
Yin and Yang of NADPH Oxidases in Myocardial Ischemia-Reperfusion


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Oxidative Stress and Myocardial I/R Injury
2.1. Roles of Oxidative Stress in Myocardial I/R Injury
2.2. Sources of Oxidative StressiIn Myocardial I/R Injury
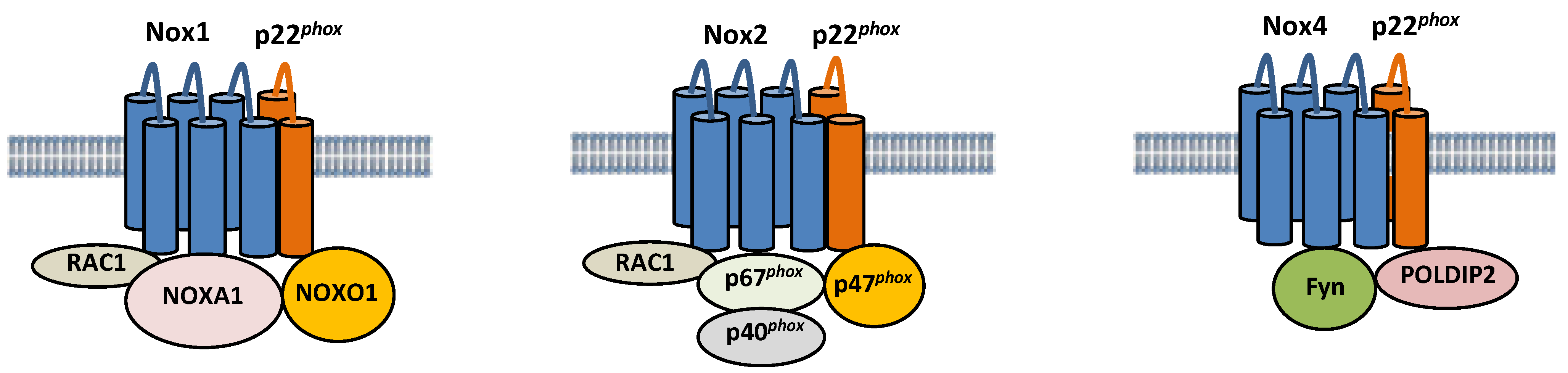
3. Pathophysiological Roles, Regulation, and Subcellular Localization of Nox Isoforms in the Heart
4. Detrimental Roles of Nox Isoforms-Derived ROS in Myocardial I/R Injury
4.1. Detrimental Roles of Nox Isoforms during Myocardial I/R Injury
4.2. Cell-Specific Roles of Nox Isoforms in Myocardial I/R Injury
4.3. ROS Production by Nox Isoforms during Myocardial I/R Injury
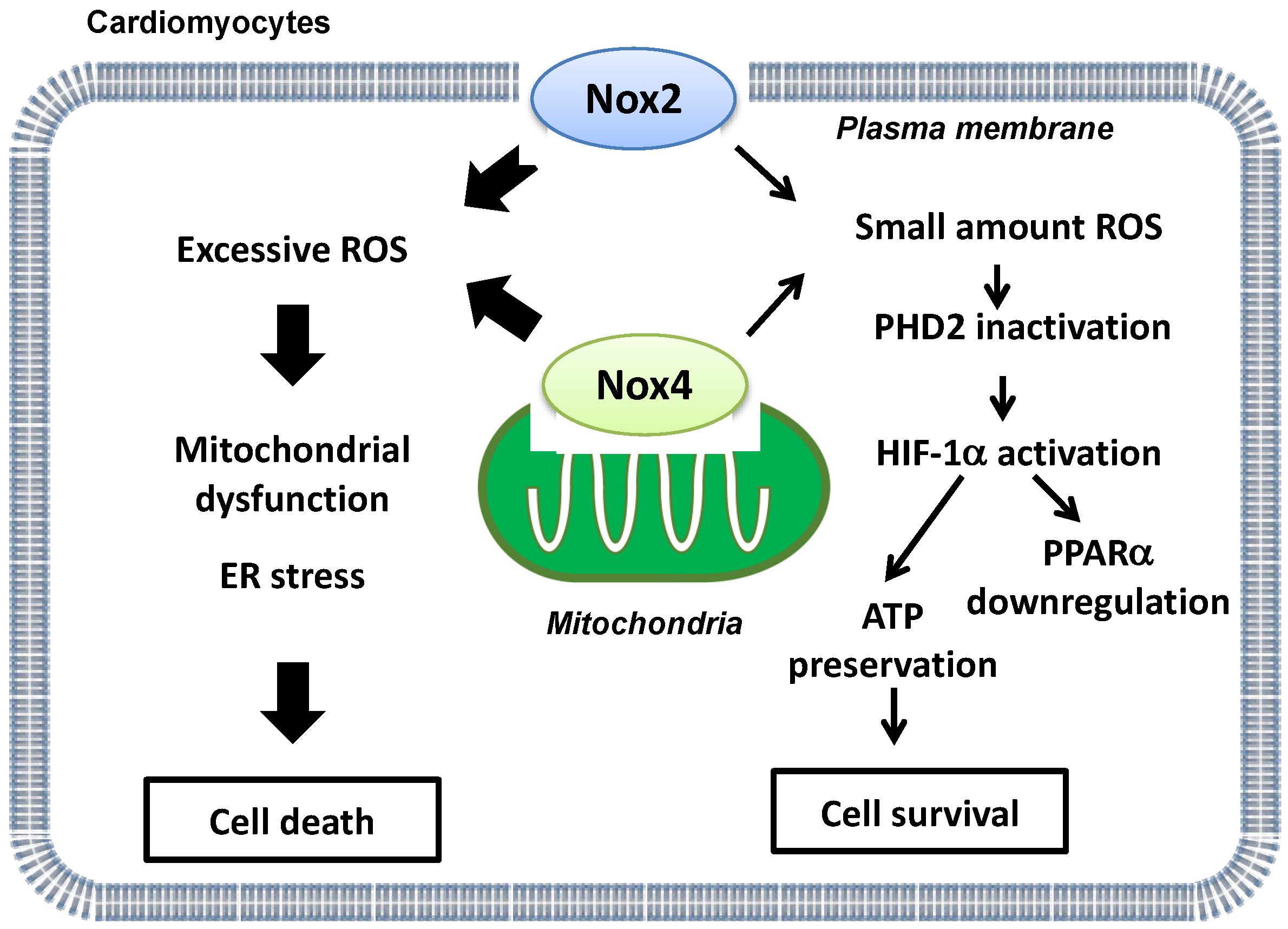
5. Beneficial Aspects of Nox Isoforms in Myocardial I/R Injury
5.1. The Physiological Level of ROS Derived from Noxs during Myocardial I/R Injury
5.2. Reductive Stress in Myocardial I/R Injury
5.3. The Role of Nox4 in ER in Myocardial I/R Injury
5.4. The Role of Nox4 at MAMs during Myocardial I/R Injury
6. Novel Regulatory Mechanisms of Nox and Its Crosstalk with Other Oxidases
7. Therapeutic Approach of Intervention into Nox Isoforms in I/R Injury
8. Future Perspectives
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic. Biol. Med. 2018, 117, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Levraut, J.; Iwase, H.; Shao, Z.H.; Vanden Hoek, T.L.; Schumacker, P.T. Cell death during ischemia: Relationship to mitochondrial depolarization and ROS generation. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H549–H558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Montero, J.; Brito, R.; Gajardo, A.I.; Rodrigo, R. Myocardial reperfusion injury and oxidative stress: Therapeutic opportunities. World J. Cardiol. 2018, 10, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Braunersreuther, V.; Jaquet, V. Reactive oxygen species in myocardial reperfusion injury: From physiopathology to therapeutic approaches. Curr. Pharm. Biotechnol. 2012, 13, 97–114. [Google Scholar] [CrossRef]
- Henry, T.D.; Archer, S.L.; Nelson, D.; Weir, E.K.; From, A.H. Enhanced chemiluminescence as a measure of oxygen-derived free radical generation during ischemia and reperfusion. Circ. Res. 1990, 67, 1453–1461. [Google Scholar] [CrossRef] [Green Version]
- Maulik, G.; Cordis, G.A.; Das, D.K. Oxidative damage to myocardial proteins and DNA during ischemia and reperfusion. Ann. N. Y. Acad. Sci. 1996, 793, 431–436. [Google Scholar] [CrossRef]
- Shintani-Ishida, K.; Inui, M.; Yoshida, K. Ischemia-reperfusion induces myocardial infarction through mitochondrial Ca2+ overload. J. Mol. Cell. Cardiol. 2012, 53, 233–239. [Google Scholar] [CrossRef]
