
Auranofin and Pharmacologic Ascorbate as Radiomodulators in the Treatment of Pancreatic Cancer

 , ,
, , 
Abstract
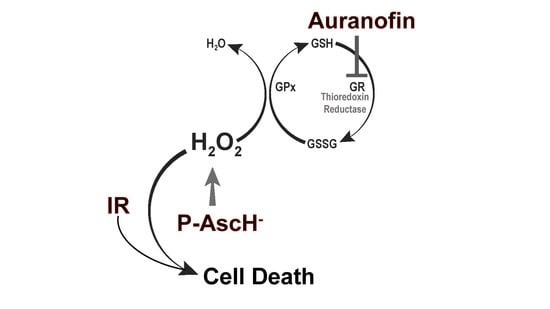
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Pancreatic Cancer
2. Pharmacologic Ascorbate
2.1. Pharmacologic Ascorbate Use in Cancer
2.2. Pharmacologic Ascorbate Increases Radiation Toxicity in Pancreatic Cancer While Protecting Normal Tissue
2.3. Long-Term Survival following P-AscH− in Pancreatic Cancer
3. Pathways for Hydrogen Peroxide Removal
4. Auranofin
4.1. Auranofin Use in Cancer
4.2. Au Inhibits Thioredoxin Reductase Activity in Pancreatic Cancer Cells
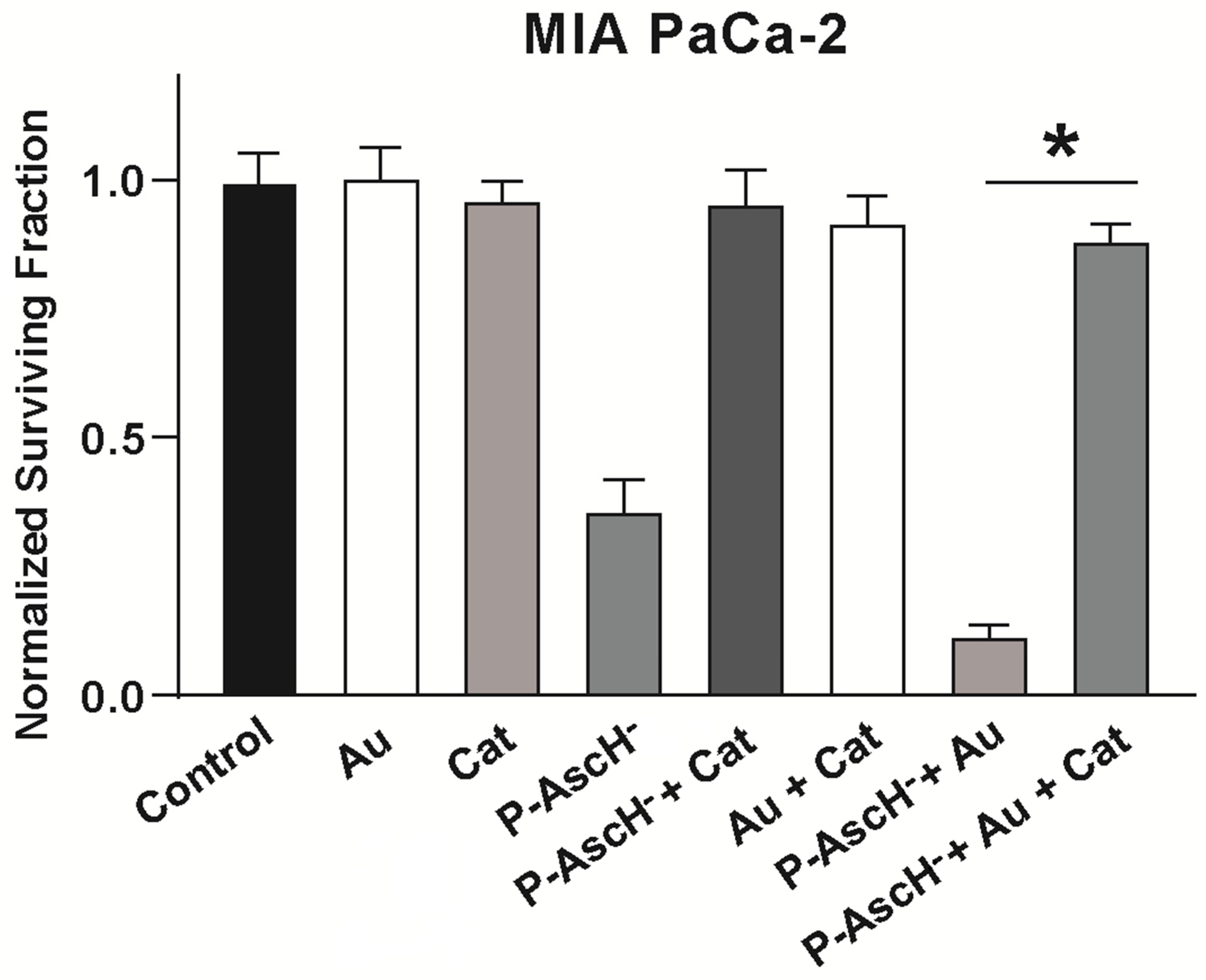
4.3. Auranofin Sensitizes Pancreatic Cancer Cells to P-AscH−
4.4. Au Combined with P-AscH− Sensitizes Pancreatic Cancer Cells to Ionizing Radiation
4.5. Au and P-AscH− Potential in Cancer Therapy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Low, F.M.; Hampton, M.B.; Peskin, A.V.; Winterbourn, C.C. Peroxiredoxin 2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte. Blood 2007, 109, 2611–2617. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.X.; Zhao, C.F.; Chen, W.B.; Liu, Q.C.; Li, Q.W.; Lin, Y.Y.; Gao, F. Pancreatic cancer: A review of epidemiology, trend, and risk factors. World J. Gastroenterol. 2021, 27, 4298–4321. [Google Scholar] [CrossRef] [PubMed]
- Network, N.C.C. NCCN Clinical Practice Guidelines in Oncology: Pancreatic Adenocarcinoma. 2022. Available online: https://www.nccn.org/ (accessed on 1 April 2022).
- Li, D.; Xie, K.; Wolff, R.; Abbruzzese, J.L. Pancreatic cancer. Lancet 2004, 363, 1049–1057. [Google Scholar] [CrossRef]
- Bengtsson, A.; Andersson, R.; Ansari, D. The actual 5-year survivors of pancreatic ductal adenocarcinoma based on real-world data. Sci. Rep. 2020, 10, 16425. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Chone, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Chin, V.; Nagrial, A.; Sjoquist, K.; O’Connor, C.A.; Chantrill, L.; Biankin, A.V.; Scholten, R.J.; Yip, D. Chemotherapy and radiotherapy for advanced pancreatic cancer. Cochrane Database Syst. Rev. 2018, 3, CD011044. [Google Scholar] [CrossRef]
- Spiliopoulos, S.; Zurlo, M.T.; Casella, A.; Laera, L.; Surico, G.; Surgo, A.; Fiorentino, A.; de’Angelis, N.; Calbi, R.; Memeo, R.; et al. Current status of non-surgical treatment of locally advanced pancreatic cancer. World J. Gastrointest. Oncol. 2021, 13, 2064–2075. [Google Scholar] [CrossRef]
- Goldstein, M.; Kastan, M.B. The DNA damage response: Implications for tumor responses to radiation and chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Powell, S.; McMillan, T.J. DNA damage and repair following treatment with ionizing radiation. Radiother Oncol. 1990, 19, 95–108. [Google Scholar] [CrossRef]
