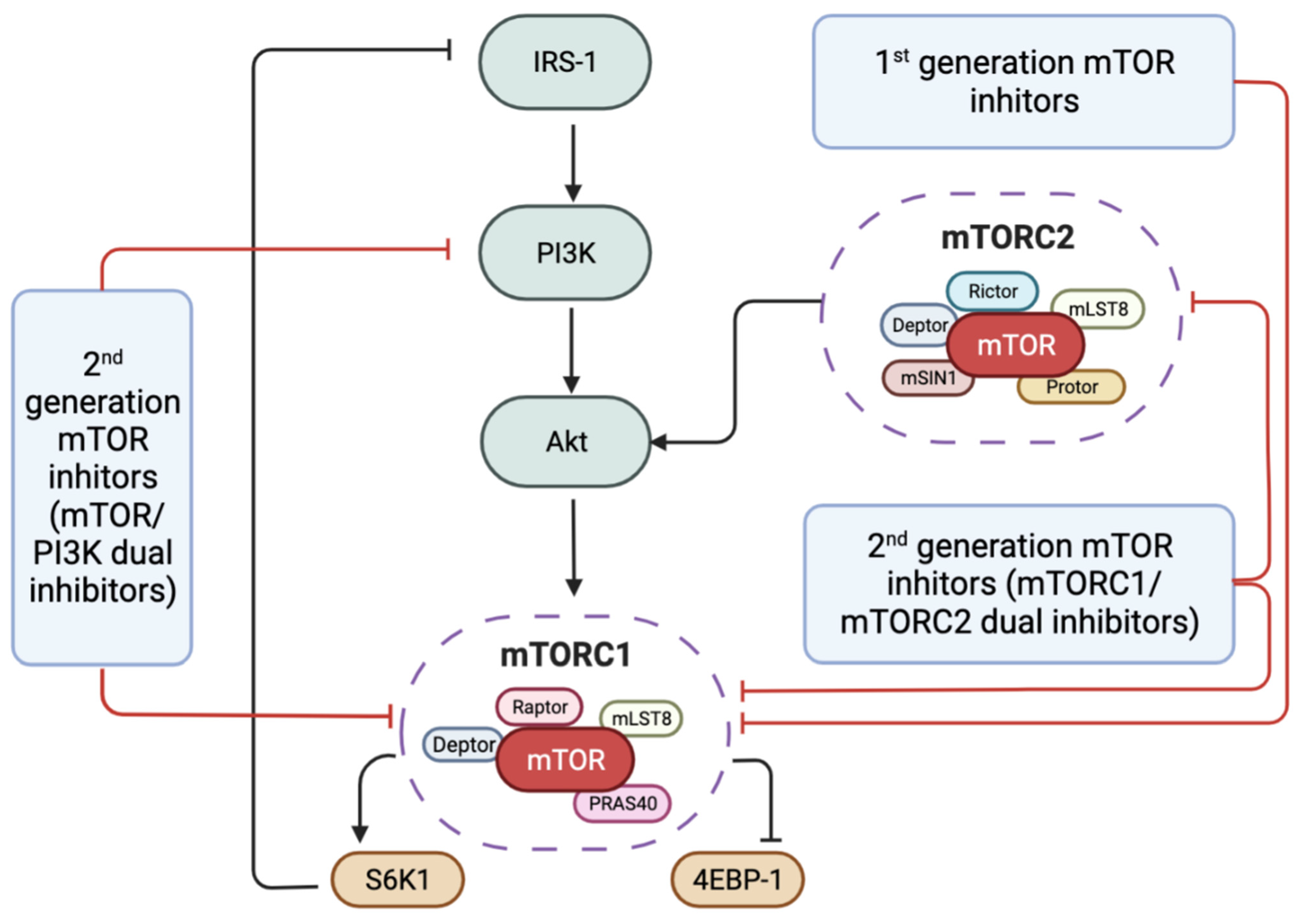
2. mTOR Signalling Pathway
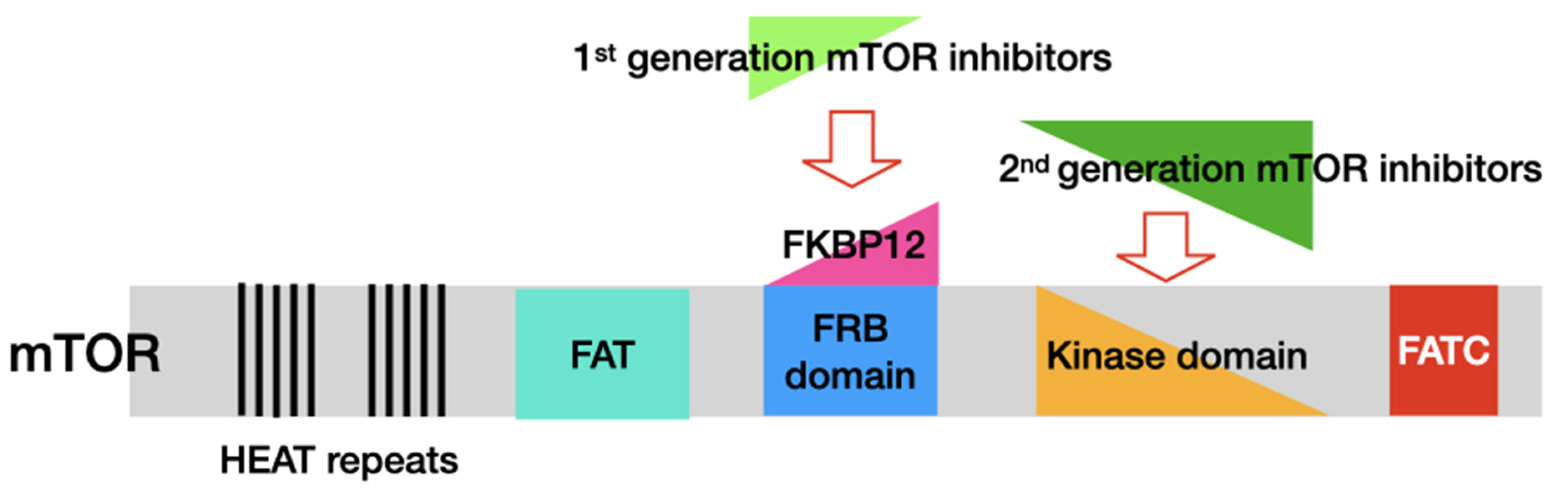
3. mTOR Inhibitors
4. mTOR in Ocular Neurodegenerative Diseases
4.1. mTOR and Diabetic Retinopathy (DR)
4.2. mTOR and Age-Related Macular Degeneration (AMD)
4.3. mTOR and Retinitis Pigmentosa (RP)
4.4. mTOR and Glaucoma
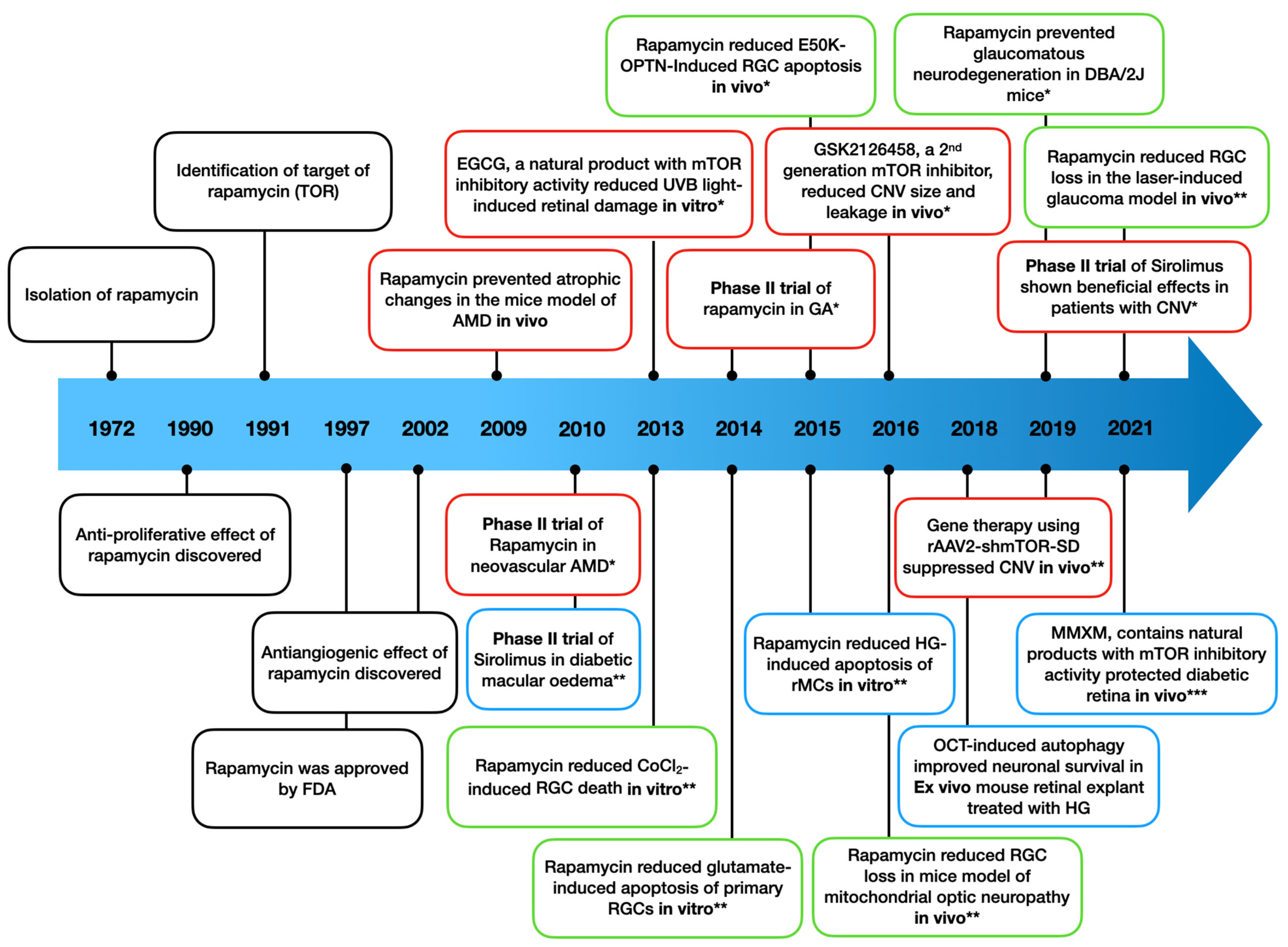
5. Clinical Trials of mTOR Inhibitors in Ocular Neurodegenerative Diseases
6. Discussion
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Laplante, M.; Sabatini, D.M. MTOR Signaling at a Glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowling, R.J.O.; Topisirovic, I.; Fonseca, B.D.; Sonenberg, N. Dissecting the Role of MTOR: Lessons from MTOR Inhibitors. Biochim. Biophys. Acta—Proteins Proteom. 2010, 1804, 433–439. [Google Scholar] [CrossRef]
- Mossmann, D.; Park, S.; Hall, M.N. MTOR Signalling and Cellular Metabolism Are Mutual Determinants in Cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Tanaka, K.; Akhavan, D.; Babic, I.; Gini, B.; Matsutani, T.; Iwanami, A.; Liu, F.; Villa, G.R.; Gu, Y.; et al. MTOR Complex 2 Controls Glycolytic Metabolism in Glioblastoma through FoxO Acetylation and Upregulation of C-Myc. Cell Metab. 2013, 18, 726–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigihara, N.; Fukunaka, A.; Hara, A.; Komiya, K.; Honda, A.; Uchida, T.; Abe, H.; Toyofuku, Y.; Tamaki, M.; Ogihara, T.; et al. Human IAPP–Induced Pancreatic β Cell Toxicity and Its Regulation by Autophagy. J. Clin. Investig. 2014, 124, 3634–3644. [Google Scholar] [CrossRef] [PubMed]
- Griffin, R.J.; Moloney, A.; Kelliher, M.; Johnston, J.A.; Ravid, R.; Dockery, P.; O’Connor, R.; O’Neill, C. Activation of Akt/PKB, Increased Phosphorylation of Akt Substrates and Loss and Altered Distribution of Akt and PTEN Are Features of Alzheimer’s Disease Pathology. J. Neurochem. 2005, 93, 105–117. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and Cancer: Lessons, Challenges and Opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.; Chen, Z.; Ding, Y.; Chen, Q.; Su, S.; Liu, H.; Ni, R.; Tang, Z. Rapamycin Attenuated Zinc-Induced Tau Phosphorylation and Oxidative Stress in Rats: Involvement of Dual MTOR/P70S6K and Nrf2/HO-1 Pathways. Front. Immunol. 2022, 13, 218. [Google Scholar] [CrossRef]
- Ha, J.-Y.; Kim, J.-S.; Kang, Y.-H.; Bok, E.; Kim, Y.-S.; Son, J.H. Tnfaip8 L1/Oxi-β Binds to FBXW5, Increasing Autophagy through Activation of TSC2 in a Parkinson’s Disease Model. J. Neurochem. 2014, 129, 527–538. [Google Scholar] [CrossRef]
- Hodgson, J.G.; Agopyan, N.; Gutekunst, C.-A.; Leavitt, B.R.; LePiane, F.; Singaraja, R.; Smith, D.J.; Bissada, N.; McCutcheon, K.; Nasir, J.; et al. A YAC Mouse Model for Huntington’s Disease with Full-Length Mutant Huntingtin, Cytoplasmic Toxicity, and Selective Striatal Neurodegeneration. Neuron 1999, 23, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Pardue, M.T.; Allen, R.S. Neuroprotective Strategies for Retinal Disease. Prog. Retin. Eye Res. 2018, 65, 50–76. [Google Scholar] [CrossRef] [PubMed]
- Chalke, S.D.; Kale, P.P. Combinational Approaches Targeting Neurodegeneration, Oxidative Stress, and Inflammation in the Treatment of Diabetic Retinopathy. Curr. Drug Targets 2021, 22, 1810–1824. [Google Scholar] [CrossRef]
- IZZOTTI, A.; BAGNIS, A.; SACCA, S. The Role of Oxidative Stress in Glaucoma. Mutat. Res. Mutat. Res. 2006, 612, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.; Mandala, A.; Malhotra, M.; Gnana-Prakasam, J.P. Canonical Wnt Signaling in the Pathology of Iron Overload-Induced Oxidative Stress and Age-Related Diseases. Oxid. Med. Cell. Longev. 2022, 2022, 7163326. [Google Scholar] [CrossRef]
- Su, W.; Li, Z.; Jia, Y.; Zhuo, Y. Rapamycin Is Neuroprotective in a Rat Chronic Hypertensive Glaucoma Model. PLoS ONE 2014, 9, e99719. [Google Scholar] [CrossRef] [PubMed]
- He, J.N.; Zhang, S.D.; Qu, Y.; Wang, H.L.; Tham, C.C.; Pang, C.P.; Chu, W.K. Rapamycin Removes Damaged Mitochondria and Protects Human Trabecular Meshwork (TM-1) Cells from Chronic Oxidative Stress. Mol. Neurobiol. 2019, 56, 6586–6593. [Google Scholar] [CrossRef]
- Heras-Sandoval, D.; Pérez-Rojas, J.M.; Hernández-Damián, J.; Pedraza-Chaverri, J. The Role of PI3K/AKT/MTOR Pathway in the Modulation of Autophagy and the Clearance of Protein Aggregates in Neurodegeneration. Cell. Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef]
- Ishikawa, M.; Takaseki, S.; Yoshitomi, T.; Covey, D.F.; Zorumski, C.F.; Izumi, Y. The Neurosteroid Allopregnanolone Protects Retinal Neurons by Effects on Autophagy and GABRs/GABAA Receptors in Rat Glaucoma Models. Autophagy 2020, 17, 743–760. [Google Scholar] [CrossRef]
- Liegl, R.; Koenig, S.; Siedlecki, J.; Haritoglou, C.; Kampik, A.; Kernt, M. Temsirolimus Inhibits Proliferation and Migration in Retinal Pigment Epithelial and Endothelial Cells via MTOR Inhibition and Decreases VEGF and PDGF Expression. PLoS ONE 2014, 9, e88203. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Wu, S.; Zeng, W.; Chen, X.; Zheng, T.; Ren, J.; Ke, M. Protective Effects of Rapamycin on Trabecular Meshwork Cells in Glucocorticoid-Induced Glaucoma Mice. Front. Pharmacol. 2020, 11, 1006. [Google Scholar] [CrossRef]
- Niu, Z.; Shi, Y.; Li, J.; Qiao, S.; Du, S.; Chen, L.; Tian, H.; Wei, L.; Cao, H.; Wang, J.; et al. Protective Effect of Rapamycin in Models of Retinal Degeneration. Exp. Eye Res. 2021, 210, 108700. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, N.; Honjo, M.; Aihara, M. Effects of Mammalian Target of Rapamycin Inhibitors on Fibrosis after Trabeculectomy. Exp. Eye Res. 2021, 203, 108421. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.F.; Liu, Y.; Yang, W.; Wan, J.; Yao, J.; Wan, Y.; Jiang, Q. Rapamycin Sensitive MTOR Activation Mediates Nerve Growth Factor (NGF) Induced Cell Migration and pro-Survival Effects against Hydrogen Peroxide in Retinal Pigment Epithelial Cells. Biochem. Biophys. Res. Commun. 2011, 414, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chen, F.; Yan, A.; Xia, X. Role of Mammalian Target of Rapamycin in Regulating HIF-1α and Vascular Endothelial Growth Factor Signals in Glaucoma. Arch. Physiol. Biochem. 2021, 127, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Shang, P.; Valapala, M.; Grebe, R.; Hose, S.; Ghosh, S.; Bhutto, I.A.; Handa, J.T.; Lutty, G.A.; Lu, L.; Wan, J.; et al. The Amino Acid Transporter SLC36A4 Regulates the Amino Acid Pool in Retinal Pigmented Epithelial Cells and Mediates the Mechanistic Target of Rapamycin, Complex 1 Signaling. Aging Cell 2017, 16, 349–359. [Google Scholar] [CrossRef] [Green Version]