- Marin, W.; Marin, D.; Ao, X.; Liu, Y. Mitochondria as a therapeutic target for cardiac ischemia-reperfusion injury (Review). Int. J. Mol. Med. 2021, 47, 485–499. [Google Scholar] [CrossRef]
- Zhou, M.; Yu, Y.; Luo, X.; Wang, J.; Lan, X.; Liu, P.; Feng, Y.; Jian, W. Myocardial Ischemia-Reperfusion Injury: Therapeutics from a Mitochondria-Centric Perspective. Cardiology 2021, 146, 781–792. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granger, D.N. Role of xanthine oxidase and granulocytes in ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 1988, 255, H1269–H1275. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Han, M.; Bao, J.; Tu, W.; Dai, Z. A superoxide anion biosensor based on direct electron transfer of superoxide dismutase on sodium alginate sol-gel film and its application to monitoring of living cells. Anal. Chim. Acta 2012, 717, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Braunersreuther, V.; Montecucco, F.; Asrih, M.; Pelli, G.; Galan, K.; Frias, M.; Burger, F.; Quindere, A.L.; Montessuit, C.; Krause, K.H.; et al. Role of NADPH oxidase isoforms NOX1, NOX2 and NOX4 in myocardial ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 2013, 64, 99–107. [Google Scholar] [CrossRef]
- Matsushima, S.; Kuroda, J.; Ago, T.; Zhai, P.; Ikeda, Y.; Oka, S.; Fong, G.H.; Tian, R.; Sadoshima, J. Broad suppression of NADPH oxidase activity exacerbates ischemia/reperfusion injury through inadvertent downregulation of hypoxia-inducible factor-1alpha and upregulation of peroxisome proliferator-activated receptor-alpha. Circ. Res. 2013, 112, 1135–1149. [Google Scholar] [CrossRef] [Green Version]
- Kahles, T.; Brandes, R.P. Which NADPH oxidase isoform is relevant for ischemic stroke? The case for nox 2. Antioxid. Redox Signal. 2013, 18, 1400–1417. [Google Scholar] [CrossRef] [Green Version]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Babior, B.M.; Kipnes, R.S.; Curnutte, J.T. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J. Clin. Investig. 1973, 52, 741–744. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Takac, I.; Schroder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef] [Green Version]
- Sorescu, D.; Weiss, D.; Lassegue, B.; Clempus, R.E.; Szocs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiszt, M.; Kopp, J.B.; Varnai, P.; Leto, T.L. Identification of renox, an NAD(P)H oxidase in kidney. Proc. Natl. Acad. Sci. USA 2000, 97, 8010–8014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ago, T.; Kuroda, J.; Pain, J.; Fu, C.; Li, H.; Sadoshima, J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ. Res. 2010, 106, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Lardy, B.; Krause, K.H. NOX family NADPH oxidases: Not just in mammals. Biochimie 2007, 89, 1107–1112. [Google Scholar] [CrossRef]
