
Selenium and Selenoproteins at the Intersection of Type 2 Diabetes and Thyroid Pathophysiology


Abstract
:1. Introduction
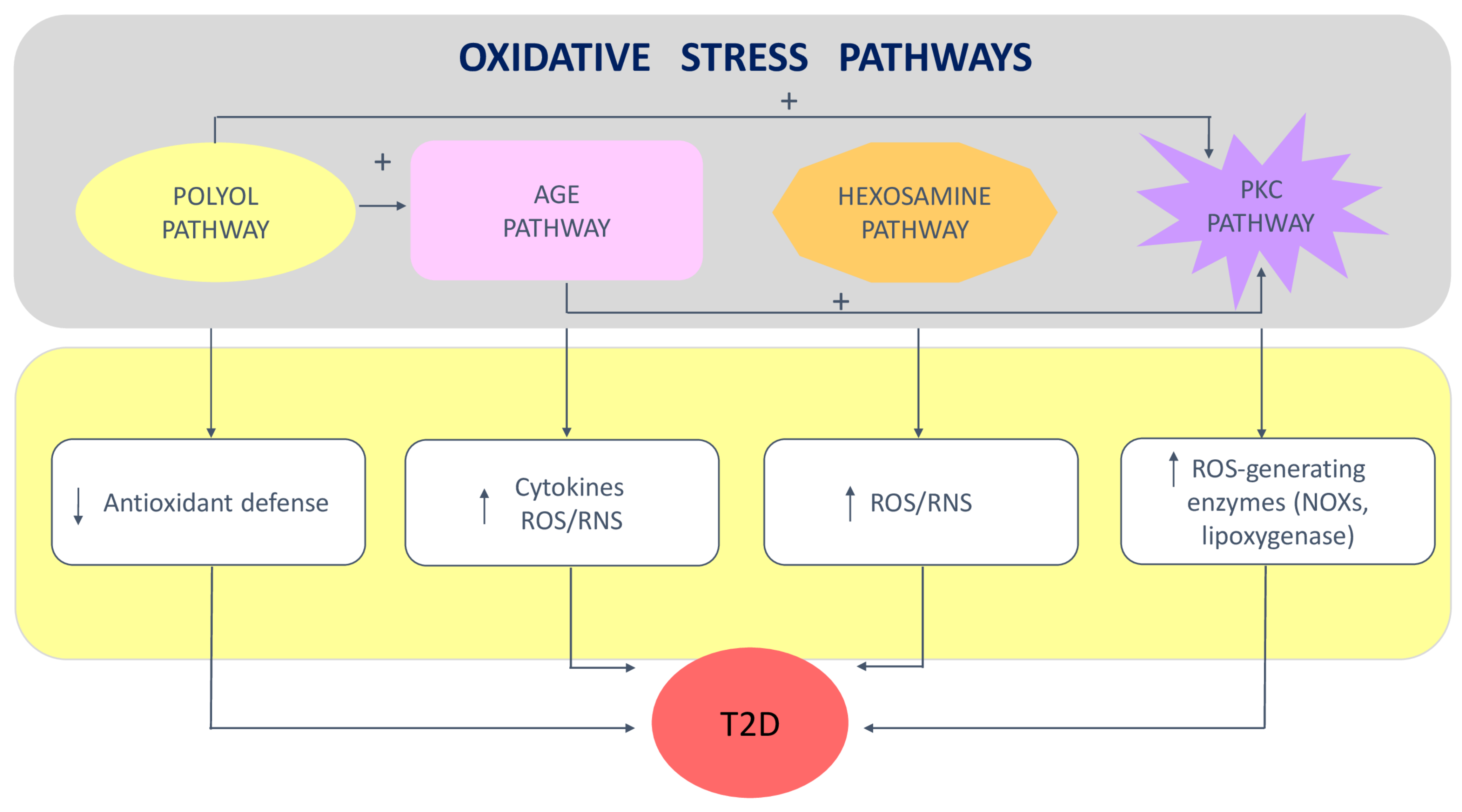
2. Role of Oxidative Stress in Insulin Resistance and Type 2 Diabetes
3. The Relationship between Selenium and Type 2 Diabetes
3.1. Experimental Studies
3.2. Epidemiological Studies
3.3. Randomized Clinical Trials
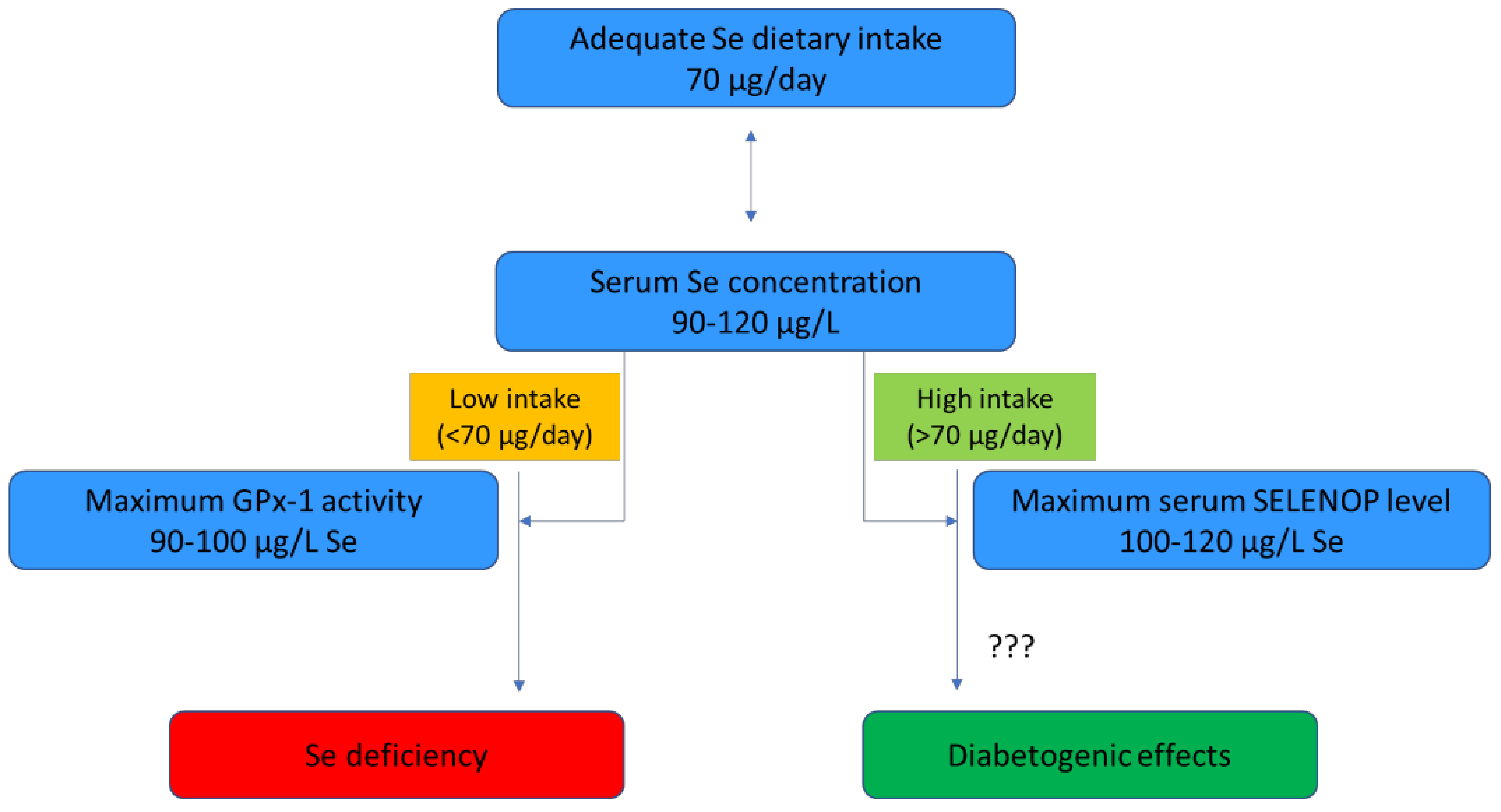
3.4. Molecular Mechanisms Underlying the Association between Selenium Exposure and Type 2 Diabetes
4. Thyroid and Type 2 Diabetes
5. Selenoprotein Effects in Thyroid Health and Type 2 Diabetes
6. Molecular Targets to Enhance the Antioxidant Defense against Type 2 Diabetes
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AGE | Advanced glycation end-product |
| Akt | Protein kinase B |
| ATP | Adenosine triphosphate |
| DIO | Deiodinase |
| eNOS | Endothelial nitric oxide synthase |
| ER | Endoplasmic reticulum |
| ERS | Endoplasmic reticulum stress |
| GSH | Reduced glutathione |
| GPx | Glutathione peroxidase |
| IR | Insulin resistance |
| IRS-1 | Insulin receptor substrate-1 |
| Keap1 | Kelch-like ECH-associated protein 1 |
| MAPK | Mitogen-activated protein kinase |
| MetS | Metabolic syndrome |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NF-κB | Nuclear factor κB |
| NO | Nitrogen oxide |
| NOX | NAPDH oxidase |
| Nrf2 | Nuclear factor erythroid 2-related factor-2 |
| PI3K | Phosphatidylinositol-3,4,5-triphosphate kinase |
| PKC | Protein kinase C |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| rT3 | Reverse triiodothyronine |
| Sec | Selenocysteine |
| SELENOK | Selenoprotein K |
| SELENOP | Selenoprotein P |
| SELENOS | Selenoprotein S |
| SELENOV | Selenoprotein V |
| SNP | Single-nucleotide polymorphism |
| SOD | Superoxide dismutase |
| T2D | Type 2 diabetes |
| T3 | Triiodothyronine |
| T4 | Thyroxine |
| TH | Thyroid hormone |
| TrxR | Thioredoxin reductase |
References
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.B.; Hashim, M.J.; King, J.K.; Govender, R.D.; Mustafa, H.; Al Kaabi, J. Epidemiology of Type 2 Diabetes—Global Burden of Disease and Forecasted Trends. J. Epidemiol. Glob. Health 2020, 10, 107–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Diabetes. 2021. Available online: https://www.who.int/health-topics/diabetes#tab=tab_1 (accessed on 3 March 2022).