- Harder, J.M.; Guymer, C.; Wood, J.P.M.; Daskalaki, E.; Chidlow, G.; Zhang, C.; Balasubramanian, R.; Cardozo, B.H.; Foxworth, N.E.; Deering, K.E.; et al. Disturbed Glucose and Pyruvate Metabolism in Glaucoma with Neuroprotection by Pyruvate or Rapamycin. Proc. Natl. Acad. Sci. USA 2020, 117, 33619–33627. [Google Scholar] [CrossRef]
- Schreiber, K.H.; Arriola Apelo, S.I.; Yu, D.; Brinkman, J.A.; Velarde, M.C.; Syed, F.A.; Liao, C.Y.; Baar, E.L.; Carbajal, K.A.; Sherman, D.S.; et al. A Novel Rapamycin Analog Is Highly Selective for MTORC1 in Vivo. Nat. Commun. 2019, 10, 3194. [Google Scholar] [CrossRef]
- Ran, Z.; Zhang, Y.; Wen, X.; Ma, J. Curcumin Inhibits High Glucose-Induced Inflammatory Injury in Human Retinal Pigment Epithelial Cells through the ROS-PI3K/AKT/MTOR Signaling Pathway. Mol. Med. Rep. 2019, 19, 1024–1031. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The Mammalian Target of Rapamycin (MTOR) Partner, Raptor, Binds the MTOR Substrates P70 S6 Kinase and 4E-BP1 through Their TOR Signaling (TOS) Motif. J. Biol. Chem. 2003, 278, 15461–15464. [Google Scholar] [CrossRef] [Green Version]
- Hwang, Y.; Kim, L.C.; Song, W.; Edwards, D.N.; Cook, R.S.; Chen, J. Disruption of the Scaffolding Function of MLST8 Selectively Inhibits MTORC2 Assembly and Function and Suppresses MTORC2-Dependent Tumor Growth In Vivo. Cancer Res. 2019, 79, 3178–3184. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Harris, T.E.; Roth, R.A.; Lawrence, J.C. PRAS40 Regulates MTORC1 Kinase Activity by Functioning as a Direct Inhibitor of Substrate Binding. J. Biol. Chem. 2007, 282, 20036–20044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 Maintains Rictor-MTOR Complex Integrity and Regulates Akt Phosphorylation and Substrate Specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultze, S.M.; Hemmings, B.A.; Niessen, M.; Tschopp, O. PI3K/AKT, MAPK and AMPK Signalling: Protein Kinases in Glucose Homeostasis. Expert Rev. Mol. Med. 2012, 14, e1. [Google Scholar] [CrossRef] [Green Version]
- Yoon, M.-S. The Role of Mammalian Target of Rapamycin (MTOR) in Insulin Signaling. Nutrients 2017, 9, 1176. [Google Scholar] [CrossRef]
- Rehbein, U.; Prentzell, M.T.; Cadena Sandoval, M.; Heberle, A.M.; Henske, E.P.; Opitz, C.A.; Thedieck, K. The TSC Complex-MTORC1 Axis: From Lysosomes to Stress Granules and Back. Front. Cell Dev. Biol. 2021, 9, 751892. [Google Scholar] [CrossRef]
- Valvezan, A.J.; Klein, P.S. GSK-3 and Wnt Signaling in Neurogenesis and Bipolar Disorder. Front. Mol. Neurosci. 2012, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Budanov, A.V.; Karin, M. P53 Target Genes Sestrin1 and Sestrin2 Connect Genotoxic Stress and MTOR Signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Sherratt, P.J.; Pickett, C.B. Regulatory Mechanisms Controlling Gene Expression Mediated by the Antioxidant Response Element. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 233–260. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.C.; Kang, K.A.; Zhang, R.; Piao, M.J.; Kim, G.Y.; Kang, M.Y.; Lee, S.J.; Lee, N.H.; Surh, Y.-J.; Hyun, J.W. Up-Regulation of Nrf2-Mediated Heme Oxygenase-1 Expression by Eckol, a Phlorotannin Compound, through Activation of Erk and PI3K/Akt. Int. J. Biochem. Cell Biol. 2010, 42, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-S.; Zhang, X.; Wu, Q.; Li, W.; Zhang, Q.-R.; Wang, C.-X.; Zhou, X.-M.; Li, H.; Shi, J.-X.; Zhou, M.-L. Astaxanthin Alleviates Early Brain Injury Following Subarachnoid Hemorrhage in Rats: Possible Involvement of Akt/Bad Signaling. Mar. Drugs 2014, 12, 4291–4310. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, N.; Takahashi, R.; Yoshino, K.; Tanimura, K.; Nakashima, A.; Eguchi, S.; Miyamoto, T.; Hara, K.; Takehana, K.; Avruch, J.; et al. The Proline-Rich Akt Substrate of 40 KDa (PRAS40) Is a Physiological Substrate of Mammalian Target of Rapamycin Complex 1. J. Biol. Chem. 2007, 282, 20329–20339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schalm, S.S.; Blenis, J. Identification of a Conserved Motif Required for MTOR Signaling. Curr. Biol. 2002, 12, 632–639. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Xu, G. Autophagy: A Role in the Apoptosis, Survival, Inflammation, and Development of the Retina. Ophthalmic Res. 2019, 61, 65–72. [Google Scholar] [CrossRef]
- Cherra 3rd, S.J.; Chu, C.T. Autophagy in Neuroprotection and Neurodegeneration: A Question of Balance. Future Neurol. 2008, 3, 309–323. [Google Scholar] [CrossRef]
- Park, H.Y.L.; Kim, J.H.; Park, C.K. Different Contributions of Autophagy to Retinal Ganglion Cell Death in the Diabetic and Glaucomatous Retinas. Sci. Rep. 2018, 8, 13321. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.J.; Quijano, C.; Chen, E.; Liu, H.; Cao, L.; Fergusson, M.M.; Rovira, I.I.; Gutkind, S.; Daniels, M.P.; Komatsu, M.; et al. Mitochondrial Dysfunction and Oxidative Stress Mediate the Physiological Impairment Induced by the Disruption of Autophagy. Aging 2009, 1, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Tal, M.C.; Sasai, M.; Lee, H.K.; Yordy, B.; Shadel, G.S.; Iwasaki, A. Absence of Autophagy Results in Reactive Oxygen Species-Dependent Amplification of RLR Signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 2770–2775. [Google Scholar] [CrossRef] [Green Version]
- Murata, H.; Takamatsu, H.; Liu, S.; Kataoka, K.; Huh, N.; Sakaguchi, M. NRF2 Regulates PINK1 Expression under Oxidative Stress Conditions. PLoS ONE 2015, 10, e0142438. [Google Scholar] [CrossRef] [PubMed]
- Losiewicz, M.K.; Elghazi, L.; Fingar, D.C.; Rajala, R.V.S.; Lentz, S.I.; Fort, P.E.; Abcouwer, S.F.; Gardner, T.W. MTORC1 and MTORC2 Expression in Inner Retinal Neurons and Glial Cells. Exp. Eye Res. 2020, 197, 108131. [Google Scholar] [CrossRef] [PubMed]
- Ballou, L.M.; Lin, R.Z. Rapamycin and MTOR Kinase Inhibitors. J. Chem. Biol. 2008, 1, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellmunt, J.; Szczylik, C.; Feingold, J.; Strahs, A.; Berkenblit, A. Temsirolimus Safety Profile and Management of Toxic Effects in Patients with Advanced Renal Cell Carcinoma and Poor Prognostic Features. Ann. Oncol. 2008, 19, 1387–1392. [Google Scholar] [CrossRef]
- Sellami, D.B.; Urva, S.R.; Grosch, K.; Cheung, W.K.; Anak, O. Meta-Analysis on the Relationship between Everolimus Exposure and Safety and Efficacy. J. Clin. Oncol. 2012, 30, 3099. [Google Scholar] [CrossRef]
- Hartford, C.M.; Desai, A.A.; Janisch, L.; Karrison, T.; Rivera, V.M.; Berk, L.; Loewy, J.W.; Kindler, H.; Stadler, W.M.; Knowles, H.L.; et al. A Phase I Trial to Determine the Safety, Tolerability, and Maximum Tolerated Dose of Deforolimus in Patients with Advanced Malignancies. Clin. Cancer Res. 2009, 15, 1428–1434. [Google Scholar] [CrossRef] [Green Version]