- Byrne, J.A.; Grieve, D.J.; Bendall, J.K.; Li, J.M.; Gove, C.; Lambeth, J.D.; Cave, A.C.; Shah, A.M. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ. Res. 2003, 93, 802–805. [Google Scholar] [CrossRef]
- Zhang, M.; Brewer, A.C.; Schroder, K.; Santos, C.X.; Grieve, D.J.; Wang, M.; Anilkumar, N.; Yu, B.; Dong, X.; Walker, S.J.; et al. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 18121–18126. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, J.; Ago, T.; Matsushima, S.; Zhai, P.; Schneider, M.D.; Sadoshima, J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 15565–15570. [Google Scholar] [CrossRef] [Green Version]
- Anilkumar, N.; Sirker, A.; Shah, A.M. Redox sensitive signaling pathways in cardiac remodeling, hypertrophy and failure. Front Biosci 2009, 14, 3168–3187. [Google Scholar] [CrossRef]
- Serrander, L.; Cartier, L.; Bedard, K.; Banfi, B.; Lardy, B.; Plastre, O.; Sienkiewicz, A.; Forro, L.; Schlegel, W.; Krause, K.H. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem. J. 2007, 406, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Leto, T.L.; Morand, S.; Hurt, D.; Ueyama, T. Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid. Redox Signal. 2009, 11, 2607–2619. [Google Scholar] [CrossRef]
- Cheng, G.; Diebold, B.A.; Hughes, Y.; Lambeth, J.D. Nox1-dependent reactive oxygen generation is regulated by Rac1. J. Biol. Chem. 2006, 281, 17718–17726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cave, A.C.; Brewer, A.C.; Narayanapanicker, A.; Ray, R.; Grieve, D.J.; Walker, S.; Shah, A.M. NADPH oxidases in cardiovascular health and disease. Antioxid. Redox Signal. 2006, 8, 691–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altenhofer, S.; Radermacher, K.A.; Kleikers, P.W.; Wingler, K.; Schmidt, H.H. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Kuroda, J.; Ago, T.; Zhai, P.; Park, J.Y.; Xie, L.H.; Tian, B.; Sadoshima, J. Increased oxidative stress in the nucleus caused by Nox4 mediates oxidation of HDAC4 and cardiac hypertrophy. Circ. Res. 2013, 112, 651–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, K.L.; Lotz, C.; Ping, P.; Cai, H. Netrin-1 abrogates ischemia/reperfusion-induced cardiac mitochondrial dysfunction via nitric oxide-dependent attenuation of NOX4 activation and recoupling of NOS. J. Mol. Cell. Cardiol. 2015, 78, 174–185. [Google Scholar] [CrossRef] [Green Version]
- Lyle, A.N.; Deshpande, N.N.; Taniyama, Y.; Seidel-Rogol, B.; Pounkova, L.; Du, P.; Papaharalambus, C.; Lassegue, B.; Griendling, K.K. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ. Res. 2009, 105, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Matsushima, S.; Kuroda, J.; Zhai, P.; Liu, T.; Ikeda, S.; Nagarajan, N.; Oka, S.; Yokota, T.; Kinugawa, S.; Hsu, C.P.; et al. Tyrosine kinase FYN negatively regulates NOX4 in cardiac remodeling. J. Clin. Investig. 2016, 126, 3403–3416. [Google Scholar] [CrossRef]