- World Health Organization. Global Report on Diabetes. 2021. Available online: https://apps.who.int/iris/bitstream/handle/10665/204871/9789241565257_eng.pdf;jsessionid=F57B0CDDF4D02AA4BCF8F920D4B3AEBB?sequence=1 (accessed on 3 March 2022).
- Liu, J.; Ren, Z.H.; Qiang, H.; Wu, J.; Shen, M.; Zhang, L.; Lyu, J. Trends in the incidence of diabetes mellitus: Results from the Global Burden of Disease Study 2017 and implications for diabetes mellitus prevention. BMC Public Health 2020, 20, 1415. [Google Scholar] [CrossRef]
- Lin, X.; Xu, Y.; Pan, X.; Hu, J.; Ding, Y.; Sun, X.; Song, X.; Ren, Y.; Shan, P.F. Global, regional, and national burden and trend of diabetes in 195 countries and territories: An analysis from 1990 to 2025. Sci. Rep. 2020, 10, 14790. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. IDF Diabetes Atlas Committee. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Key Facts. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/diabetes (accessed on 4 March 2022).
- Rong, F.; Dai, H.; Wu, Y.; Li, J.; Liu, G.; Chen, H.; Zhang, X. Association between thyroid dysfunction and type 2 diabetes: A meta-analysis of prospective observational studies. BMC Med. 2021, 19, 257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yan, C.; Yang, Z.; Zhang, W.; Niu, Y.; Li, X.; Qin, L.; Su, Q. Alterations of serum trace elements in patients with type 2 diabetes. J. Trace Elem. Med. Biol. 2017, 40, 91–96. [Google Scholar] [CrossRef]
- Dubey, P.; Thakur, V.; Chattopadhyay, M. Role of Minerals and Trace Elements in Diabetes and Insulin Resistance. Nutrients 2020, 12, 1864. [Google Scholar] [CrossRef]
- Rayman, M.P.; Stranges, S. Epidemiology of selenium and type 2 diabetes: Can we make sense of it? Free Radic. Biol. Med. 2013, 65, 1557–1564. [Google Scholar] [CrossRef]
- Kim, J.; Chung, H.S.; Choi, M.K.; Roh, Y.K.; Yoo, H.J.; Park, J.H.; Kim, D.S.; Yu, J.M.; Moon, S. Association between Serum Selenium Level and the Presence of Diabetes Mellitus: A Meta-Analysis of Observational Studies. Diabetes Metab. J. 2019, 43, 447–460. [Google Scholar] [CrossRef]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorini, F.; Sabatino, L.; Pingitore, A.; Vassalle, C. Selenium: An Element of Life Essential for Thyroid Function. Molecules 2021, 26, 7084. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Rayman, M.P.; Lv, H.; Schomburg, L.; Cui, B.; Gao, C.; Chen, P.; Zhuang, G.; Zhang, Z.; Peng, X.; et al. Low Population Selenium Status Is Associated with Increased Prevalence of Thyroid Disease. J. Clin. Endocrinol. Metab. 2015, 100, 4037–4047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roa Dueñas, O.H.; Van der Burgh, A.C.; Ittermann, T.; Ligthart, S.; Ikram, M.A.; Peeters, R.; Chaker, L. Thyroid Function and the Risk of Prediabetes and Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2022, 17, 1789–1798. [Google Scholar] [CrossRef]
- Asmat, U.; Abad, K.; Ismail, K. Diabetes mellitus and oxidative stress—A concise review. Saudi Pharm. J. 2016, 24, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Mechanistic Insight into Oxidative Stress-Triggered Signaling Pathways and Type 2 Diabetes. Molecules 2022, 27, 950. [Google Scholar] [CrossRef]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Hurrle, S.; Hsu, W.H. The etiology of oxidative stress in insulin resistance. Biomed. J. 2017, 40, 257–262. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Lenzen, S.; Drinkgern, J.; Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic. Biol. Med. 1996, 20, 463–466. [Google Scholar] [CrossRef]
- Tiedge, M.; Lortz, S.; Drinkgern, J.; Lenzen, S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 1997, 46, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Drews, G.; Krippeit-Drews, P.; Düfer, M. Oxidative stress and beta-cell dysfunction. Pflug. Arch. 2010, 460, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Luc, K.; Schramm-Luc, A.; Guzik, T.J.; Mikolajczyk, T.P. Oxidative stress and inflammatory markers in prediabetes and diabetes. J. Physiol. Pharmacol. 2019, 70, 809–824. [Google Scholar] [CrossRef]
- Rochette, L.; Lorin, J.; Zeller, M.; Guilland, J.C.; Lorgis, L.; Cottin, Y.; Vergely, C. Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: Possible therapeutic targets? Pharmacol. Ther. 2013, 140, 239–257. [Google Scholar] [CrossRef]
- Lakey, J.R.; Suarez-Pinzon, W.L.; Strynadka, K.; Korbutt, G.S.; Rajotte, R.V.; Mabley, J.G.; Szabó, C.; Rabinovitch, A. Peroxynitrite is a mediator of cytokine-induced destruction of human pancreatic islet beta cells. Lab Investig. 2001, 81, 1683–1692. [Google Scholar] [CrossRef]
- Ighodaro, O.M. Molecular pathways associated with oxidative stress in diabetes mellitus. Biomed. Pharmacother. 2018, 108, 656–662. [Google Scholar] [CrossRef]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and mithocondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [Green Version]
- Al-Aubaidy, H.A.; Jelinek, H.F. Oxidative stress and triglycerides as predictors of subclinical atherosclerosis in prediabetes. Redox Rep. 2014, 19, 87–91. [Google Scholar] [CrossRef]
- Jiménez-Osorio, A.S.; Picazo, A.; González-Reyes, S.; Barrera-Oviedo, D.; Rodríguez-Arellano, M.E.; Pedraza-Chaverri, J. Nrf2 and redox status in prediabetic and diabetic patients. Int. J. Mol. Sci. 2014, 15, 20290–20305. [Google Scholar] [CrossRef]
- Maschirow, L.; Khalaf, K.; Al-Aubaidy, H.A.; Jelinek, H.F. Inflammation, coagulation, endothelial dysfunction and oxidative stress in prediabetes—Biomarkers as a possible tool for early disease detection for rural screening. Clin. Biochem. 2015, 48, 581–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagishita, Y.; Fukutomi, T.; Sugawara, A.; Kawamura, H.; Takahashi, T.; Pi, J.; Uruno, A.; Yamamoto, M. Nrf2 protects pancreatic β-cells from oxidative and nitrosative stress in diabetic model mice. Diabetes 2014, 63, 605–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodiha, M.; Stochaj, U. Nuclear transport: A switch for the oxidative stress–Signaling circuit? J. Signal. Transduct. 2012, 2012, 208650. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Shao, W.; Chiang, Y.; Foltz, W.; Zhang, Z.; Ling, W.; Fantus, I.G.; Jin, T. Oltipraz upregulates the nuclear factor (erythroid-derived 2)-like 2 [corrected](NRF2) antioxidant system and prevents insulin resistance and obesity induced by a high-fat diet in C57BL/6J mice. Diabetologia 2011, 54, 922–934. [Google Scholar] [CrossRef] [Green Version]
- Negi, G.; Kumar, A.; Joshi, R.P.; Sharma, S.S. Oxidative stress and Nrf2 in the pathophysiology of diabetic neuropathy: Old perspective with a new angle. Biochem. Biophys. Res. Commun. 2011, 408, 1–5. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [Green Version]