- Banerji, U.; Dean, E.J.; Gonzalez, M.; Greystoke, A.P.; Basu, B.; Krebs, M.; Puglisi, M.; Grinsted, L.; Oelmann, E.; Burke, W.; et al. First-in-Human Phase I Trial of the Dual MTORC1 and MTORC2 Inhibitor AZD2014 in Solid Tumors. J. Clin. Oncol. 2012, 30, 3004. [Google Scholar] [CrossRef]
- Tan, D.S.; Dumez, H.; Olmos, D.; Sandhu, S.K.; Hoeben, A.; Stephens, A.W.; Poondru, S.; Gedrich, R.; Kaye, S.B.; Schoffski, P. First-in-Human Phase I Study Exploring Three Schedules of OSI-027, a Novel Small Molecule TORC1/TORC2 Inhibitor, in Patients with Advanced Solid Tumors and Lymphoma. J. Clin. Oncol. 2010, 28, 3006. [Google Scholar] [CrossRef]
- Tabernero, J.; Cervantes, A.; Gordon, M.S.; Chiorean, E.G.; Burris, H.A.; Macarulla, T.; Perez-Fidalgo, A.; Martin, M.; Jessen, K.; Liu, Y.; et al. Abstract CT-02: A Phase I, Open Label, Dose Escalation Study of Oral Mammalian Target of Rapamycin Inhibitor INK128 Administered by Intermittent Dosing Regimens in Patients with Advanced Malignancies. In Proceedings of the Clinical Trials; American Association for Cancer Research: Philadelphia, PA, USA, 2012; p. CT-02-CT-02. [Google Scholar]
- Banerji, U.; Ranson, M.; Schellens, J.H.; Esaki, T.; Dean, E.; Zivi, A.; van der Noll, R.; Stockman, P.K.; Marotti, M.; Garrett, M.D.; et al. Abstract LB-66: Results of Two Phase I Multicenter Trials of AZD5363, an Inhibitor of AKT1, 2 and 3: Biomarker and Early Clinical Evaluation in Western and Japanese Patients with Advanced Solid Tumors. In Proceedings of the Clinical Trials; American Association for Cancer Research: Philadelphia, PA, USA, 2013; p. LB-66-LB-66. [Google Scholar]
- Spencer, A.; Yoon, S.-S.; Harrison, S.J.; Morris, S.; Smith, D.; Freedman, S.J.; Brigandi, R.; Oliff, A.; Opalinska, J.B.; Chen, C. Novel AKT Inhibitor GSK2110183 Shows Favorable Safety, Pharmacokinetics, and Clinical Activity in Multiple Myeloma. Preliminary Results From a Phase I First-Time-In-Human Study. Blood 2011, 118, 1856. [Google Scholar] [CrossRef]
- Tabernero, J.; Saura, C.; Roda Perez, D.; Dienstmann, R.; Rosello, S.; Prudkin, L.; Perez-Fidalgo, J.A.; Graña, B.; Jones, C.; Musib, L.; et al. First-in-Human Phase I Study Evaluating the Safety, Pharmacokinetics (PK), and Intratumor Pharmacodynamics (PD) of the Novel, Oral, ATP-Competitive Akt Inhibitor GDC-0068. J. Clin. Oncol. 2011, 29, 3022. [Google Scholar] [CrossRef]
- Ochiai, T.; Gunji, Y.; Nagata, M.; Komori, A.; Asano, T.; Isono, K. Effects of Rapamycin in Experimental Organ Allografting. Transplantation 1993, 56, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Bové, J.; Martínez-Vicente, M.; Vila, M. Fighting Neurodegeneration with Rapamycin: Mechanistic Insights. Nat. Rev. Neurosci. 2011, 12, 437–452. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Banerjee, S.; Huang, W.; Li, Y.; Wang, Z.; Kim, H.-R.C.; Sarkar, F.H. Mammalian Target of Rapamycin Repression by 3,3′-Diindolylmethane Inhibits Invasion and Angiogenesis in Platelet-Derived Growth Factor-D–Overexpressing PC3 Cells. Cancer Res. 2008, 68, 1927–1934. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Houghton, P.J. Resistance to Rapamycin: A Novel Anticancer Drug. Cancer Metastasis Rev. 2001, 20, 69–78. [Google Scholar] [CrossRef]
- Simamora, P.; Alvarez, J.M.; Yalkowsky, S.H. Solubilization of Rapamycin. Int. J. Pharm. 2001, 213, 25–29. [Google Scholar] [CrossRef]
- Elit, L. CCI-779 Wyeth. Curr. Opin. Investig. Drugs 2002, 3, 1249–1253. [Google Scholar]
- Dumont, F.J. Everolimus. Novartis. Curr. Opin. Investig. Drugs 2001, 2, 1220–1234. [Google Scholar]
- Mita, M.; Sankhala, K.; Abdel-Karim, I.; Mita, A.; Giles, F. Deforolimus (AP23573) a Novel MTOR Inhibitor in Clinical Development. Expert Opin. Investig. Drugs 2008, 17, 1947–1954. [Google Scholar] [CrossRef]
- Kwitkowski, V.E.; Prowell, T.M.; Ibrahim, A.; Farrell, A.T.; Justice, R.; Mitchell, S.S.; Sridhara, R.; Pazdur, R. FDA Approval Summary: Temsirolimus as Treatment for Advanced Renal Cell Carcinoma. Oncologist 2010, 15, 428–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chresta, C.M.; Davies, B.R.; Hickson, I.; Harding, T.; Cosulich, S.; Critchlow, S.E.; Vincent, J.P.; Ellston, R.; Jones, D.; Sini, P.; et al. AZD8055 Is a Potent, Selective, and Orally Bioavailable ATP-Competitive Mammalian Target of Rapamycin Kinase Inhibitor with in Vitro and in Vivo Antitumor Activity. Cancer Res. 2010, 70, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Xue, Q.; Hopkins, B.; Perruzzi, C.; Udayakumar, D.; Sherris, D.; Benjamin, L.E. Palomid 529, a Novel Small-Molecule Drug, Is a TORC1/TORC2 Inhibitor That Reduces Tumor Growth, Tumor Angiogenesis, and Vascular Permeability. Cancer Res. 2008, 68, 9551–9557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-Competitive Mammalian Target of Rapamycin Inhibitor Reveals Rapamycin-Resistant Functions of MTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrik-Outmezguine, V.S.; Chandarlapaty, S.; Pagano, N.C.; Poulikakos, P.I.; Scaltriti, M.; Moskatel, E.; Baselga, J.; Guichard, S.; Rosen, N. MTOR Kinase Inhibition Causes Feedback-Dependent Biphasic Regulation of AKT Signaling. Cancer Discov. 2011, 1, 248–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carew, J.S.; Kelly, K.R.; Nawrocki, S.T. Mechanisms of MTOR Inhibitor Resistance in Cancer Therapy. Target. Oncol. 2011, 6, 17–27. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and Molecular Cell Biology of Diabetic Complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Sifuentes-Franco, S.; Pacheco-Moisés, F.P.; Rodríguez-Carrizalez, A.D.; Miranda-Díaz, A.G. The Role of Oxidative Stress, Mitochondrial Function, and Autophagy in Diabetic Polyneuropathy. J. Diabetes Res. 2017, 2017, 2760716. [Google Scholar] [CrossRef] [Green Version]
- Zafar, S.; Sachdeva, M.; Frankfort, B.J.; Channa, R. Retinal Neurodegeneration as an Early Manifestation of Diabetic Eye Disease and Potential Neuroprotective Therapies. Curr. Diab. Rep. 2019, 19, 17. [Google Scholar] [CrossRef]
- Simó, R.; Stitt, A.W.; Gardner, T.W. Neurodegeneration in Diabetic Retinopathy: Does It Really Matter? Diabetologia 2018, 61, 1902–1912. [Google Scholar] [CrossRef] [Green Version]
- Hernández, C.; Simó, R. Neurodegeneration in Diabetic Retinopathy: Current Concepts and Therapeutic Implications. Av. Diabetol. 2014, 30, 72–79. [Google Scholar] [CrossRef]
- Wang, W.; Tam, K.C.; Ng, T.C.; Goit, R.K.; Chan, K.L.S.; Lo, A.C.Y. Long-Term Lutein Administration Attenuates Retinal Inflammation and Functional Deficits in Early Diabetic Retinopathy Using the Ins2 Akita/+ Mice. BMJ Open Diabetes Res. Care 2020, 8, e001519. [Google Scholar] [CrossRef]