- Sutherland, B.M.; Bennett, P.V.; Sidorkina, O.; Laval, J. Clustered DNA damages induced in isolated DNA and in human cells by low doses of ionizing radiation. Proc. Natl. Acad. Sci. USA 2000, 97, 103–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, B.M.; Bennett, P.V.; Sutherland, J.C.; Laval, J. Clustered DNA damages induced by x rays in human cells. Radiat. Res. 2002, 157, 611–616. [Google Scholar] [CrossRef]
- Kobayashi, J.; Iwabuchi, K.; Miyagawa, K.; Sonoda, E.; Suzuki, K.; Takata, M.; Tauchi, H. Current topics in DNA double-strand break repair. J. Radiat. Res. 2008, 49, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Tempero, M.A.; Malafa, M.P.; Al-Hawary, M.; Asbun, H.; Bain, A.; Behrman, S.W.; Benson, A.B.; Binder, E.; Cardin, D.B.; Cha, C.; et al. Pancreatic Adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2017, 15, 1028–1061. [Google Scholar] [CrossRef]
- Hammel, P.; Huguet, F.; van Laethem, J.L.; Goldstein, D.; Glimelius, B.; Artru, P.; Borbath, I.; Bouche, O.; Shannon, J.; Andre, T.; et al. Effect of Chemoradiotherapy vs Chemotherapy on Survival in Patients with Locally Advanced Pancreatic Cancer Controlled After 4 Months of Gemcitabine With or Without Erlotinib: The LAP07 Randomized Clinical Trial. JAMA 2016, 315, 1844–1853. [Google Scholar] [CrossRef]
- Tempero, M.A.; Malafa, M.P.; Chiorean, E.G.; Czito, B.; Scaife, C.; Narang, A.K.; Fountzilas, C.; Wolpin, B.M.; Al-Hawary, M.; Asbun, H.; et al. NCCN Guidelines Insights: Pancreatic Adenocarcinoma, Version 1.2019: Featured Updates to the NCCN Guidelines. J. Natl. Compr. Cancer Netw. 2019, 17, 202–210. [Google Scholar] [CrossRef] [Green Version]
- Abi Jaoude, J.; Thunshelle, C.P.; Kouzy, R.; Nguyen, N.D.; Lin, D.; Prakash, L.; Bumanlag, I.M.; Noticewala, S.S.; Niedzielski, J.S.; Beddar, S.; et al. Stereotactic Versus Conventional Radiation Therapy for Patients with Pancreatic Cancer in the Modern Era. Adv. Radiat. Oncol. 2021, 6, 100763. [Google Scholar] [CrossRef]
- Ghaly, M.; Gogineni, E.; Herman, J.; Saif, M.W. New Potential Options for SBRT in Pancreatic Cancer. Cancer Med. J. 2021, 4, 41–50. [Google Scholar]
- Pavic, M.; Niyazi, M.; Wilke, L.; Corradini, S.; Vornhulz, M.; Mansmann, U.; Al Tawil, A.; Fritsch, R.; Horner-Rieber, J.; Debus, J.; et al. MR-guided adaptive stereotactic body radiotherapy (SBRT) of primary tumor for pain control in metastatic pancreatic ductal adenocarcinoma (mPDAC): An open randomized, multicentric, parallel group clinical trial (MASPAC). Radiat. Oncol. 2022, 17, 18. [Google Scholar] [CrossRef]
- Qing, S.; Gu, L.; Zhang, H. Phase I study of dose-escalated stereotactic body radiation therapy for locally advanced pancreatic head cancers: Initial clinical results. Cancer Med. 2021, 10, 6736–6743. [Google Scholar] [CrossRef] [PubMed]