- Steinbrenner, H. Interference of selenium and selenoproteins with the insulin-regulated carbohydrate and lipid metabolism. Free Radic. Biol. Med. 2013, 65, 1538–1547. [Google Scholar] [CrossRef]
- Blanco, C.L.; McGill-Vargas, L.L.; Gastaldelli, A.; Seidner, S.R.; McCurnin, D.C.; Leland, M.M.; Anzueto, D.G.; Johnson, M.C.; Liang, H.; DeFronzo, R.A.; et al. Peripheral insulin resistance and impaired insulin signaling contribute to abnormal glucose metabolism in preterm baboons. Endocrinology 2015, 156, 813–823. [Google Scholar] [CrossRef] [Green Version]
- Evans, J.L.; Maddux, B.A.; Goldfine, I.D. The molecular basis for oxidative stress-induced insulin resistance. Antioxid. Redox Signal. 2005, 7, 1040–1052. [Google Scholar] [CrossRef]
- Jheng, H.F.; Tsai, P.J.; Guo, S.M.; Kuo, L.H.; Chang, C.S.; Su, I.J.; Chang, C.R.; Tsai, Y.S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell. Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Liu, Z.X.; Choi, C.S.; Tian, L.; Kibbey, R.; Dong, J.; Cline, G.W.; Wood, P.A.; Shulman, G.I. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 17075–17080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneto, H.; Matsuoka, T.A. Involvement of oxidative stress in suppression of insulin biosynthesis under diabetic conditions. Int. J. Mol. Sci. 2012, 13, 13680–13690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.L.; Yang, T.B.; Wei, J.; Lei, G.H.; Zeng, C. Association between serum selenium level and type 2 diabetes mellitus: A non-linear dose-response meta-analysis of observational studies. Nutr. J. 2016, 15, 48. [Google Scholar] [CrossRef] [Green Version]
- Steinbrenner, H.; Sies, H. Protection against reactive oxygen species by selenoproteins. Biochim. Biophys. Acta 2009, 1790, 1478–1485. [Google Scholar] [CrossRef]
- Bermingham, E.N.; Hesketh, J.E.; Sinclair, B.R.; Koolaard, J.P.; Roy, N.C. Selenium-enriched foods are more effective at increasing glutathione peroxidase (GPx) activity compared with selenomethionine: A meta-analysis. Nutrients 2014, 6, 4002–4031. [Google Scholar] [CrossRef]
- Bates, J.M.; Spate, V.L.; Morris, J.S.; St Germain, D.L.; Galton, V.A. Effects of selenium deficiency on tissue selenium content, deiodinase activity, and thyroid hormone economy in the rat during development. Endocrinology 2000, 141, 2490–2500. [Google Scholar] [CrossRef]
- Saito, Y. Selenoprotein P as an in vivo redox regulator: Disorders related to its deficiency and excess. J. Clin. Biochem. Nutr. 2020, 66, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Turanov, A.A.; Shchedrina, V.A.; Everley, R.A.; Lobanov, A.V.; Yim, S.H.; Marino, S.M.; Gygi, S.P.; Hatfield, D.L.; Gladyshev, V.N. Selenoprotein S is involved in maintenance and transport of multiprotein complexes. Biochem. J. 2014, 462, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Lin, S.L.; Wang, C.W.; Yao, H.D.; Zhang, Z.W.; Xu, S.W. Effect of selenium on selenoprotein expression in the adipose tissue of chickens. Biol. Trace Elem. Res. 2014, 160, 41–48. [Google Scholar] [CrossRef]
- Steinbrenner, H.; Hotze, A.L.; Speckmann, B.; Pinto, A.; Sies, H.; Schott, M.; Ehlers, M.; Scherbaum, W.A.; Schinner, S. Localization and regulation of pancreatic selenoprotein P. J. Mol. Endocrinol. 2012, 50, 31–42. [Google Scholar] [CrossRef] [Green Version]
- EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA). Scientific Opinion on Dietary Reference Values for selenium. EFSA J. 2014, 12, 3846. [Google Scholar] [CrossRef]
- Fairweather-Tait, S.J.; Bao, Y.; Broadley, M.R.; Collings, R.; Ford, D.; Hesketh, J.E.; Hurst, R. Selenium in human health and disease. Antioxid. Redox Signal. 2011, 14, 1337–1383. [Google Scholar] [CrossRef]
- Vinceti, M.; Crespi, C.M.; Malagoli, C.; Del Giovane, C.; Krogh, V. Friend or foe? The current epidemiologic evidence on selenium and human cancer risk. J. Environ. Sci. Health Part C 2013, 31, 305–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoffaneller, R.; Morse, N.L. A review of dietary selenium intake and selenium status in Europe and the Middle East. Nutrients 2015, 7, 1494–1537. [Google Scholar] [CrossRef] [PubMed]
- Ashton, K.; Hooper, L.; Harvey, L.J.; Hurst, R.; Casgrain, A.; Fairweather-Tait, S.J. Methods of assessment of selenium status in humans: A systematic review. Am. J. Clin. Nutr. 2009, 89, 2025S–2039S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurst, R.; Armah, C.N.; Dainty, J.R.; Hart, D.J.; Teucher, B.; Goldson, A.J.; Broadley, M.R.; Motley, A.K.; Fairweather-Tait, S.J. Establishing optimal selenium status: Results of a randomized, double-blind, placebo-controlled trial. Am. J. Clin. Nutr. 2010, 91, 923–931. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Hill, K.E.; Li, P.; Xu, J.; Zhou, D.; Motley, A.K.; Wang, L.; Byrne, D.W.; Burk, R.F. Optimization of selenoprotein P and other plasma selenium biomarkers for the assessment of the selenium nutritional requirement: A placebo-controlled, double-blind study of selenomethionine supplementation in selenium-deficient Chinese subjects. Am. J. Clin. Nutr. 2010, 92, 525–531. [Google Scholar] [CrossRef] [Green Version]
- Kohler, L.N.; Foote, J.; Kelley, C.P.; Florea, A.; Shelly, C.; Chow, H.S.; Hsu, P.; Batai, K.; Ellis, N.; Saboda, K.; et al. Selenium and Type 2 Diabetes: Systematic Review. Nutrients 2018, 10, 1924. [Google Scholar] [CrossRef] [Green Version]
- Ogawa-Wong, A.N.; Berry, M.J.; Seale, L.A. Selenium and Metabolic Disorders: An Emphasis on Type 2 Diabetes Risk. Nutrients 2016, 8, 80. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Huang, K.; Lei, X.G. Selenium and diabetes—Evidence from animal studies. Free Radic. Biol. Med. 2013, 65, 1548–1556. [Google Scholar] [CrossRef] [Green Version]