- Sergeys, J.; Etienne, I.; Van Hove, I.; Lefevere, E.; Stalmans, I.; Feyen, J.H.M.; Moons, L.; Van Bergen, T. Longitudinal In Vivo Characterization of the Streptozotocin-Induced Diabetic Mouse Model: Focus on Early Inner Retinal Responses. Investig. Ophthalmol. Vis. Sci. 2019, 60, 807–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, T.S.; Berkowitz, B.A. Photoreceptors in Diabetic Retinopathy. J. Diabetes Investig. 2015, 6, 371–380. [Google Scholar] [CrossRef] [Green Version]
- de Hoz, R.; Rojas, B.; Ramírez, A.I.; Salazar, J.J.; Gallego, B.I.; Triviño, A.; Ramírez, J.M. Retinal Macroglial Responses in Health and Disease. Biomed Res. Int. 2016, 2016, 2954721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altmann, C.; Schmidt, M.H.H. The Role of Microglia in Diabetic Retinopathy: Inflammation, Microvasculature Defects and Neurodegeneration. Int. J. Mol. Sci. 2018, 19, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Shi, K.; Lu, H.; Lu, L.; Qiu, B. Mingmu Xiaomeng Tablets Restore Autophagy and Alleviate Diabetic Retinopathy by Inhibiting PI3K/Akt/MTOR Signaling. Front. Pharmacol. 2021, 12, 632040. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, H.; Su, S.B. Neuroinflammatory Responses in Diabetic Retinopathy. J. Neuroinflam. 2015, 12, 141. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Cang, X.; Zhu, L.; Zhu, M.; Li, A.; Wang, Z.; Zhang, Y.; Wang, X.; Song, E. PPP1CA/YAP/GS/Gln/MTORC1 Pathway Activates Retinal Müller Cells during Diabetic Retinopathy. Exp. Eye Res. 2021, 210, 108703. [Google Scholar] [CrossRef]
- De Faria, J.M.L.; Duarte, D.A.; Montemurro, C.; Papadimitriou, A.; Consonni, S.R.; De Faria, J.B.L. Defective Autophagy in Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4356–4366. [Google Scholar] [CrossRef] [Green Version]
- Taki, K.; Horie, T.; Kida, T.; Mimura, M.; Ikeda, T.; Oku, H. Impairment of Autophagy Causes Superoxide Formation and Caspase Activation in 661 w Cells, a Cell Line for Cone Photoreceptors, under Hyperglycemic Conditions. Int. J. Mol. Sci. 2020, 21, 4240. [Google Scholar] [CrossRef]
- Amato, R.; Catalani, E.; Dal Monte, M.; Cammalleri, M.; Di Renzo, I.; Perrotta, C.; Cervia, D.; Casini, G. Autophagy-Mediated Neuroprotection Induced by Octreotide in an Ex Vivo Model of Early Diabetic Retinopathy. Pharmacol. Res. 2018, 128, 167–178. [Google Scholar] [CrossRef]
- Madrakhimov, S.B.; Yang, J.Y.; Kim, J.H.; Han, J.W.; Park, T.K. MTOR-Dependent Dysregulation of Autophagy Contributes to the Retinal Ganglion Cell Loss in Streptozotocin-Induced Diabetic Retinopathy. Cell Commun. Signal. 2021, 19, 29. [Google Scholar] [CrossRef]
- Liu, Y.; Zheng, Y.; Zhou, Y.; Liu, Y.; Xie, M.; Meng, W.; An, M. The Expression and Significance of MTORC1 in Diabetic Retinopathy. BMC Ophthalmol. 2020, 20, 297. [Google Scholar] [CrossRef] [PubMed]
- Fox, T.E.; Young, M.M.; Pedersen, M.M.; Han, X.; Gardner, T.W.; Kester, M. Diabetes Diminishes Phosphatidic Acid in the Retina: A Putative Mediator for Reduced MTOR Signaling and Increased Neuronal Cell Death. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7257–7267. [Google Scholar] [CrossRef] [PubMed]
- Fort, P.E.; Losiewicz, M.K.; Pennathur, S.; Jefferson, L.S.; Kimball, S.R.; Abcouwer, S.F.; Gardner, T.W. MTORC1-Independent Reduction of Retinal Protein Synthesis in Type 1 Diabetes. Diabetes 2014, 63, 3077–3090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Yang, C.; Iyaswamy, A.; Krishnamoorthi, S.; Sreenivasmurthy, S.G.; Liu, J.; Wang, Z.; Tong, B.C.-K.; Song, J.; Lu, J.; et al. Balancing MTOR Signaling and Autophagy in the Treatment of Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 728. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.H.C.; Fung, F.K.C.; Lai, A.K.W.; Wong, I.Y.H.; Shih, K.C.; Lo, A.C.Y. Autophagic Upregulation Is Cytoprotective in Ischemia/Reperfusion-Injured Retina and Retinal Progenitor Cells. Int. J. Mol. Sci. 2021, 22, 8446. [Google Scholar] [CrossRef]
- Ozdemir, G.; Kılınç, M.; Ergün, Y.; Sahin, E. Rapamycin Inhibits Oxidative and Angiogenic Mediators in Diabetic Retinopathy. Can. J. Ophthalmol. 2014, 49, 443–449. [Google Scholar] [CrossRef]
- Chauhan, A.; Sharma, U.; Jagannathan, N.R.; Reeta, K.H.; Gupta, Y.K. Rapamycin Protects against Middle Cerebral Artery Occlusion Induced Focal Cerebral Ischemia in Rats. Behav. Brain Res. 2011, 225, 603–609. [Google Scholar] [CrossRef]
- Yang, X.; Wu, S.; Feng, Z.; Yi, G.; Zheng, Y.; Xia, Z. Combination Therapy with Semaglutide and Rosiglitazone as a Synergistic Treatment for Diabetic Retinopathy in Rodent Animals. Life Sci. 2021, 269, 119013. [Google Scholar] [CrossRef]
- Steinmetz, J.D.; Bourne, R.R.A.; Briant, P.S.; Flaxman, S.R.; Taylor, H.R.B.; Jonas, J.B.; Abdoli, A.A.; Abrha, W.A.; Abualhasan, A.; Abu-Gharbieh, E.G.; et al. Causes of Blindness and Vision Impairment in 2020 and Trends over 30 Years, and Prevalence of Avoidable Blindness in Relation to VISION 2020: The Right to Sight: An Analysis for the Global Burden of Disease Study. Lancet Glob. Health 2021, 9, e144–e160. [Google Scholar] [CrossRef]
- Evans, J.R. 28 000 Cases of Age Related Macular Degeneration Causing Visual Loss in People Aged 75 Years and above in the United Kingdom May Be Attributable to Smoking. Br. J. Ophthalmol. 2005, 89, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Bowes Rickman, C.; Farsiu, S.; Toth, C.A.; Klingeborn, M. Dry Age-Related Macular Degeneration: Mechanisms, Therapeutic Targets, and Imaging. Investig. Opthalmol. Vis. Sci. 2013, 54, ORSF68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollyfield, J.G.; Bonilha, V.L.; Rayborn, M.E.; Yang, X.; Shadrach, K.G.; Lu, L.; Ufret, R.L.; Salomon, R.G.; Perez, V.L. Oxidative Damage–Induced Inflammation Initiates Age-Related Macular Degeneration. Nat. Med. 2008, 14, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I. Oxidative Stress in Age-Related Macular Degeneration: Nrf2 as Therapeutic Target. Front. Pharmacol. 2018, 9, 1280. [Google Scholar] [CrossRef] [PubMed]
- Mulfaul, K.; Ozaki, E.; Fernando, N.; Brennan, K.; Chirco, K.R.; Connolly, E.; Greene, C.; Maminishkis, A.; Salomon, R.G.; Linetsky, M.; et al. Toll-like Receptor 2 Facilitates Oxidative Damage-Induced Retinal Degeneration. Cell Rep. 2020, 30, 2209–2224.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid. Med. Cell. Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef] [Green Version]
- Piippo, N.; Korhonen, E.; Hytti, M.; Kinnunen, K.; Kaarniranta, K.; Kauppinen, A. Oxidative Stress Is the Principal Contributor to Inflammasome Activation in Retinal Pigment Epithelium Cells with Defunct Proteasomes and Autophagy. Cell. Physiol. Biochem. 2018, 49, 359–367. [Google Scholar] [CrossRef]