- Oberstein, P.E.; Olive, K.P. Pancreatic cancer: Why is it so hard to treat? Therap. Adv. Gastroenterol. 2013, 6, 321–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buettner, G.R. The pecking order of free radicals and antioxidants: Lipid peroxidation, alpha-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef]
- Padayatty, S.J.; Sun, H.; Wang, Y.; Riordan, H.D.; Hewitt, S.M.; Katz, A.; Wesley, R.A.; Levine, M. Vitamin C pharmacokinetics: Implications for oral and intravenous use. Ann. Intern. Med. 2004, 140, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.; Wang, Y.; Padayatty, S.J.; Morrow, J. A new recommended dietary allowance of vitamin C for healthy young women. Proc. Natl. Acad. Sci. USA 2001, 98, 9842–9846. [Google Scholar] [CrossRef] [Green Version]
- Vera, J.C.; Rivas, C.I.; Velasquez, F.V.; Zhang, R.H.; Concha, I.I.; Golde, D.W. Resolution of the facilitated transport of dehydroascorbic acid from its intracellular accumulation as ascorbic acid. J. Biol. Chem. 1995, 270, 23706–23712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savini, I.; Rossi, A.; Pierro, C.; Avigliano, L.; Catani, M.V. SVCT1 and SVCT2: Key proteins for vitamin C uptake. Amino. Acids. 2008, 34, 347–355. [Google Scholar] [CrossRef]
- Levine, M.; Conry-Cantilena, C.; Wang, Y.; Welch, R.W.; Washko, P.W.; Dhariwal, K.R.; Park, J.B.; Lazarev, A.; Graumlich, J.F.; King, J.; et al. Vitamin C pharmacokinetics in healthy volunteers: Evidence for a recommended dietary allowance. Proc. Natl. Acad. Sci. USA 1996, 93, 3704–3709. [Google Scholar] [CrossRef] [Green Version]
- Verrax, J.; Calderon, P.B. Pharmacologic concentrations of ascorbate are achieved by parenteral administration and exhibit antitumoral effects. Free Radic. Biol. Med. 2009, 47, 32–40. [Google Scholar] [CrossRef]
- Du, J.; Martin, S.M.; Levine, M.; Wagner, B.A.; Buettner, G.R.; Wang, S.H.; Taghiyev, A.F.; Du, C.; Knudson, C.M.; Cullen, J.J. Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenfeld, J.D.; Sibenaller, Z.A.; Mapuskar, K.A.; Wagner, B.A.; Cramer-Morales, K.L.; Furqan, M.; Sandhu, S.; Carlisle, T.L.; Smith, M.C.; Abu Hejleh, T.; et al. O2− and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell 2017, 31, 487–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, E.; Campbell, A.; Jack, T. The orthomolecular treatment of cancer. III. Reticulum cell sarcoma: Double complete regression induced by high-dose ascorbic acid therapy. Chem. Biol. Interact. 1975, 11, 387–393. [Google Scholar] [CrossRef]
- Cameron, E.; Pauling, L. Supplemental ascorbate in the supportive treatment of cancer: Prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. USA 1976, 73, 3685–3689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, E.; Pauling, L. Supplemental ascorbate in the supportive treatment of cancer: Reevaluation of prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. USA 1978, 75, 4538–4542. [Google Scholar] [CrossRef] [Green Version]