- Seale, L.A.; Hashimoto, A.C.; Kurokawa, S.; Gilman, C.L.; Seyedali, A.; Bellinger, F.P.; Raman, A.V.; Berry, M.J. Disruption of the selenocysteine lyase-mediated selenium recycling pathway leads to metabolic syndrome in mice. Mol. Cell. Biol. 2012, 32, 4141–4154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, R.P.; Harmon, J.S. Pancreatic islet beta-cell and oxidative stress: The importance of glutathione peroxidase. FEBS Lett. 2007, 581, 3743–3748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, S.C.; Aldibbiat, A.; Marriott, C.E.; Landy, C.; Ali, T.; Ferris, W.F.; Butler, C.S.; Shaw, J.A.; Macfarlane, W.M. Selenium stimulates pancreatic beta-cell gene expression and enhances islet function. FEBS Lett. 2008, 582, 2333–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.D.; Vatamaniuk, M.Z.; Wang, S.K.; Roneker, C.A.; Simmons, R.A.; Lei, X.G. Molecular mechanisms for hyperinsulinaemia induced by overproduction of selenium-dependent glutathione peroxidase-1 in mice. Diabetologia 2008, 51, 1515–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmon, J.S.; Bogdani, M.; Parazzoli, S.D.; Mak, S.S.; Oseid, E.A.; Berghmans, M.; Leboeuf, R.C.; Robertson, R.P. beta-Cell-specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology 2009, 150, 4855–4862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, A.S.; Klomann, S.D.; Wolf, N.M.; Schneider, S.; Schmidt, R.; Spielmann, J.; Stangl, G.; Eder, K.; Pallauf, J. Redox regulation of protein tyrosine phosphatase 1B by manipulation of dietary selenium affects the triglyceride concentration in rat liver. J. Nutr. 2008, 138, 2328–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbrenner, H.; Speckmann, B.; Pinto, A.; Sies, H. High selenium intake and increased diabetes risk: Experimental evidence for interplay between selenium and carbohydrate metabolism. J. Clin. Biochem. Nutr. 2011, 48, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Misu, H.; Takamura, T.; Takayama, H.; Hayashi, H.; Matsuzawa-Nagata, N.; Kurita, S.; Ishikura, K.; Ando, H.; Takeshita, Y.; Ota, T.; et al. A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell. Metab. 2010, 12, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Kornhauser, C.; Garcia-Ramirez, J.R.; Wrobel, K.; Pérez-Luque, E.L.; Garay-Sevilla, M.E.; Wrobel, K. Serum selenium and glutathione peroxidase concentrations in type 2 diabetes mellitus patients. Prim. Care Diabetes 2008, 2, 81–85. [Google Scholar] [CrossRef]
- Sedighi, O.; Makhlough, A.; Shokrzadeh, M.; Hoorshad, S. Association between plasma selenium and glutathione peroxidase levels and severity of diabetic nephropathy in patients with type two diabetes mellitus. Nephrourol. Mon. 2014, 6, e21355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, B.; Ramesh, A.; Suresh, S.; Prasad, B.R. A comparative evaluation of antioxidant enzymes and selenium in the serum of periodontitis patients with diabetes mellitus type 2. Contemp. Clin. Dent. 2013, 4, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.; Prasad, B.R.; Kumari, N.S.; Radhakrishna, V.; Ramesh, A. A comparative evaluation of the micronutrient profile in the serum of diabetes mellitus Type II patients and healthy individuals with periodontitis. J. Indian Soc. Periodontol. 2019, 23, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Aziz, F.; AlHazmi, A.; Aljameil, N.; Mahmood, I.; Tabassum, H.; Mushfiq, S.; Hijazy, S. Serum Selenium and Lead Levels: A Possible Link with Diabetes and Associated Proteinuria. Biol. Trace Elem. Res. 2020, 193, 342–347. [Google Scholar] [CrossRef]
- Vinceti, M.; Filippini, T.; Rothman, K.J. Selenium exposure and the risk of type 2 diabetes: A systematic review and meta-analysis. Eur. J. Epidemiol. 2018, 33, 789–810. [Google Scholar] [CrossRef]
- Vinceti, M.; Filippini, T.; Wise, L.A.; Rothman, K.J. A systematic review and dose-response meta-analysis of exposure to environmental selenium and the risk of type 2 diabetes in nonexperimental studies. Environ. Res. 2021, 197, 111210. [Google Scholar] [CrossRef]
- Filippini, T.; Ferrari, A.; Michalke, B.; Grill, P.; Vescovi, L.; Salvia, C.; Malagoli, C.; Malavolti, M.; Sieri, S.; Krogh, V.; et al. Toenail selenium as an indicator of environmental exposure: A cross-sectional study. Mol. Med. Rep. 2017, 15, 3405–3412. [Google Scholar] [CrossRef] [Green Version]
- Gu, Q.; Cui, X.; Du, K.; Wang, B.; Cai, W.; Tang, Q.; Shen, X. Higher toenail selenium is associated with increased insulin resistance risk in omnivores, but not in vegetarians. Nutr. Metab. 2020, 17, 62. [Google Scholar] [CrossRef]
- Stranges, S.; Marshall, J.R.; Natarajan, R.; Donahue, R.P.; Trevisan, M.; Combs, G.F.; Cappuccio, F.P.; Ceriello, A.; Reid, M.E. Effects of long-term selenium supplementation on the incidence of type 2 diabetes: A randomized trial. Ann. Intern. Med. 2007, 147, 217–223. [Google Scholar] [CrossRef]
- Algotar, A.M.; Stratton, M.S.; Stratton, S.P.; Hsu, C.H.; Ahmann, F.R. No effect of selenium supplementation on serum glucose levels in men with prostate cancer. Am. J. Med. 2010, 123, 765–768. [Google Scholar] [CrossRef] [Green Version]
- Alizadeh, M.; Safaeiyan, A.; Ostadrahimi, A.; Estakhri, R.; Daneghian, S.; Ghaffari, A.; Gargari, B.P. Effect of L-arginine and selenium added to a hypocaloric diet enriched with legumes on cardiovascular disease risk factors in women with central obesity: A randomized, double-blind, placebo-controlled trial. Ann. Nutr. Metab. 2012, 60, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Lippman, S.M.; Klein, E.A.; Goodman, P.J.; Lucia, M.S.; Thompson, I.M.; Ford, L.G.; Parnes, H.L.; Minasian, L.M.; Gaziano, J.M.; Hartline, J.A.; et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009, 30, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, E.A.; Thompson, I.M., Jr.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Rayman, M.P.; Blundell-Pound, G.; Pastor-Barriuso, R.; Guallar, E.; Steinbrenner, H.; Stranges, S. A randomized trial of selenium supplementation and risk of type-2 diabetes, as assessed by plasma adiponectin. PLoS ONE 2012, 7, e45269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, P.A.; Ashbeck, E.L.; Roe, D.J.; Fales, L.; Buckmeier, J.; Wang, F.; Bhattacharyya, A.; Hsu, C.H.; Chow, H.H.; Ahnen, D.J.; et al. Selenium Supplementation for Prevention of Colorectal Adenomas and Risk of Associated Type 2 Diabetes. J. Natl. Cancer Inst. 2016, 108, djw152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayman, M.P. Selenium and human health. Lancet 2012, 379, 1256–1268. [Google Scholar] [CrossRef]
- Vinceti, M.; Chiari, A.; Eichmüller, M.; Rothman, K.J.; Filippini, T.; Malagoli, C.; Weuve, J.; Tondelli, M.; Zamboni, G.; Nichelli, P.F.; et al. A selenium species in cerebrospinal fluid predicts conversion to Alzheimer’s dementia in persons with mild cognitive impairment. Alzheimers Res. Ther. 2017, 9, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, M.I.; Cao, J.; Zeng, H.; Uthus, E.; Combs, G.F., Jr. S-adenosylmethionine-dependent protein methylation is required for expression of selenoprotein P and gluconeogenic enzymes in HepG2 human hepatocytes. J. Biol. Chem. 2012, 287, 36455–36464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalhan, S.C.; Edmison, J.; Marczewski, S.; Dasarathy, S.; Gruca, L.L.; Bennett, C.; Duenas, C.; Lopez, R. Methionine and protein metabolism in non-alcoholic steatohepatitis: Evidence for lower rate of transmethylation of methionine. Clin. Sci. 2011, 121, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.J.; Hwang, S.Y.; Choi, H.Y.; Yoo, H.J.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Baik, S.H.; Choi, D.S.; Choi, K.M. Serum selenoprotein P levels in patients with type 2 diabetes and prediabetes: Implications for insulin resistance, inflammation, and atherosclerosis. J. Clin. Endocrinol. Metab. 2011, 96, E1325–E1329. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Rizk, N.M.; Ibrahim, S.; Younes, N.; Uppal, A.; Dennis, K.; Karve, T.; Blakeslee, K.; Kwagyan, J.; Zirie, M.; et al. iTRAQ-based quantitative protein expression profiling and MRM verification of markers in type 2 diabetes. J. Proteome Res. 2012, 11, 5527–5539. [Google Scholar] [CrossRef] [PubMed]