- Ushio-Fukai, M. Localizing NADPH oxidase-derived ROS. Sci. STKE 2006, 2006, re8. [Google Scholar] [CrossRef]
- Cave, A. Selective targeting of NADPH oxidase for cardiovascular protection. Curr. Opin. Pharmacol. 2009, 9, 208–213. [Google Scholar] [CrossRef]
- Yu, Q.; Lee, C.F.; Wang, W.; Karamanlidis, G.; Kuroda, J.; Matsushima, S.; Sadoshima, J.; Tian, R. Elimination of NADPH oxidase activity promotes reductive stress and sensitizes the heart to ischemic injury. J. Am. Heart Assoc. 2014, 3, e000555. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.L.; Takano, H.; Rizvi, A.; Turrens, J.F.; Qiu, Y.; Wu, W.J.; Zhang, Q.; Bolli, R. Oxidant species trigger late preconditioning against myocardial stunning in conscious rabbits. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H281–H291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Sauve, A.A. NAD+ metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta Proteins Proteom. 2016, 1864, 1787–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal. 2008, 10, 179–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gores, G.J.; Flarsheim, C.E.; Dawson, T.L.; Nieminen, A.L.; Herman, B.; Lemasters, J.J. Swelling, reductive stress, and cell death during chemical hypoxia in hepatocytes. Am. J. Physiol. Cell Physiol. 1989, 257, C347–C354. [Google Scholar] [CrossRef]
- Loscalzo, J. Adaptions to Hypoxia and Redox Stress: Essential Concepts Confounded by Misleading Terminology. Circ. Res. 2016, 119, 511–513. [Google Scholar] [CrossRef] [Green Version]
- Sarsour, E.H.; Kumar, M.G.; Chaudhuri, L.; Kalen, A.L.; Goswami, P.C. Redox control of the cell cycle in health and disease. Antioxid. Redox Signal. 2009, 11, 2985–3011. [Google Scholar] [CrossRef]
- Mokhtari, B.; Badalzadeh, R. The potentials of distinct functions of autophagy to be targeted for attenuation of myocardial ischemia/reperfusion injury in preclinical studies: An up-to-date review. J. Physiol. Biochem. 2021, 77, 377–404. [Google Scholar] [CrossRef]
- Kubli, D.A.; Gustafsson, A.B. Mitochondria and mitophagy: The yin and yang of cell death control. Circ. Res. 2012, 111, 1208–1221. [Google Scholar] [CrossRef] [Green Version]
- Sciarretta, S.; Zhai, P.; Shao, D.; Zablocki, D.; Nagarajan, N.; Terada, L.S.; Volpe, M.; Sadoshima, J. Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2alpha/activating transcription factor 4 pathway. Circ. Res. 2013, 113, 1253–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forte, M.; Palmerio, S.; Yee, D.; Frati, G.; Sciarretta, S. Functional Role of Nox4 in Autophagy. In Mitochondrial Dynamics in Cardiovascular Medicine; Springer: Cham, Switzerland, 2017; Volume 982, pp. 307–326. [Google Scholar] [CrossRef]