- Creagan, E.T.; Moertel, C.G.; O’Fallon, J.R.; Schutt, A.J.; O’Connell, M.J.; Rubin, J.; Frytak, S. Failure of high-dose vitamin C (ascorbic acid) therapy to benefit patients with advanced cancer. A controlled trial. N. Engl. J. Med. 1979, 301, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Moertel, C.G.; Fleming, T.R.; Creagan, E.T.; Rubin, J.; O’Connell, M.J.; Ames, M.M. High-dose vitamin C versus placebo in the treatment of patients with advanced cancer who have had no prior chemotherapy. A randomized double-blind comparison. N. Engl. J. Med. 1985, 312, 137–141. [Google Scholar] [CrossRef]
- Ghanem, A.; Melzer, A.M.; Zaal, E.; Neises, L.; Baltissen, D.; Matar, O.; Glennemeier-Marke, H.; Almouhanna, F.; Theobald, J.; Abu El Maaty, M.A.; et al. Ascorbate kills breast cancer cells by rewiring metabolism via redox imbalance and energy crisis. Free Radic. Biol. Med. 2021, 163, 196–209. [Google Scholar] [CrossRef]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109. [Google Scholar] [CrossRef] [Green Version]
- Cieslak, J.A.; Strother, R.K.; Rawal, M.; Du, J.; Doskey, C.M.; Schroeder, S.R.; Button, A.; Wagner, B.A.; Buettner, G.R.; Cullen, J.J. Manganoporphyrins and ascorbate enhance gemcitabine cytotoxicity in pancreatic cancer. Free Radic. Biol. Med. 2015, 83, 227–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doskey, C.M.; Buranasudja, V.; Wagner, B.A.; Wilkes, J.G.; Du, J.; Cullen, J.J.; Buettner, G.R. Tumor cells have decreased ability to metabolize H2O2: Implications for pharmacological ascorbate in cancer therapy. Redox Biol. 2016, 10, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espey, M.G.; Chen, P.; Chalmers, B.; Drisko, J.; Sun, A.Y.; Levine, M.; Chen, Q. Pharmacologic ascorbate synergizes with gemcitabine in preclinical models of pancreatic cancer. Free Radic. Biol. Med. 2011, 50, 1610–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Chen, P.; Drisko, J.A.; Khabele, D.; Godwin, A.K.; Chen, Q. Pharmacological ascorbate induces ‘BRCAness’ and enhances the effects of Poly(ADP-Ribose) polymerase inhibitors against BRCA1/2 wild-type ovarian cancer. Oncol. Lett. 2020, 19, 2629–2638. [Google Scholar] [CrossRef] [Green Version]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Liu, J.; Hinkhouse, M.M.; Sun, W.; Weydert, C.J.; Ritchie, J.M.; Oberley, L.W.; Cullen, J.J. Redox regulation of pancreatic cancer cell growth: Role of glutathione peroxidase in the suppression of the malignant phenotype. Hum. Gene Ther. 2004, 15, 239–250. [Google Scholar] [CrossRef]