- Chopra, I.; Li, H.F.; Wang, H.; Webster, K.A. Phosphorylation of the insulin receptor by AMP-activated protein kinase (AMPK) promotes ligand-independent activation of the insulin signalling pathway in rodent muscle. Diabetologia 2012, 55, 783–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellwege, J.N.; Palmer, N.D.; Ziegler, J.T.; Langefeld, C.D.; Lorenzo, C.; Norris, J.M.; Takamura, T.; Bowden, D.W. Genetic variants in selenoprotein P plasma 1 gene (SEPP1) are associated with fasting insulin and first phase insulin response in Hispanics. Gene 2014, 534, 33–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oo, S.M.; Misu, H.; Saito, Y.; Tanaka, M.; Kato, S.; Kita, Y.; Takayama, H.; Takeshita, Y.; Kanamori, T.; Nagano, T.; et al. Serum selenoprotein P, but not selenium, predicts future hyperglycemia in a general Japanese population. Sci. Rep. 2018, 8, 16727. [Google Scholar] [CrossRef] [PubMed]
- Zubčić, Ž.; Šestak, A.; Mihalj, H.; Kotromanović, Ž.; Včeva, A.; Prpić, T.; Rezo, M.; Milanković, S.G.; Bogović, V.; Abičić, I. The AsSociation Between Type 2 Diabetes Mellitus, Hypothyroidism, and Thyroid Cancer. Acta Clin. Croat. 2020, 59, 129–135. [Google Scholar] [CrossRef]
- Bardugo, A.; Derazne, E.; Zucker, I.; Bendor, C.D.; Puris, G.; Lutski, M.; Pinhas-Hamiel, O.; Cukierman-Yaffe, T.; Mosenzon, O.; Schechter, M.; et al. Adolescent Thyroid Disorders and Risk for Type 2 Diabetes in Young Adulthood. J. Clin. Endocrinol. Metab. 2021, 106, e3426–e3435. [Google Scholar] [CrossRef]
- Wang, J.J.; Zhuang, Z.H.; Yu, C.Q.; Wang, W.Y.; Wang, W.X.; Zhang, K.; Meng, X.B.; Gao, J.; Tian, J.; Zheng, J.L.; et al. Assessment of causal direction between thyroid function and cardiometabolic health: A Mendelian randomization study. J. Geriatr. Cardiol. 2022, 19, 61–70. [Google Scholar] [CrossRef]
- Oda, T.; Taneichi, H.; Takahashi, K.; Togashi, H.; Hangai, M.; Nakagawa, R.; Ono, M.; Matsui, M.; Sasai, T.; Nagasawa, K.; et al. Positive association of free triiodothyronine with pancreatic β-cell function in people with prediabetes. Diabet. Med. 2015, 32, 213–219. [Google Scholar] [CrossRef]
- Matsuda, H.; Mullapudi, S.T.; Zhang, Y.; Hesselson, D.; Stainier, D.Y.R. Thyroid Hormone Coordinates Pancreatic Islet Maturation During the Zebrafish Larval-to-Juvenile Transition to Maintain Glucose Homeostasis. Diabetes 2017, 66, 2623–2635. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.K.; Lee, J.S.; Jung, H.S.; Ha, T.K.; Kim, S.M.; Han, N.; Lee, E.J.; Kim, T.N.; Kwon, M.J.; Lee, S.H.; et al. Triiodothyronine induces proliferation of pancreatic β-cells through the MAPK/ERK pathway. Exp. Clin. Endocrinol. Diabetes 2014, 122, 240–245. [Google Scholar] [CrossRef]
- Eom, Y.S.; Wilson, J.R.; Bernet, V.J. Links between Thyroid Disorders and Glucose Homeostasis. Diabetes Metab. J. 2022, 46, 239–256. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Boelen, A.; Bisschop, P.H.; Kalsbeek, A.; Fliers, E. Hypothalamic effects of thyroid hormone. Mol. Cell. Endocrinol. 2017, 458, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Sieglaff, D.H.; York, J.P.; Suh, J.H.; Ayers, S.D.; Winnier, G.E.; Kharitonenkov, A.; Pin, C.; Zhang, P.; Webb, P.; et al. Thyroid hormone receptor regulates most genes independently of fibroblast growth factor 21 in liver. J. Endocrinol. 2015, 224, 289–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Jiang, Y.; Meltzer, P.; Yen, P.M. Thyroid hormone regulation of hepatic genes in vivo detected by complementary DNA microarray. Mol. Endocrinol. 2000, 14, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Merchan-Ramirez, E.; Sanchez-Delgado, G.; Arrizabalaga-Arriazu, C.; Acosta, F.M.; Arias-Tellez, M.J.; Muñoz-Torres, M.; Garcia-Lario, J.V.; Llamas-Elvira, J.M.; Ruiz, J.R. Circulating concentrations of free triiodothyronine are associated with central adiposity and cardiometabolic risk factors in young euthyroid adults. J. Physiol. Biochem. 2022. [Google Scholar] [CrossRef] [PubMed]
- Elgazar, E.H.; Esheba, N.E.; Shalaby, S.A.; Mohamed, W.F. Thyroid dysfunction prevalence and relation to glycemic control in patients with type 2 diabetes mellitus. Diabetes Metab. Syndr. 2019, 13, 2513–2517. [Google Scholar] [CrossRef] [PubMed]
- Jali, M.V.; Kambar, S.; Jali, S.M.; Pawar, N.; Nalawade, P. Prevalence of thyroid dysfunction among type 2 diabetes mellitus patients. Diabetes Metab. Syndr. 2017, 11, S105–S108. [Google Scholar] [CrossRef]
- Bech, K.; Damsbo, P.; Eldrup, E.; Beck-Nielsen, H.; Røder, M.E.; Hartling, S.G.; Vølund, A.; Madsbad, S. beta-cell function and glucose and lipid oxidation in Graves’ disease. Clin. Endocrinol. 1996, 44, 59–66. [Google Scholar] [CrossRef]
- Ağbaht, K.; Erdogan, M.F.; Emral, R.; Baskal, N.; Güllü, S. Circulating glucagon to ghrelin ratio as a determinant of insulin resistance in hyperthyroidism. Endocrine 2014, 45, 106–113. [Google Scholar] [CrossRef]
- Liang, B.; Liu, L.; Huang, H.; Li, L.; Zhou, J. High T3 Induces β-Cell Insulin Resistance via Endoplasmic Reticulum Stress. Mediat. Inflamm. 2020, 2020, 5287108. [Google Scholar] [CrossRef]
- Brenta, G. Why can insulin resistance be a natural consequence of thyroid dysfunction? J. Thyroid Res. 2011, 2011, 152850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handisurya, A.; Pacini, G.; Tura, A.; Gessl, A.; Kautzky-Willer, A. Effects of T4 replacement therapy on glucose metabolism in subjects with subclinical (SH) and overt hypothyroidism (OH). Clin. Endocrinol. 2008, 69, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Unnikrishnan, A.G.; Sahay, R. The hypoglycemic side of hypothyroidism. Indian J. Endocrinol. Metab. 2014, 18, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Maratou, E.; Hadjidakis, D.J.; Kollias, A.; Tsegka, K.; Peppa, M.; Alevizaki, M.; Mitrou, P.; Lambadiari, V.; Boutati, E.; Nikzas, D.; et al. Studies of insulin resistance in patients with clinical and subclinical hypothyroidism. Eur. J. Endocrinol. 2009, 160, 785–790. [Google Scholar] [CrossRef] [Green Version]
- Blanc, E.; Ponce, C.; Brodschi, D.; Nepote, A.; Barreto, A.; Schnitman, M.; Fossati, P.; Salgado, P.; Cejas, C.; Faingold, C.; et al. Association between worse metabolic control and increased thyroid volume and nodular disease in elderly adults with metabolic syndrome. Metab. Syndr. Relat. Disord. 2015, 13, 221–226. [Google Scholar] [CrossRef]