- Kim, S.-Y.; Kambhampati, S.P.; Bhutto, I.A.; McLeod, D.S.; Lutty, G.A.; Kannan, R.M. Evolution of Oxidative Stress, Inflammation and Neovascularization in the Choroid and Retina in a Subretinal Lipid Induced Age-Related Macular Degeneration Model. Exp. Eye Res. 2021, 203, 108391. [Google Scholar] [CrossRef]
- Abokyi, S.; To, C.-H.; Lam, T.T.; Tse, D.Y. Central Role of Oxidative Stress in Age-Related Macular Degeneration: Evidence from a Review of the Molecular Mechanisms and Animal Models. Oxid. Med. Cell. Longev. 2020, 2020, 7901270. [Google Scholar] [CrossRef] [Green Version]
- Imamura, Y.; Noda, S.; Hashizume, K.; Shinoda, K.; Yamaguchi, M.; Uchiyama, S.; Shimizu, T.; Mizushima, Y.; Shirasawa, T.; Tsubota, K. Drusen, Choroidal Neovascularization, and Retinal Pigment Epithelium Dysfunction in SOD1-Deficient Mice: A Model of Age-Related Macular Degeneration. Proc. Natl. Acad. Sci. USA 2006, 103, 11282–11287. [Google Scholar] [CrossRef] [Green Version]
- Justilien, V.; Pang, J.-J.; Renganathan, K.; Zhan, X.; Crabb, J.W.; Kim, S.R.; Sparrow, J.R.; Hauswirth, W.W.; Lewin, A.S. SOD2 Knockdown Mouse Model of Early AMD. Investig. Opthalmol. Vis. Sci. 2007, 48, 4407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Jia, L.; Khan, N.; Lin, C.; Mitter, S.K.; Boulton, M.E.; Dunaief, J.L.; Klionsky, D.J.; Guan, J.L.; Thompson, D.A.; et al. Deletion of Autophagy Inducer RB1CC1 Results in Degeneration of the Retinal Pigment Epithelium. Autophagy 2015, 11, 939–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W.; Ding, J.; et al. Dysregulated Autophagy in the RPE Is Associated with Increased Susceptibility to Oxidative Stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [Green Version]
- Stone, W.L.; Farnsworth, C.C.; Dratz, E.A. A Reinvestigation of the Fatty Acid Content of Bovine, Rat and Frog Retinal Rod Outer Segments. Exp. Eye Res. 1979, 28, 387–397. [Google Scholar] [CrossRef]
- Fernandes, S.A.; Demetriades, C. The Multifaceted Role of Nutrient Sensing and MTORC1 Signaling in Physiology and Aging. Front. Aging 2021, 2, 38. [Google Scholar] [CrossRef]
- Arjamaa, O.; Nikinmaa, M.; Salminen, A.; Kaarniranta, K. Regulatory Role of HIF-1α in the Pathogenesis of Age-Related Macular Degeneration (AMD). Ageing Res. Rev. 2009, 8, 349–358. [Google Scholar] [CrossRef]
- Wang, M.; Li, Y.J.; Ding, Y.; Zhang, H.N.; Sun, T.; Zhang, K.; Yang, L.; Guo, Y.Y.; Liu, S.B.; Zhao, M.G.; et al. Silibinin Prevents Autophagic Cell Death upon Oxidative Stress in Cortical Neurons and Cerebral Ischemia-Reperfusion Injury. Mol. Neurobiol. 2016, 53, 932–943. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, H.; Zhang, Y.F.; Zhou, Z.; Wu, S. MicroRNA-29 Enhances Autophagy and Cleanses Exogenous Mutant AB-Crystallin in Retinal Pigment Epithelial Cells. Exp. Cell Res. 2019, 374, 231–248. [Google Scholar] [CrossRef]
- Park, T.K.; Lee, S.H.; Choi, J.S.; Nah, S.K.; Kim, H.J.; Park, H.Y.; Lee, H.; Lee, S.H.S.; Park, K. Adeno-Associated Viral Vector-Mediated MTOR Inhibition by Short Hairpin RNA Suppresses Laser-Induced Choroidal Neovascularization. Mol. Ther.—Nucleic Acids 2017, 8, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Ebeling, M.C.; Polanco, J.R.; Qu, J.; Tu, C.; Montezuma, S.R.; Ferrington, D.A. Improving Retinal Mitochondrial Function as a Treatment for Age-Related Macular Degeneration. Redox Biol. 2020, 34, 101552. [Google Scholar] [CrossRef]
- Cheng, L.; Cheng, L.; Bi, H.; Zhang, Z.; Yao, J.; Zhou, X.; Jiang, Q. Alpha-Melanocyte Stimulating Hormone Protects Retinal Pigment Epithelium Cells from Oxidative Stress through Activation of Melanocortin 1 Receptor-Akt-MTOR Signaling. Biochem. Biophys. Res. Commun. 2014, 443, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Josifovska, N.; Albert, R.; Nagymihály, R.; Lytvynchuk, L.; Moe, M.C.; Kaarniranta, K.; Veréb, Z.J.; Petrovski, G. Resveratrol as Inducer of Autophagy, pro-Survival, and Anti-Inflammatory Stimuli in Cultured Human RPE Cells. Int. J. Mol. Sci. 2020, 21, 813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.L.; Chen, Y.H.; Liang, C.M.; Tai, M.C.; Lu, D.W.; Chen, J.T. Glucosamine-Induced Autophagy through AMPK–MTOR Pathway Attenuates Lipofuscin-like Autofluorescence in Human Retinal Pigment Epithelial Cells in Vitro. Int. J. Mol. Sci. 2018, 19, 1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Sun, Y.; López, F.J.; Adamson, P.; Kurali, E.; Lashkari, K. Blockage of PI3K/MTOR Pathways Inhibits Laser-Induced Choroidal Neovascularization and Improves Outcomes Relative to VEGF-A Suppression Alone. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3138–3144. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.S.; Chang, H.S.; Kim, H.J.; Choi, J.S.; Kim, J.; Kim, J.H.; Woo, H.N.; Nah, S.K.; Jung, S.J.; Lee, J.Y.; et al. Effects of Stuffer DNA on the Suppression of Choroidal Neovascularization by a RAAV Expressing a MTOR-Inhibiting ShRNA. Mol. Ther.—Methods Clin. Dev. 2019, 14, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Cruz, P.M. Mutation of the Receptor Tyrosine Kinase Gene Mertk in the Retinal Dystrophic RCS Rat. Hum. Mol. Genet. 2000, 9, 645–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gal, A.; Li, Y.; Thompson, D.A.; Weir, J.; Orth, U.; Jacobson, S.G.; Apfelstedt-Sylla, E.; Vollrath, D. Mutations in MERTK, the Human Orthologue of the RCS Rat Retinal Dystrophy Gene, Cause Retinitis Pigmentosa. Nat. Genet. 2000, 26, 270–271. [Google Scholar] [CrossRef]
- Xue, G.; Kohler, R.; Tang, F.; Hynx, D.; Wang, Y.; Orso, F.; Prêtre, V.; Ritschard, R.; Hirschmann, P.; Cron, P.; et al. MTORC1/Autophagy-Regulated MerTK in Mutant BRAFV600 Melanoma with Acquired Resistance to BRAF Inhibition. Oncotarget 2017, 8, 69204–69218. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.-L.; Zou, T.-D.; Yang, F.; Yang, Z.-L.; Zhang, H.-B. Inhibition of MTOR Signaling by Rapamycin Protects Photoreceptors from Degeneration in Rd1 Mice. Zool. Res. 2021, 42, 482–486. [Google Scholar] [CrossRef]
- Resnikoff, S.; Pascolini, D.; Etya’ale, D.; Kocur, I.; Pararajasegaram, R.; Pokharel, G.P.; Mariotti, S.P. Global Data on Visual Impairment in the Year 2002. Bull. World Health Organ. 2004, 82, 844–851. [Google Scholar]
- Ventura, L.M.; Sorokac, N.; Santos, R.D.L.; Feuer, W.J.; Porciatti, V. The Relationship between Retinal Ganglion Cell Function and Retinal Nerve Fiber Thickness in Early Glaucoma. Investig. Opthalmol. Vis. Sci. 2006, 47, 3904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, B.M.; Crawley, L.; Pahlitzsch, M.; Javaid, F.; Cordeiro, M.F. Glaucoma: The Retina and Beyond. Acta Neuropathol. 2016, 132, 807–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belforte, N.; Agostinone, J.; Alarcon-Martinez, L.; Villafranca-Baughman, D.; Dotigny, F.; Cueva Vargas, J.L.; Di Polo, A. AMPK Hyperactivation Promotes Dendrite Retraction, Synaptic Loss, and Neuronal Dysfunction in Glaucoma. Mol. Neurodegener. 2021, 16, 43. [Google Scholar] [CrossRef] [PubMed]