- Monti, D.A.; Mitchell, E.; Bazzan, A.J.; Littman, S.; Zabrecky, G.; Yeo, C.J.; Pillai, M.V.; Newberg, A.B.; Deshmukh, S.; Levine, M. Phase I evaluation of intravenous ascorbic acid in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. PLoS ONE 2012, 7, e29794. [Google Scholar] [CrossRef]
- Welsh, J.L.; Wagner, B.A.; van’t Erve, T.J.; Zehr, P.S.; Berg, D.J.; Halfdanarson, T.R.; Yee, N.S.; Bodeker, K.L.; Du, J.; Roberts, L.J., 2nd; et al. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): Results from a phase I clinical trial. Cancer Chemother. Pharmacol. 2013, 71, 765–775. [Google Scholar] [CrossRef] [Green Version]
- Di Marco, M.; Di Cicilia, R.; Macchini, M.; Nobili, E.; Vecchiarelli, S.; Brandi, G.; Biasco, G. Metastatic pancreatic cancer: Is gemcitabine still the best standard treatment? (Review). Oncol. Rep. 2010, 23, 1183–1192. [Google Scholar] [CrossRef] [Green Version]
- Polireddy, K.; Dong, R.; Reed, G.; Yu, J.; Chen, P.; Williamson, S.; Violet, P.C.; Pessetto, Z.; Godwin, A.K.; Fan, F.; et al. High Dose Parenteral Ascorbate Inhibited Pancreatic Cancer Growth and Metastasis: Mechanisms and a Phase I/IIa study. Sci. Rep. 2017, 7, 17188. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Cieslak, J.A., 3rd; Welsh, J.L.; Sibenaller, Z.A.; Allen, B.G.; Wagner, B.A.; Kalen, A.L.; Doskey, C.M.; Strother, R.K.; Button, A.M.; et al. Pharmacological Ascorbate Radiosensitizes Pancreatic Cancer. Cancer Res. 2015, 75, 3314–3326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, M.S.; Wilkes, J.G.; Schroeder, S.R.; Buettner, G.R.; Wagner, B.A.; Du, J.; Gibson-Corley, K.; O’Leary, B.R.; Spitz, D.R.; Buatti, J.M.; et al. Pharmacologic Ascorbate Reduces Radiation-Induced Normal Tissue Toxicity and Enhances Tumor Radiosensitization in Pancreatic Cancer. Cancer Res. 2018, 78, 6838–6851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, M.S.; O’Leary, B.R.; Wilkes, J.G.; Gibson, A.R.; Wagner, B.A.; Du, J.; Sarsour, E.; Hwang, R.F.; Buettner, G.R.; Cullen, J.J. Enhanced Pharmacological Ascorbate Oxidation Radiosensitizes Pancreatic Cancer. Radiat. Res. 2019, 191, 43–51. [Google Scholar] [CrossRef]
- Perrakis, N.; Athanassiou, E.; Vamvakopoulou, D.; Kyriazi, M.; Kappos, H.; Vamvakopoulos, N.C.; Nomikos, I. Practical approaches to effective management of intestinal radiation injury: Benefit of resectional surgery. World J. Gastroenterol. 2011, 17, 4013–4016. [Google Scholar] [CrossRef]
- Ngo, B.; Van Riper, J.M.; Cantley, L.C.; Yun, J. Targeting cancer vulnerabilities with high-dose vitamin C. Nat. Rev. Cancer 2019, 19, 271–282. [Google Scholar] [CrossRef]
- Wang, H.; Liu, J.; Xia, G.; Lei, S.; Huang, X.; Huang, X. Survival of pancreatic cancer patients is negatively correlated with age at diagnosis: A population-based retrospective study. Sci. Rep. 2020, 10, 7048. [Google Scholar] [CrossRef]