- Tang, Y.; Yan, T.; Wang, G.; Chen, Y.; Zhu, Y.; Jiang, Z.; Yang, M.; Li, C.; Li, Z.; Yu, P.; et al. Correlation between Insulin Resistance and Thyroid Nodule in Type 2 Diabetes Mellitus. Int. J. Endocrinol. 2017, 2017, 1617458. [Google Scholar] [CrossRef] [Green Version]
- Rawal, S.; Tsai, M.Y.; Hinkle, S.N.; Zhu, Y.; Bao, W.; Lin, Y.; Panuganti, P.; Albert, P.S.; Ma, R.C.W.; Zhang, C. A Longitudinal Study of Thyroid Markers Across Pregnancy and the Risk of Gestational Diabetes. J. Clin. Endocrinol. Metab. 2018, 103, 2447–2456. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.D.; Gou, X.Y.; Pang, T.T.; Li, P.S.; Zhou, Z.X.; Lin, D.X.; Fan, D.Z.; Guo, X.L.; Wang, L.J.; Liu, Z.P. Associations between thyroid function and gestational diabetes mellitus in Chinese pregnant women: A retrospective cohort study. BMC Endocr. Disord. 2022, 22, 44. [Google Scholar] [CrossRef]
- Kushchayeva, Y.; Kushchayev, S.; Jensen, K.; Brown, R.J. Impaired Glucose Metabolism, Anti-Diabetes Medications, and Risk of Thyroid Cancer. Cancers 2022, 14, 555. [Google Scholar] [CrossRef]
- García-Sáenz, M.; Lobaton-Ginsberg, M.; Ferreira-Hermosillo, A. Metformin in Differentiated Thyroid Cancer: Molecular Pathways and Its Clinical Implications. Biomolecules 2022, 12, 574. [Google Scholar] [CrossRef]
- Zhang, X.; Sheng, X.; Miao, T.; Yao, K.; Yao, D. Effect of insulin on thyroid cell proliferation, tumor cell migration, and potentially related mechanisms. Endocr. Res. 2019, 44, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Heydarzadeh, S.; Moshtaghie, A.A.; Daneshpoor, M.; Hedayati, M. Regulators of glucose uptake in thyroid cancer cell lines. Cell Commun. Signal. 2020, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Shchedrina, V.A.; Everley, R.A.; Zhang, Y.; Gygi, S.P.; Hatfield, D.L.; Gladyshev, V.N. Selenoprotein K binds multiprotein complexes and is involved in the regulation of endoplasmic reticulum homeostasis. J. Biol. Chem. 2011, 286, 42937–42948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addinsall, A.B.; Wright, C.R.; Andrikopoulos, S.; van der Poel, C.; Stupka, N. Emerging roles of endoplasmic reticulum-resident selenoproteins in the regulation of cellular stress responses and the implications for metabolic disease. Biochem. J. 2018, 475, 1037–1057. [Google Scholar] [CrossRef]
- Liu, J.; Rozovsky, S. Membrane-bound selenoproteins. Antioxid. Redox Signal. 2015, 23, 795–813. [Google Scholar] [CrossRef]
- Ventura, M.; Melo, M.; Carrilho, F. Selenium and Thyroid Disease: From Pathophysiology to Treatment. Int. J. Endocrinol. 2017, 2017, 1297658. [Google Scholar] [CrossRef] [Green Version]
- Bos, M.M.; Smit, R.A.J.; Trompet, S.; van Heemst, D.; Noordam, R. Thyroid Signaling, Insulin Resistance, and 2 Diabetes Mellitus: A Mendelian Randomization Study. J. Clin. Endocrinol. Metab. 2017, 102, 1960–1970. [Google Scholar] [CrossRef]
- Wang, X.; Chen, K.; Zhang, C.; Wang, H.; Li, J.; Wang, C.; Teng, W.; Shan, Z.; Lai, Y. The Type 2 Deiodinase Thr92Ala Polymorphism Is Associated with Higher Body Mass Index and Fasting Glucose Levels: A Systematic Review and Meta-Analysis. Biomed. Res. Int. 2021, 2021, 9914009. [Google Scholar] [CrossRef]
- Weltman, N.Y.; Ojamaa, K.; Schlenker, E.H.; Chen, Y.F.; Zucchi, R.; Saba, A.; Colligiani, D.; Rajagopalan, V.; Pol, C.J.; Gerdes, A.M. Low-dose T3 replacement restores depressed cardiac T3 levels, preserves coronary microvasculature and attenuates cardiac dysfunction in experimental diabetes mellitus. Mol. Med. 2014, 20, 302–312. [Google Scholar] [CrossRef]
- Beckett, G.J.; Arthur, J.R. Selenium and endocrine systems. J. Endocrinol. 2005, 184, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Metere, A.; Frezzotti, F.; Graves, C.E.; Vergine, M.; De Luca, A.; Pietraforte, D.; Giacomelli, L. A possible role for selenoprotein glutathione peroxidase (GPx1) and thioredoxin reductases (TrxR1) in thyroid cancer: Our experience in thyroid surgery. Cancer Cell Int. 2018, 18, 7. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Q.; Zhou, J.C.; Wu, Y.Y.; Ren, F.Z.; Lei, X.G. Role of glutathione peroxidase 1 in glucose and lipid metabolism-related diseases. Free Radic. Biol. Med. 2018, 127, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Schmutzler, C.; Mentrup, B.; Schomburg, L.; Hoang-Vu, C.; Herzog, V.; Köhrle, J. Selenoproteins of the thyroid gland: Expression, localization and possible function of glutathione peroxidase 3. Biol. Chem. 2007, 388, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Baez-Duarte, B.G.; Mendoza-Carrera, F.; García-Zapién, A.; Flores-Martínez, S.E.; Sánchez-Corona, J.; Zamora-Ginez, I.; Torres-Rasgado, E.; León-Chávez, B.A.; Pérez-Fuentes, R. Multidisciplinary Research Group on Diabetes of the Instituto Mexicano del Seguro Social. Glutathione peroxidase 3 serum levels and GPX3 gene polymorphisms in subjects with metabolic syndrome. Arch. Med. Res. 2014, 45, 375–382. [Google Scholar] [CrossRef]
- Roumeliotis, A.; Roumeliotis, S.; Tsetsos, F.; Georgitsi, M.; Georgianos, P.I.; Stamou, A.; Vasilakou, A.; Kotsa, K.; Tsekmekidou, X.; Paschou, P.; et al. Oxidative Stress Genes in Diabetes Mellitus Type 2: Association with Diabetic Kidney Disease. Oxid. Med. Cell. Longev. 2021, 2021, 2531062. [Google Scholar] [CrossRef]
- Ling, P.; Shan, W.; Zhai, G.; Qiu, C.; Liu, Y.; Xu, Y.; Yang, X. Association between glutathione peroxidase-3 activity and carotid atherosclerosis in patients with type 2 diabetes mellitus. Brain Behav. 2020, 10, e01773. [Google Scholar] [CrossRef]
- Hauffe, R.; Stein, V.; Chudoba, C.; Flore, T.; Rath, M.; Ritter, K.; Schell, M.; Wardelmann, K.; Deubel, S.; Kopp, J.F.; et al. GPx3 dysregulation impacts adipose tissue insulin receptor expression and sensitivity. JCI Insight 2020, 5, e136283. [Google Scholar] [CrossRef]
- Shi, J.; Wu, P.; Sheng, L.; Sun, W.; Zhang, H. Ferroptosis-related gene signature predicts the prognosis of papillary thyroid carcinoma. Cancer Cell Int. 2021, 21, 669. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, T.; Hu, S.; Sun, L. Association of serum lipid peroxidation and glutathione peroxidase 4 levels with clinical outcomes and metabolic abnormalities among patients with gestational diabetes mellitus: A case-control study in the Chinese population. Front. Biosci. 2022, 27, 68. [Google Scholar] [CrossRef]
- Huang, J.; Chen, G.; Wang, J.; Liu, S.; Su, J. Platycodin D regulates high glucose-induced ferroptosis of HK-2 cells through glutathione peroxidase 4 (GPX4). Bioengineered 2022, 13, 6627–6637. [Google Scholar] [CrossRef]