- Del Olmo-Aguado, S.; Núñez-Álvarez, C.; Ji, D.; Manso, A.G.; Osborne, N.N. RTP801 Immunoreactivity in Retinal Ganglion Cells and Its Down-Regulation in Cultured Cells Protect Them from Light and Cobalt Chloride. Brain Res. Bull. 2013, 98, 132–144. [Google Scholar] [CrossRef] [Green Version]
- Fung, F.K.C.; Law, B.Y.K.; Lo, A.C.Y. Lutein Attenuates Both Apoptosis and Autophagy upon Cobalt (II) Chloride-Induced Hypoxia in Rat Muller Cells. PLoS ONE 2016, 11, e0167828. [Google Scholar] [CrossRef]
- Yao, A.; van Wijngaarden, P. Metabolic Pathways in Context: MTOR Signalling in the Retina and Optic Nerve—A Review. Clin. Exp. Ophthalmol. 2020, 48, 1072–1084. [Google Scholar] [CrossRef]
- Yu, A.K.; Datta, S.; McMackin, M.Z.; Cortopassi, G.A. Rescue of Cell Death and Inflammation of a Mouse Model of Complex 1-Mediated Vision Loss by Repurposed Drug Molecules. Hum. Mol. Genet. 2017, 26, 4929–4936. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Liton, P.B. The Autophagic Lysosomal System in Outflow Pathway Physiology and Pathophysiology. Exp. Eye Res. 2016, 144, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Abu-Hassan, D.W.; Acott, T.S.; Kelley, M.J. The Trabecular Meshwork: A Basic Review of Form and Function. J. Ocul. Biol. 2014, 2. [Google Scholar] [CrossRef]
- Porter, K.; Hirt, J.; Stamer, W.D.; Liton, P.B. Autophagic Dysregulation in Glaucomatous Trabecular Meshwork Cells. Biochim. Biophys. Acta 2015, 1852, 379–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medchalmi, S.; Tare, P.; Sayyad, Z.; Swarup, G. A Glaucoma- and ALS-Associated Mutant of OPTN Induces Neuronal Cell Death Dependent on Tbk1 Activity, Autophagy and ER Stress. FEBS J. 2021, 288, 4576–4595. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, M.L.S.; Kumari, A.; Radha, V.; Swarup, G. E50K-OPTN-Induced Retinal Cell Death Involves the Rab GTPase-Activating Protein, TBC1D17 Mediated Block in Autophagy. PLoS ONE 2014, 9, e95758. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.L.; Zhao, G.L.; Hou, Y.; Zhong, S.M.; Xu, L.J.; Li, F.; Niu, W.R.; Yuan, F.; Yang, X.L.; Wang, Z.; et al. Rac1 Conditional Deletion Attenuates Retinal Ganglion Cell Apoptosis by Accelerating Autophagic Flux in a Mouse Model of Chronic Ocular Hypertension. Cell Death Dis. 2020, 11, 734. [Google Scholar] [CrossRef] [PubMed]
- Kitaoka, Y.; Munemasa, Y.; Kojima, K.; Hirano, A.; Ueno, S.; Takagi, H. Axonal Protection by Nmnat3 Overexpression with Involvement of Autophagy in Optic Nerve Degeneration. Cell Death Dis. 2013, 4, e860. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.H.; Turturro, S.S.; Nguyen, T.T.; Shen, X.X.; Zelkha, R.R.; Johnson, E.E.C.; Morrison, J.J.C.; Yue, B.B.Y.J.T. Induction of Autophagy in Rats upon Overexpression of Wild-Type and Mutant Optineurin Gene. BMC Cell Biol. 2015, 16, 14. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Shim, K.S.; Kim, C.Y.; Park, T.K. Characterization of the Role of Autophagy in Retinal Ganglion Cell Survival over Time Using a Rat Model of Chronic Ocular Hypertension. Sci. Rep. 2021, 11, 5767. [Google Scholar] [CrossRef]
- Krishnadev, N.; Forooghian, F.; Cukras, C.; Wong, W.; Saligan, L.; Chew, E.Y.; Nussenblatt, R.; Ferris, F.; Meyerle, C. Subconjunctival Sirolimus in the Treatment of Diabetic Macular Edema. Graefe’s Arch. Clin. Exp. Ophthalmol. 2011, 249, 1627. [Google Scholar] [CrossRef] [Green Version]
- Dugel, P.U.; Blumenkranz, M.S.; Haller, J.A.; Williams, G.A.; Solley, W.A.; Kleinman, D.M.; Naor, J. A Randomized, Dose-Escalation Study of Subconjunctival and Intravitreal Injections of Sirolimus in Patients with Diabetic Macular Edema. Ophthalmology 2012, 119, 124–131. [Google Scholar] [CrossRef]
- Wong, W.T.; Dresner, S.; Forooghian, F.; Glaser, T.; Doss, L.; Zhou, M.; Cunningham, D.; Shimel, K.; Harrington, M.; Hammel, K.; et al. Treatment of Geographic Atrophy With Subconjunctival Sirolimus: Results of a Phase I/II Clinical Trial. Investig. Opthalmol. Vis. Sci. 2013, 54, 2941. [Google Scholar] [CrossRef] [Green Version]
- Petrou, P.A.; Cunningham, D.; Shimel, K.; Harrington, M.; Hammel, K.; Cukras, C.A.; Ferris, F.L.; Chew, E.Y.; Wong, W.T. Intravitreal Sirolimus for the Treatment of Geographic Atrophy: Results of a Phase I/II Clinical Trial. Investig. Ophthalmol. Vis. Sci. 2014, 56, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Gensler, G.; Clemons, T.E.; Domalpally, A.; Danis, R.P.; Blodi, B.; Wells, J.; Rauser, M.; Hoskins, J.; Hubbard, G.B.; Elman, M.J.; et al. Treatment of Geographic Atrophy with Intravitreal Sirolimus: The Age-Related Eye Disease Study 2 Ancillary Study. Ophthalmol. Retin. 2018, 2, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Kirubakaran, S.; Hur, W.; Niepel, M.; Westover, K.; Thoreen, C.C.; Wang, J.; Ni, J.; Patricelli, M.P.; Vogel, K.; et al. Kinome-Wide Selectivity Profiling of ATP-Competitive Mammalian Target of Rapamycin (MTOR) Inhibitors and Characterization of Their Binding Kinetics. J. Biol. Chem. 2012, 287, 9742–9752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussenblatt, R.B.; Byrnes, G.; Sen, H.N.; Yeh, S.; Faia, L.; Meyerle, C.; Wroblewski, K.; Li, Z.; Liu, B.; Chew, E.; et al. A Randomized Pilot Study of Systemic Immunosuppression in the Treatment of Age-Related Macular Degeneration with Choroidal Neovascularization. Retina 2010, 30, 1579–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minturn, R.J.; Bracha, P.; Klein, M.J.; Chhablani, J.; Harless, A.M.; Maturi, R.K. Intravitreal Sirolimus for Persistent, Exudative Age-Related Macular Degeneration: A Pilot Study. Int. J. Retin. Vitr. 2021, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Dalal, M.; Jacobs-El, N.; Nicholson, B.; Tuo, J.; Chew, E.; Chan, C.C.; Nussenblatt, R.; Ferris, F.; Meyerle, C. Subconjunctival Palomid 529 in the Treatment of Neovascular Age-Related Macular Degeneration. Graefe’s Arch. Clin. Exp. Ophthalmol. 2013, 251, 2705–2709. [Google Scholar] [CrossRef]
- Pravin, N.J.D. A Multicenter, Randomized, Double-Masked, Dose-Ranging, Placebo-Controlled Phase 2 Study to Assess Sirolimus in the Treatment of Patients With Diabetic Macular Edema (DIAMOND Study). Investig. Ophthalmol. Vis. Sci 2010, 51, 4244. [Google Scholar]
- Phase 1/2 Study of an Ocular Sirolimus (Rapamycin) Formulation in Patients with Age-Related Macular Degeneration; 2010. Available online: https://www.clinicaltrials.gov/ct2/show/NCT00712491 (accessed on 30 May 2022).