- Petrelli, F.; Coinu, A.; Borgonovo, K.; Cabiddu, M.; Barni, S. Progression-free survival as surrogate endpoint in advanced pancreatic cancer: Meta-analysis of 30 randomized first-line trials. Hepatobiliary Pancreat. Dis. Int. 2015, 14, 124–131. [Google Scholar] [CrossRef]
- Du, J.; Carroll, R.S.; Steers, G.J.; Wagner, B.A.; O’Leary, B.R.; Jensen, C.S.; Buettner, G.R.; Cullen, J.J. Catalase Modulates the Radio-Sensitization of Pancreatic Cancer Cells by Pharmacological Ascorbate. Antioxidants 2021, 10, 614. [Google Scholar] [CrossRef]
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 2012, 1826, 443–457. [Google Scholar] [CrossRef] [Green Version]
- Oberley, T.D.; Oberley, L.W. Antioxidant enzyme levels in cancer. Histol. Histopathol. 1997, 12, 525–535. [Google Scholar] [PubMed]
- Sasaki, K.; Bannai, S.; Makino, N. Kinetics of hydrogen peroxide elimination by human umbilical vein endothelial cells in culture. Biochim. Biophys. Acta 1998, 1380, 275–288. [Google Scholar] [CrossRef]
- Mueller, S.; Riedel, H.D.; Stremmel, W. Direct evidence for catalase as the predominant H2O2-removing enzyme in human erythrocytes. Blood 1997, 90, 4973–4978. [Google Scholar] [CrossRef] [PubMed]
- Flohe, L. The glutathione peroxidase reaction: Molecular basis of the antioxidant function of selenium in mammals. Curr. Top Cell Regul. 1985, 27, 473–478. [Google Scholar] [CrossRef]
- Schoenfeld, J.D.; Alexander, M.S.; Waldron, T.J.; Sibenaller, Z.A.; Spitz, D.R.; Buettner, G.R.; Allen, B.G.; Cullen, J.J. Pharmacological Ascorbate as a Means of Sensitizing Cancer Cells to Radio-Chemotherapy While Protecting Normal Tissue. Semin. Radiat. Oncol. 2019, 29, 25–32. [Google Scholar] [CrossRef]
- Larsson, A. Enzymatic Synthesis of Deoxyribonucleotides. VII. Studies on the Hydrogen Transfer with Tritiated Water. Biochemistry 1965, 4, 1984–1993. [Google Scholar] [CrossRef]
- Moore, E.C. A thioredoxin—thioredoxin reductase system from rat tumor. Biochem. Biophys. Res. Commun. 1967, 29, 264–268. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Deisseroth, A.; Dounce, A.L. Catalase: Physical and chemical properties, mechanism of catalysis, and physiological role. Physiol. Rev. 1970, 50, 319–375. [Google Scholar] [CrossRef]
- Ogusucu, R.; Rettori, D.; Munhoz, D.C.; Netto, L.E.; Augusto, O. Reactions of yeast thioredoxin peroxidases I and II with hydrogen peroxide and peroxynitrite: Rate constants by competitive kinetics. Free Radic. Biol. Med. 2007, 42, 326–334. [Google Scholar] [CrossRef]
- Holmgren, A.; Lu, J. Thioredoxin and thioredoxin reductase: Current research with special reference to human disease. Biochem. Biophys. Res. Commun. 2010, 396, 120–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoeib, T.; Atkinson, D.W.; Sharp, B.L. Structural analysis of the anti-arthritic drug Auranofin: Its complexes with cysteine, selenocysteine and their fragmentation products. Inorg. Chim. Acta 2010, 363, 184–192. [Google Scholar] [CrossRef]