- Wang, S.; Yang, S.; Vlantis, A.C.; Liu, S.Y.; Ng, E.K.; Chan, A.B.; Wu, J.; Du, J.; Wei, W.; Liu, X.; et al. Expression of Antioxidant Molecules and Heat Shock Protein 27 in Thyroid Tumors. J. Cell. Biochem. 2016, 117, 2473–2481. [Google Scholar] [CrossRef] [PubMed]
- Hanschmann, E.M.; Petry, S.F.; Eitner, S.; Maresch, C.C.; Lingwal, N.; Lillig, C.H.; Linn, T. Paracrine regulation and improvement of β-cell function by thioredoxin. Redox Biol. 2020, 34, 101570. [Google Scholar] [CrossRef] [PubMed]
- Stancill, J.S.; Corbett, J.A. The Role of Thioredoxin/Peroxiredoxin in the β-Cell Defense Against Oxidative Damage. Front. Endocrinol. 2021, 12, 718235. [Google Scholar] [CrossRef] [PubMed]
- Tinkov, A.A.; Bjørklund, G.; Skalny, A.V.; Holmgren, A.; Skalnaya, M.G.; Chirumbolo, S.; Aaseth, J. The role of the thioredoxin/thioredoxin reductase system in the metabolic syndrome: Towards a possible prognostic marker? Cell. Mol. Life Sci. 2018, 75, 1567–1586. [Google Scholar] [CrossRef]
- Ramus, S.M.; Cilensek, I.; Petrovic, M.G.; Soucek, M.; Kruzliak, P.; Petrovic, D. Single nucleotide polymorphisms in the Trx2/TXNIP and TrxR2 genes of the mitochondrial thioredoxin antioxidant system and the risk of diabetic retinopathy in patients with Type 2 diabetes mellitus. J. Diabetes Complicat. 2016, 30, 192–198. [Google Scholar] [CrossRef]
- Kariž, S.; Mankoč, S.; Petrovič, D. Association of thioredoxin reductase 2 (TXNRD2) gene polymorphisms with myocardial infarction in Slovene patients with type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2015, 108, 323–328. [Google Scholar] [CrossRef]
- Sun, Q.; Mehl, S.; Renko, K.; Seemann, P.; Görlich, C.L.; Hackler, J.; Minich, W.B.; Kahaly, G.J.; Schomburg, L. Natural Autoimmunity to Selenoprotein P Impairs Selenium Transport in Hashimoto’s Thyroiditis. Int. J. Mol. Sci. 2021, 22, 13088. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, P.; Lv, H.J.; Wu, Y.; Liu, S.; Deng, X.; Shi, B.; Fu, J. Comprehensive Analysis of Expression and Prognostic Value of Selenoprotein Genes in Thyroid Cancer. Genet. Test. Mol. Biomark. 2022, 26, 159–173. [Google Scholar] [CrossRef]
- Santos, L.R.; Neves, C.; Melo, M.; Soares, P. Selenium and Selenoproteins in Immune Mediated Thyroid Disorders. Diagnostics 2018, 8, 70. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.S.; Men, L.L.; Wu, J.L.; Huang, L.W.; Xing, Q.; Yao, J.J.; Wang, Y.B.; Song, G.R.; Guo, H.S.; Sun, G.H.; et al. The source of circulating selenoprotein S and its association with type 2 diabetes mellitus and atherosclerosis: A preliminary study. Cardiovasc. Diabetol. 2016, 15, 70. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.S.; Du, J.L. Selenoprotein S: A therapeutic target for diabetes and macroangiopathy? Cardiovasc. Diabetol. 2017, 16, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Zheng, Y.Y.; Chen, Y.; Ma, Y.T.; Yang, Y.N.; Li, X.M.; Ma, X.; Xie, X. Association of genetic polymorphisms of SelS with Type 2 diabetes in a Chinese population. Biosci. Rep. 2018, 38, BSR20181696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.; Guo, Y.; Wang, L.; Jing, L.; Chen, Z.; Lu, S.; Fu, R.; Tian, L. High glucose and TGF-β1 reduce expression of endoplasmic reticulum-resident selenoprotein S and selenoprotein N in human mesangial cells. Ren. Fail. 2019, 41, 762–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.L.; Huang, J.Q.; Wu, Y.Y.; Chen, L.B.; Li, S.P.; Zhang, X.; Wu, S.; Ren, F.Z.; Lei, X.G. Loss of Selenov predisposes mice to extra fat accumulation and attenuated energy expenditure. Redox Biol. 2021, 45, 102048. [Google Scholar] [CrossRef]
- Mullur, R.; Liu, Y.Y.; Brent, G.A. Thyroid hormone regulation of metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef] [Green Version]
- Leiria, L.B.; Dora, J.M.; Wajner, S.M.; Estivalet, A.A.; Crispim, D.; Maia, A.L. The rs225017 polymorphism in the 3’UTR of the human DIO2 gene is associated with increased insulin resistance. PLoS ONE 2014, 9, e103960. [Google Scholar] [CrossRef] [Green Version]
- Dora, J.M.; Machado, W.E.; Rheinheimer, J.; Crispim, D.; Maia, A.L. Association of the type 2 deiodinase Thr92Ala polymorphism with type 2 diabetes: Case-control study and meta-analysis. Eur. J. Endocrinol. 2010, 163, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Martinez-deMena, R.; Calvo, R.M.; Garcia, L.; Obregon, M.J. Effect of glucocorticoids on the activity, expression and proximal promoter of type II deiodinase in rat brown adipocytes. Mol. Cell. Endocrinol. 2016, 428, 58–67. [Google Scholar] [CrossRef]
- McClung, J.P.; Roneker, C.A.; Mu, W.; Lisk, D.J.; Langlais, P.; Liu, F.; Lei, X.G. Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase. Proc. Natl. Acad. Sci. USA 2004, 101, 8852–8857. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Vatamaniuk, M.Z.; Roneker, C.A.; Pepper, M.P.; Hu, L.G.; Simmons, R.A.; Lei, X.G. Knockouts of SOD1 and GPX1 exert different impacts on murine islet function and pancreatic integrity. Antioxid. Redox Signal. 2011, 14, 391–401. [Google Scholar] [CrossRef] [Green Version]
- Loh, K.; Deng, H.; Fukushima, A.; Cai, X.; Boivin, B.; Galic, S.; Bruce, C.; Shields, B.J.; Skiba, B.; Ooms, L.M.; et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009, 10, 260–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vats, P.; Sagar, N.; Singh, T.P.; Banerjee, M. Association of Superoxide dismutases (SOD1 and SOD2) and Glutathione peroxidase 1 (GPx1) gene polymorphisms with type 2 diabetes mellitus. Free Radic. Res. 2015, 49, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Vats, P.; Kushwah, A.S.; Srivastava, N. Interaction of antioxidant gene variants and susceptibility to type 2 diabetes mellitus. Br. J. Biomed. Sci. 2019, 76, 166–171. [Google Scholar] [CrossRef]
- Katunga, L.A.; Gudimella, P.; Efird, J.T.; Abernathy, S.; Mattox, T.A.; Beatty, C.; Darden, T.M.; Thayne, K.A.; Alwair, H.; Kypson, A.P.; et al. Obesity in a model of gpx4 haploinsufficiency uncovers a causal role for lipid-derived aldehydes in human metabolic disease and cardiomyopathy. Mol. Metab. 2015, 4, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Takamura, T. Hepatokine Selenoprotein P-Mediated Reductive Stress Causes Resistance to Intracellular Signal Transduction. Antioxid. Redox Signal. 2020, 33, 517–524. [Google Scholar] [CrossRef]
- Pitts, M.W.; Hoffmann, P.R. Endoplasmic reticulum-resident selenoproteins as regulators of calcium signaling and homeostasis. Cell Calcium 2018, 70, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Hoffmann, F.W.; Kumar, M.; Huang, Z.; Roe, K.; Nguyen-Wu, E.; Hashimoto, A.S.; Hoffmann, P.R. Selenoprotein K knockout mice exhibit deficient calcium flux in immune cells and impaired immune responses. J. Immunol. 2011, 186, 2127–2137. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.L.; Huang, J.Q.; Xiao, Y.; Wu, Y.Y.; Ren, F.Z.; Lei, X.G. Knockout of Selenoprotein V Affects Regulation of Selenoprotein Expression by Dietary Selenium and Fat Intakes in Mice. J. Nutr. 2020, 150, 483–491. [Google Scholar] [CrossRef]