- Beretta, M.; Santos, C.X.; Molenaar, C.; Hafstad, A.D.; Miller, C.C.; Revazian, A.; Betteridge, K.; Schroder, K.; Streckfuss-Bomeke, K.; Doroshow, J.H.; et al. Nox4 regulates InsP3 receptor-dependent Ca2+ release into mitochondria to promote cell survival. EMBO J. 2020, 39, e103530. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Yuan, W.; Cao, M.; Chen, R.; Wu, X.; Yan, J. Cyclophilin A Protects Cardiomyocytes against Hypoxia/Reoxygenation-Induced Apoptosis via the AKT/Nox2 Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 2717986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, L.; Gu, Y.; Ren, G.; Chen, L.; Liu, L.; Wang, X.; Gao, L. miRNA-146a Mimic Inhibits NOX4/P38 Signalling to Ameliorate Mouse Myocardial Ischaemia Reperfusion (I/R) Injury. Oxid. Med. Cell. Longev. 2021, 2021, 6366254. [Google Scholar] [CrossRef]
- Wang, F.; Wang, H.; Liu, X.; Yu, H.; Huang, X.; Huang, W.; Wang, G. Neuregulin-1 alleviate oxidative stress and mitigate inflammation by suppressing NOX4 and NLRP3/caspase-1 in myocardial ischaemia-reperfusion injury. J. Cell. Mol. Med. 2021, 25, 1783–1795. [Google Scholar] [CrossRef]
- Olejnik, A.; Banaszkiewicz, M.; Krzywonos-Zawadzka, A.; Bil-Lula, I. The Klotho protein supports redox balance and metabolic functions of cardiomyocytes during ischemia/reperfusion injury. Cardiol. J. 2021. [Google Scholar] [CrossRef]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef]
- Pendyala, S.; Gorshkova, I.A.; Usatyuk, P.V.; He, D.; Pennathur, A.; Lambeth, J.D.; Thannickal, V.J.; Natarajan, V. Role of Nox4 and Nox2 in hyperoxia-induced reactive oxygen species generation and migration of human lung endothelial cells. Antioxid. Redox Signal. 2009, 11, 747–764. [Google Scholar] [CrossRef] [Green Version]
- Chalupsky, K.; Cai, H. Endothelial dihydrofolate reductase: Critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2005, 102, 9056–9061. [Google Scholar] [CrossRef] [Green Version]
- Abramov, A.Y.; Scorziello, A.; Duchen, M.R. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J. Neurosci. 2007, 27, 1129–1138. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Chen, Q.; Moghaddas, S.; Hassan, M.O.; Tandler, B.; Hoppel, C.L. Blockade of electron transport during ischemia protects cardiac mitochondria. J. Biol. Chem. 2004, 279, 47961–47967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadenas, E.; Boveris, A.; Ragan, C.I.; Stoppani, A.O. Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch. Biochem. Biophys. 1977, 180, 248–257. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stovell, M.G.; Mada, M.O.; Helmy, A.; Carpenter, T.A.; Thelin, E.P.; Yan, J.L.; Guilfoyle, M.R.; Jalloh, I.; Howe, D.J.; Grice, P.; et al. The effect of succinate on brain NADH/NAD+ redox state and high energy phosphate metabolism in acute traumatic brain injury. Sci. Rep. 2018, 8, 11140. [Google Scholar] [CrossRef]