- Abraham, P.; Yue, H.; Wilson, L. Randomized, Double-Masked, Sham-Controlled Trial of Ranibizumab for Neovascular Age-Related Macular Degeneration: PIER Study Year 2. Am. J. Ophthalmol. 2010, 150, 315–324.e1. [Google Scholar] [CrossRef]
- Fang, Y.; Westbrook, R.; Hill, C.; Boparai, R.K.; Arum, O.; Spong, A.; Wang, F.; Javors, M.A.; Chen, J.; Sun, L.Y.; et al. Duration of Rapamycin Treatment Has Differential Effects on Metabolism in Mice. Cell Metab. 2013, 17, 456–462. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Zheng, X.; Jia, X.; Binderiya, U.; Wang, Y.; Bao, W.; Bao, L.; Zhao, K.; Fu, Y.; Hao, H.; et al. A Quantitative Transcriptomic Analysis of the Physiological Significance of MTOR Signaling in Goat Fetal Fibroblasts. BMC Genomics 2016, 17, 879. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.T.; Zhang, J.R.; Kapupara, K.; Tsai, R.K. MTORC2 Activation Protects Retinal Ganglion Cells via Akt Signaling after Autophagy Induction in Traumatic Optic Nerve Injury. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Target | Potency (IC50, nM) | Pros/Cons | Development Status | Adverse Effects |
|---|---|---|---|---|---|
| 1st generation mTOR inhibitors | |||||
| Rapamycin | mTOR/FKBP12 | 0.1 | 1st FDA-approved mTOR inhibitor/ low biological utilisation due to its poor water solubility and stability | FDA-approved | Hyperglycaemia, fatigue, nausea/vomiting, anaemia, stomatitis, mucositis, pulmonary and metabolic toxicities [54,55,56] |
| Temsirolimus | mTOR/FKBP12 | 1.76 | Relatively high water solubility and stability, intravenous administration only | FDA-approved | |
| Everolimus | mTOR/FKBP12 | 1.6–2.4 | Relatively high water solubility and stability, low toxicity and high efficacy for some types of tumours | FDA-approved | |
| Ridaforolimus | mTOR/FKBP12 | 0.2–5.6 | Latest developed rapalogs, well-tolerated in children | FDA-approved | |
| 2nd generation mTOR inhibitors | |||||
| Torin1 | mTORC1/mTORC2 | 0.29 (mTORC1)/5 (mTOR) | Strong anti-proliferation activity/poor stability and low oral bioavailability | Preclinical | Hyperglycaemia, fatigue, nausea/vomiting, stomatitis, mucositis, diarrhoea, decreased appetite, liver dysfunction, pneumonia [57,58,59] |
| PP242 | mTORC1/mTORC2 | 8 (mTOR) | Relatively strong selectivity to mTOR | Preclinical | |
| AZD8055 | mTORC1/mTORC2 | 10 (mTORC1)/2.8 (mTOR) | Potent anti-proliferation and apoptosis induction activity/relatively high liver toxicity | Phase I | |
| OSI-027 | mTORC1/mTORC2 | 4 (mTORC1)/22.6 (mTOR) | Strong inhibitory effects on mTOR, dose-dependent manner in patients with some types of tumours | Phase I | |
| PI-103 | mTOR/PI3K | 3–3.6 (PI3K) | 1st developed mTOR/PI3K dual inhibitor/poor drug properties | Preclinical | Hyperglycaemia, fatigue, nausea/vomiting, mucositis, diarrhoea, decreased appetite, rash [60,61,62] |
| GSK2126458 | mTOR/PI3K | 0.18 (mTORC1)/0.019–0.13 (PI3K) | Confirmed target engagement in blood and lungs/affect insulin release and blood glucose level | Preclinical | |
| NVP-BEZ235 | mTOR/PI3K | mTOR (20.7)/4–75 (PI3K) | Potent PI3K inhibitory effects on PI3K | Phase I |
| Target Cells or Tissue | Disease Model | mTOR Regulator | Autophagy-Related Markers | Related Pathways | Effects of Regulated mTOR | References |
|---|---|---|---|---|---|---|
| R28 cells | Hypoxia-induced AMD model | Insulin | LC3A↓ | PI3K/AKT/mTOR↑ | Oxidative stress↓ VEGF↓ | [49] |
| rMC-1 | HG | Rapamycin | Beclin1↑ p62↓ | mTOR↓ | Apoptosis↓ VEGF↓ | [90] |
| ARPE-19 | HG | Curcumin | - | PI3K/AKT/mTOR↓ | TNF-α/ IL-1β/IL-6↓ | |
| 661W cells | HG | 3-MA | LC3B2↓ p62↑ | PI3K/AKT/mTOR↑ | ROS↑ Mitophagy↓ Apoptosis↑ | [91] |
| Ex vivo mouse retinal explants | HG | Octreotide | LC3-II↑ LC3-II net flux↑ | mTOR/S6K1↓ | Apoptosis↑ | [92] |
| RGCs | STZ-induced diabetic rats | 3-MA | LC3B↓ Beclin-1↑ | AMPK↓/mTOR↑ | Apoptosis↑ | [28] |
| RGCs | STZ-induced diabetic mice | Rapamycin | - | mTOR/S6K1↓ | GLUT1↓ GFAP↓ | [93] |
| RMCs | STZ-induced diabetic rats/HG | PPP1CA | - | YAP/GS/Gln/ mTORC1↑ | RMCs activation/ proliferation↑ | [89] |
| RMCs | STZ-induced diabetic rats | MMXM | LC3-II↑ p62↓ | PI3K/AKT/mTOR↓ | IL-1β/ IL-6↓ VEGF↓ GFAP↓ | [87] |
| Retina tissue | STZ-induced diabetic rats | Rapamycin | - | mTORC1/S6K1↓ | VEGF↓ PEDF↓ HRCECs proliferation/migration↓ | [94] |
| Retina tissue | STZ-induced diabetic rats | Phosphatidic acid | - | mTOR/S6K1↑ | Apoptosis↓ | [95] |
| Retina tissue | STZ-induced diabetic rats/Ins2Akita mice | Insulin/phloridzin | - | AKT/mTORC2↑ mTORC1/S6K1/ 4E-BP1 ↔ | Retinal protein synthesis↑ | [96] |
| Target Cells or Tissue | Disease Model | mTOR Regulator | Autophagy-related Markers | Related Pathways | Effects of Regulated mTOR | Reference |
|---|---|---|---|---|---|---|
| ARPE-19 | H2O2-induced RPE cell injury model | Silibinin | LC3A↓ | PI3K/AKT/mTOR↓ | Oxidative stress↓ VEGF↓ | [46] |
| ARPE-19/hRPE | H2O2-induced RPE cell injury model | a-MSH | - | PI3K/AKT/mTOR↑ | Oxidative stress↓ Apoptosis↓ | [123] |
| hRPE/HUVEC | Hypoxia-induced RPE cell injury model | Temsirolimus | - | mTOR↓ | VEGF↓ PEDF↓ | [19] |
| hRPE | Human AMD patient | Rapamycin | LC3-II/I↑ | mTOR↓ | Mt function↑ Mitophagy↑ | [122] |
| ARPE-19/hRPE | H2O2-induced RPE cell injury model (acute/chronic) | Rapamycin | LC3 puncta↑ | mTOR↓ | Oxidative stress↓ ROS↓ Lipofuscin-like deposit↓ | [115] |
| ARPE-19 | H2O2-induced RPE cell injury model | Resveratrol | LC3-II/I↑ p62↓ | mTOR↓ | Apoptosis↓ VEGFA↓ IL-6/ IL-8↓ | [124] |
| ARPE-19 | Lipid-peroxidation-induced RPE injury model | Glucosamine | LC3-II/I↑ p62 ↗↘ | AMPK↑/mTOR↓ | Lipofuscin-like deposit↓ | [125] |
| ARPE-19/hRPE | αB-crystallin R120G-mutation-induced protein aggregation model | miR-29 | LC3-II/I↑ p62↓ | mTOR↓ | Protein aggregation↓ | [120] |
| Retina tissue | Laser-induced model of CNV | GSK2126458 | - | PI3K/mTOR↓ | Vascular leakage↓ CNV lesions↓ Apoptosis↓ Serum glucose level↑ | [126] |
| Retina tissue | Laser-induced model of CNV | rAAV-mTOR shRNA | LC3B↑ ATG7↑ | PI3K/mTOR↓ | Vascular leakage↓ CNV lesions↓ Apoptosis↓ | [127] |
| Retina tissue | Laser-induced model of CNV | rAAV2-shmTOR-SD | - | mTOR↓ | CNV lesions↓ Apoptosis↓ | [127] |
| Retina tissue | NaIO3-induced retinal degeneration | Rapamycin | - | mTOR↓ | Oxidative stress↓ Apoptosis↓ GFAP↓ IL-6/ MCP-1/TNF-α↓ | [21] |
| Target Cells or Tissue | Disease Model | mTOR Regulator | Autophagy-Related Markers | Related Pathways | Effects of Regulated mTOR | References |
|---|---|---|---|---|---|---|
| NSC-34 /661W cells | 2bpIns-OPTN-induced cell death | Rapamycin | LC LC3-II/I↑↑ LC3↑ ATG5↑ | mTOR↓ | Apoptosis↓ ER stress↓ | [144] |