- Zhang, X.; Xiong, W.; Chen, L.L.; Huang, J.Q.; Lei, X.G. Selenoprotein V protects against endoplasmic reticulum stress and oxidative injury inuced by pro-oxidants. Free Radic. Biol. Med. 2020, 160, 670–679. [Google Scholar] [CrossRef]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef]
- Ma, X.; Chen, Z.; Wang, L.; Wang, G.; Wang, Z.; Dong, X.; Wen, B.; Zhang, Z. The Pathogenesis of Diabetes Mellitus by Oxidative Stress and Inflammation: Its Inhibition by Berberine. Front. Pharmacol. 2018, 9, 782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y. Selenium Transport Mechanism via Selenoprotein P—Its Physiological Role and Related Diseases. Front. Nutr. 2021, 8, 685517. [Google Scholar] [CrossRef] [PubMed]
- Takayama, H.; Misu, H.; Iwama, H.; Chikamoto, K.; Saito, Y.; Murao, K.; Teraguchi, A.; Lan, F.; Kikuchi, A.; Saito, R.; et al. Metformin suppresses expression of the selenoprotein P gene via an AMP-activated kinase (AMPK)/FoxO3a pathway in H4IIEC3 hepatocytes. J. Biol. Chem. 2014, 289, 335–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajima-Shirasaki, N.; Ishii, K.A.; Takayama, H.; Shirasaki, T.; Iwama, H.; Chikamoto, K.; Saito, Y.; Iwasaki, Y.; Teraguchi, A.; Lan, F.; et al. Eicosapentaenoic acid down-regulates expression of the selenoprotein P gene by inhibiting SREBP-1c protein independently of the AMP-activated protein kinase pathway in H4IIEC3 hepatocytes. J. Biol. Chem. 2017, 292, 10791–10800. [Google Scholar] [CrossRef] [Green Version]
- Mita, Y.; Nakayama, K.; Inari, S.; Nishito, Y.; Yoshioka, Y.; Sakai, N.; Sotani, K.; Nagamura, T.; Kuzuhara, Y.; Inagaki, K.; et al. Selenoprotein P-neutralizing antibodies improve insulin secretion and glucose sensitivity in type 2 diabetes mouse models. Nat. Commun. 2017, 8, 1658. [Google Scholar] [CrossRef]
- Burgos-Morón, E.; Abad-Jiménez, Z.; Marañón, A.M.; Iannantuoni, F.; Escribano-López, I.; López-Domènech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I.; et al. Relationship Between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Molecule | Acronym | Function | Reference |
|---|---|---|---|
| Nicotinamide adenine dinucleotide phosphate oxidases | NOXs | Production of superoxide; mitochondria-induced stress response | [19,22] |
| Endothelial nitric oxide synthase | eNOS | Production of nitrix oxide | [27] |
| Tetrahydrobiopterin | - | Formation of oxygen-derived radicals | [28] |
| Nitric oxide | NO | Reaction with superoxide which generates peroxynitrite | [29] |
| Kelch-like ECH-associated protein 1 | Keap1 | Negative regulator of Nrf2 | [33] |
| Nuclear factor erythroid 2-related factor-2 | Nrf2 | Control of antioxidant response | [35,36,37,38,39] |
| Hydrogen peroxide | H2O2 | Downregulation of GLUT4; stimulation of NF-κB, c-Jun N-terminal kinase and p38 MAPKs | [22] |
| Glucose transporter 4 | GLUT4 | Insulin-stimulated glucose uptake by adipose tissue and skeletal muscle | [42] |
| Pancreatic and duodenal homeobox 1 | PDX-1 | β-cell transcription factor | [46] |
| MAF BZIP Transcription Factor A | MAFA | β-cell transcription factor | [46] |
| Selenoprotein (Acronym) | Types/ Isoenzymes | Main Functions | Effects Linked to Thyroid | Reference | Effects Linked to T2D | Reference |
|---|---|---|---|---|---|---|
| Iodothyronine deiodinase (DIO) | DIO1 DIO2 DIO3 | T4 to T3 conversion T4 to T3 conversion T4 to rT3 conversion | Conversion of TH Conversion of TH Deactivation of TH | [129] [129] [129] | DIO1 polymorphism (rs7527713) associated with IR DIO2 polymorphism (Thr92Ala) associated with BMI and FBG Increased DIO3 associated with cardiac dysfunction in diabetic heart | [130] [131] [132] |
| Glutathione peroxidase (GPx) | GPx1 | Cytosolic antioxidant | Thyrocyte protection from peroxidative damage | [133,134] | Overexpression of GPx1 associated with T2D-like phenotypes. Human GPX1 polymorphism related to risks of diabetes and obesity | [135] |
| GPx2 | Extracellular antioxidant | Thyrocyte protection from peroxidative damage | [133,136] | GPx3 involved in MetS, IR and T2D complications | [137,138,139,140] | |
| GPx3 | Membrane phospholipid antioxidant | Antioxidant protecting the membrane, regulation of cellular death | [141] | GPx4 levels associated with clinical outcomes and metabolic abnormalities among patients with gestational diabetes mellitus. GPX4 involvement in high-glucose-induced ferroptosis (programmed cell death dependent on iron) | [142,143] | |
| Thioredoxin reductase (TrxR) | TrxR1 TrxR2 | Redox state regulation and antioxidant actions | Thyrocyte protection from peroxidative damage Antioxidant | [133,134,144] | Improvement of survival and function of pancreatic β-cells TRx2 polymorphisms associated with T2D complications | [145,146,147] [148,149] |
| Selenoprotein P (SELENOP) | - | Selenium transport and storage, antioxidant defense | Selenium supply to thyroid | [150] | Relationship with IR and T2D | [51,97] |
| Selenoprotein S (SELENOS) | - | Protection against ERS | Protection against ERS and oxidative injury | [151,152] | SELENOS levels associated with T2D and T2D macrovascular complications. SELENOS polymorphisms associated with the risk for developing T2D and macroangiopathy. | [153,154,155] |
| Selenoprotein K (SELENOK) | - | Quality control within the ER | Protection against ERS and oxidative injury | [152] | SELENOK expression downregulated by glucose | [156] |
| Selenoprotein V (SELENOV) | - | Modulation of redox processes and ER calcium homeostasis, cell adhesion and angiogenesis | Protection against ERS and oxidative injury | [130] | SELENOV as modulator of body fat accumulation and energy expenditure, and regulator of O-GlcNAcylation (protein associated with various metabolic diseases including T2D and obesity) | [157] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorini, F.; Vassalle, C. Selenium and Selenoproteins at the Intersection of Type 2 Diabetes and Thyroid Pathophysiology. Antioxidants 2022, 11, 1188. https://doi.org/10.3390/antiox11061188
Gorini F, Vassalle C. Selenium and Selenoproteins at the Intersection of Type 2 Diabetes and Thyroid Pathophysiology. Antioxidants. 2022; 11(6):1188. https://doi.org/10.3390/antiox11061188
Chicago/Turabian StyleGorini, Francesca, and Cristina Vassalle. 2022. "Selenium and Selenoproteins at the Intersection of Type 2 Diabetes and Thyroid Pathophysiology" Antioxidants 11, no. 6: 1188. https://doi.org/10.3390/antiox11061188