- Szekeres, F.L.M.; Walum, E.; Wikstrom, P.; Arner, A. A small molecule inhibitor of Nox2 and Nox4 improves contractile function after ischemia-reperfusion in the mouse heart. Sci. Rep. 2021, 11, 11970. [Google Scholar] [CrossRef]
- Cai, X.; Yang, C.; Shao, L.; Zhu, H.; Wang, Y.; Huang, X.; Wang, S.; Hong, L. Targeting NOX 4 by petunidin improves anoxia/reoxygenation-induced myocardium injury. Eur. J. Pharmacol. 2020, 888, 173414. [Google Scholar] [CrossRef]
- Shi, Y.; Hou, S.A. Protective effects of metformin against myocardial ischemiareperfusion injury via AMPKdependent suppression of NOX4. Mol. Med. Rep. 2021, 24, 712. [Google Scholar] [CrossRef]
- Okabe, K.; Matsushima, S.; Ikeda, S.; Ikeda, M.; Ishikita, A.; Tadokoro, T.; Enzan, N.; Yamamoto, T.; Sada, M.; Deguchi, H.; et al. DPP (Dipeptidyl Peptidase)-4 Inhibitor Attenuates Ang II (Angiotensin II)-Induced Cardiac Hypertrophy via GLP (Glucagon-Like Peptide)-1-Dependent Suppression of Nox (Nicotinamide Adenine Dinucleotide Phosphate Oxidase) 4-HDAC (Histone Deacetylase) 4 Pathway. Hypertension 2020, 75, 991–1001. [Google Scholar] [CrossRef]
- Chua, S.; Lee, F.Y.; Tsai, T.H.; Sheu, J.J.; Leu, S.; Sun, C.K.; Chen, Y.L.; Chang, H.W.; Chai, H.T.; Liu, C.F.; et al. Inhibition of dipeptidyl peptidase-IV enzyme activity protects against myocardial ischemia-reperfusion injury in rats. J. Transl. Med. 2014, 12, 357. [Google Scholar] [CrossRef] [Green Version]
- Reutens, A.T.; Jandeleit-Dahm, K.; Thomas, M.; Salim, A.; De Livera, A.M.; Bach, L.A.; Colman, P.G.; Davis, T.M.E.; Ekinci, E.I.; Fulcher, G.; et al. A physician-initiated double-blind, randomised, placebo-controlled, phase 2 study evaluating the efficacy and safety of inhibition of NADPH oxidase with the first-in-class Nox-1/4 inhibitor, GKT137831, in adults with type 1 diabetes and persistently elevated urinary albumin excretion: Protocol and statistical considerations. Contemp. Clin. Trials 2020, 90, 105892. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsushima, S.; Sadoshima, J. Yin and Yang of NADPH Oxidases in Myocardial Ischemia-Reperfusion. Antioxidants 2022, 11, 1069. https://doi.org/10.3390/antiox11061069
Matsushima S, Sadoshima J. Yin and Yang of NADPH Oxidases in Myocardial Ischemia-Reperfusion. Antioxidants. 2022; 11(6):1069. https://doi.org/10.3390/antiox11061069
Chicago/Turabian StyleMatsushima, Shouji, and Junichi Sadoshima. 2022. "Yin and Yang of NADPH Oxidases in Myocardial Ischemia-Reperfusion" Antioxidants 11, no. 6: 1069. https://doi.org/10.3390/antiox11061069