| TM-1 cells | Rotenone-induced oxidative stress model | Rapamycin | LC3-II/I↑ p62↓ | PI3K/AKT/mTOR↑ | Apoptosis↓ Oxidative stress↓Mitophagy↑ | [16] |
| RGC-5 | E50K-OPTN-induced RGC death | Rapamycin | - | mTOR↓ | Apoptosis↓ | [145] |
| Retina tissue/RGC-5 | Rat CoCl2-induced hypoxia model | Rapamycin | - | mTOR/RhoA/ROCK↓ | IOP↓ RGCs loss↓ Microglial activation↓Mitophagy↑ | [119] |
| HCF cells/TM cells | TGFβ1-induced fibrosis/rabbit model of glaucoma filtration surgery | Rapamycin/Torin-1 | - | AKT/mTOR↓ | HCF proliferation/migration↓ TM fibrosis↓ | [22] |
| RGCs/TM cells | Mouse glucocorticoid-induced glaucoma model | Rapamycin | LC3-II/I↑ Beclin-1↑ p62↓ | mTOR↓ | IOP↓ RGCs loss↓ TM fibrosis↓ Mitophagy↑ | [20] |
| RGCs | Mouse chronic hypertensive glaucoma model | Rac1 cKO | LC3-II/I↑ Beclin-1↑ p62↓ | mTOR↓ | Apoptosis↓ RGCs loss↓ | [146] |
| RGCs | Rat hypertensive glaucoma model | 3-MA | LC3B↓ Beclin-1↓ | AMPK↓/mTOR↑ | Apoptosis↑ | [48] |
| RGCs | Rat hypertensive glaucoma model | Rapamycin | LC3-II↑ p62↓ | mTOR↓ | Axon loss↓ | [147] |
| RGCs | E50K-OPTN-induced normal tension glaucoma model | Rapamycin | LC3↑ p62↓ | mTOR↓ | Apoptosis↓ Axon loss↓ | [148] |
| RGCs | DBA 2J mouse model for experimental glaucoma | Rapamycin | - | mTOR↓ | Apoptosis↓ Axon loss↓ | [26] |
| RGCs | Rat microbead occlusion model/ex vivo rat glaucoma model | Rapamycin | LC3-II/I↑ p62↓ | mTOR↓ | Apoptosis↓ RGCs loss↓ | [18] |
| RGCs | E50K-OPTN-induced RGC death | Rapamycin | LC3-II↑ p62↓ | mTOR↓ | Apoptosis↓ RGCs loss↓ TDP-43 aggregation↓ | [43] |
| RGCs | Rat laser-induced glaucoma model | Rapamycin | - | mTORC1/S6K1↓ | Apoptosis↓ RGCs loss↓ VEGFR-2↓ | [24] |
| RGCs | Mouse microbead occlusion model | Rapamycin | - | AMPK↑/mTOR↓ | RGCs loss↓ | [135] |
| RGCs | Circumlimbal-suture-induced OHT rat model | Rapamycin | LC3-II/I↑ p62↓ | AMPK↑/mTOR↓ | Apoptosis↓ RGCs loss↓ | [149] |
| BV2 microglia/primary RGCs/retina tissue | Rat chronic hypertensive glaucoma model | Rapamycin | - | AKT↔/mTOR↓ | Apoptosis↓ iNOS/TNF-a/NF-kB↓ Microglial activation↓ | [15] |
| Retina tissue | Ndufs4 KO mouse model of mitochondrial optic neuropathy | Rapamycin | - | mTOR↓ | Apoptosis↓ Microglial activation↓ Inflammation↓ | [88] |
| Study (NCT Number) | Design | Subjects | Intervention | Treatment Regimen | Results | Reference |
|---|---|---|---|---|---|---|
| Phase II trial Naor et al. 2010 (NCT00656643) | Four-arm study in US; placebo injection as control | 131 with diabetic macular oedema | Sirolimus subconjunctival injection | Two subconjunctival injections of 220, 440, 880 μg, or placebo (1:1:1:1) observation through day 180 | Awaiting results | [159] |
| Phase I/II trial Krishnadev et al. 2011 (NCT00711490) | Single-arm study in US; fellow eye as control | 5 with diabetic macular oedema | Sirolimus subconjunctival injection | 440 μg injection every 2 months for 12 months follow-up period | Safe and well-tolerated; efficacy trials required | [150] |
| Phase I trial Dugel et al. 2012 (NCT00401115) | Two-arm study in US; fellow eye as control | 50 with diabetic macular oedema (n = 25 for SCJ and IVT, respectively) | Sirolimus single subconjunctival (SCJ)/intravitreal injection (IVT) | SCJ (220, 440, 880, 1320, or 1760 μg)/IVT (44, 110, 176, 264, or 352 μg); observation through day 90 | Safe and well-tolerated (no dose-limiting toxicities); efficacy trials required | [151] |
| Phase I/II trial Naor et al. 2010 (NCT00712491) | Two-arm study in US; fellow eye as control | 20 with AMD (CNV); n = 10 for each arm | Rapamycin intravitreal injection | Three injections of 352 or 1320 μg observation through 12 months | Awaiting results | [160] |
| Phase II trial Nussenblatt et al. 2010 (NCT00304954) | Four-arm study in US; fellow eye as control | 13 with AMD (CNV) | Intravenous daclizumab/intravenous infliximab/oral rapamycin/observation with anti-VEGF therapy | Daily 2 mg oral tablet (n = 3) vs. daclizumab, vs. infliximab vs. no immunosuppression plus intraocular anti-VEGF therapy for 6 months fellow up | Safe and well-tolerated; no benefit | [156] |
| Phase II trial Abraham et al. 2010 (NCT00766337) | Three-arm study in US; placebo comparator as control | 62 with AMD (CNV) | Sirolimus in combination with ranibizumab subconjunctival injection | 440 or 1320 μg both with 500 μg ranibizumab every 2 months for 24 months fellow up | Awaiting results | [161] |
| Phase II trial Wong et al. 2013 (NCT00766649) | Single-arm study in US; fellow eye as control | 11 with AMD (GA) | Rapamycin subconjunctival injection | 440 μg injection every three months for 24 months follow-up | Safe and well-tolerated; no benefit | [152] |
| Phase I trial Dalal et al. 2013 (NCT01271270) | Single-arm study in US; fellow eye as control | 13 with AMD (CNV) | Palomid 529 subconjunctival injection | 1.9 mg injection every 4 weeks for 12 weeks follow-up | Safe and well-tolerated; efficacy trials required | [158] |
| Phase I/II trial Petrou et al. 2014 (NCT01445548) | Single-arm study in US; fellow eye as control | 6 with AMD (GA) | Rapamycin intravitreal injection | 440 μg injection every two months for 12 months follow-up | Ocular adverse events appeared; no benefit | [153] |
| Phase II trial Gensler et al. 2017 (NCT01675947) | Two-arm study in US; sham treatment as control | 52 with AMD (GA); n = 27 for rapamycin | Rapamycin intravitreal injection | 440 μg injection monthly for 24 months follow-up | Safe and well-tolerated; no benefit | [154] |
| Phase II trial Minturn et al. 2021 (NCT02357342) | Two-arm study in US; fellow eye as control | 40 with AMD (CNV); n = 20 for each arm | Sirolimus intravitreal injection/anti-VEGF therapy | 440 μg injection every two months for 6 months follow-up | Safe and well-tolerated; CST decreased by 40 μm in sirolimus group (p = 0.03) | [157] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Fung, N.S.K.; Lam, W.-C.; Lo, A.C.Y. mTOR Signalling Pathway: A Potential Therapeutic Target for Ocular Neurodegenerative Diseases. Antioxidants 2022, 11, 1304. https://doi.org/10.3390/antiox11071304
Wang Y, Fung NSK, Lam W-C, Lo ACY. mTOR Signalling Pathway: A Potential Therapeutic Target for Ocular Neurodegenerative Diseases. Antioxidants. 2022; 11(7):1304. https://doi.org/10.3390/antiox11071304
Chicago/Turabian StyleWang, Yipin, Nicholas Siu Kay Fung, Wai-Ching Lam, and Amy Cheuk Yin Lo. 2022. "mTOR Signalling Pathway: A Potential Therapeutic Target for Ocular Neurodegenerative Diseases" Antioxidants 11, no. 7: 1304. https://doi.org/10.3390/antiox11071304