- Finkelstein, A.E.; Walz, D.T.; Batista, V.; Mizraji, M.; Roisman, F.; Misher, A. Auranofin. New oral gold compound for treatment of rheumatoid arthritis. Ann. Rheum. Dis. 1976, 35, 251–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Champion, G.D.; Graham, G.G.; Ziegler, J.B. The gold complexes. Baillieres Clin. Rheumatol. 1990, 4, 491–534. [Google Scholar] [CrossRef]
- Kim, N.H.; Lee, M.Y.; Park, S.J.; Choi, J.S.; Oh, M.K.; Kim, I.S. Auranofin blocks interleukin-6 signalling by inhibiting phosphorylation of JAK1 and STAT3. Immunology 2007, 122, 607–614. [Google Scholar] [CrossRef]
- Stern, I.; Wataha, J.C.; Lewis, J.B.; Messer, R.L.; Lockwood, P.E.; Tseng, W.Y. Anti-rheumatic gold compounds as sublethal modulators of monocytic LPS-induced cytokine secretion. Toxicol. Vitr. 2005, 19, 365–371. [Google Scholar] [CrossRef]
- Kim, N.H.; Oh, M.K.; Park, H.J.; Kim, I.S. Auranofin, a gold(I)-containing antirheumatic compound, activates Keap1/Nrf2 signaling via Rac1/iNOS signal and mitogen-activated protein kinase activation. J. Pharmacol. Sci. 2010, 113, 246–254. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Kim, K.; Kim, H.; Kwon, J.; Lee, Y.H.; Lee, C.K.; Song, Y.; Lee, S.J.; Ha, N.; Kim, K. Auranofin inhibits overproduction of pro-inflammatory cytokines, cyclooxygenase expression and PGE2 production in macrophages. Arch. Pharm. Res. 2008, 31, 67–74. [Google Scholar] [CrossRef]
- Han, S.; Kim, K.; Song, Y.; Kim, H.; Kwon, J.; Lee, Y.H.; Lee, C.K.; Lee, S.J.; Ha, N.; Kim, K. Auranofin, an immunosuppressive drug, inhibits MHC class I and MHC class II pathways of antigen presentation in dendritic cells. Arch. Pharm. Res. 2008, 31, 370–376. [Google Scholar] [CrossRef]
- Park, S.J.; Kim, I.S. The role of p38 MAPK activation in auranofin-induced apoptosis of human promyelocytic leukaemia HL-60 cells. Br. J. Pharmacol. 2005, 146, 506–513. [Google Scholar] [CrossRef] [Green Version]
- Honda, Z.; Iizasa, T.; Morita, Y.; Matsuta, K.; Nishida, Y.; Miyamoto, T. Differential inhibitory effects of auranofin on leukotriene B4 and leukotriene C4 formation by human polymorphonuclear leukocytes. Biochem. Pharmacol. 1987, 36, 1475–1481. [Google Scholar] [CrossRef]
- Yamada, M.; Niki, H.; Yamashita, M.; Mue, S.; Ohuchi, K. Prostaglandin E2 production dependent upon cyclooxygenase-1 and cyclooxygenase-2 and its contradictory modulation by auranofin in rat peritoneal macrophages. J. Pharmacol. Exp. Ther. 1997, 281, 1005–1012. [Google Scholar] [PubMed]
- Yamashita, M.; Niki, H.; Yamada, M.; Watanabe-Kobayashi, M.; Mue, S.; Ohuchi, K. Dual effects of auranofin on prostaglandin E2 production by rat peritoneal macrophages. Eur. J. Pharmacol. 1997, 325, 221–227. [Google Scholar] [CrossRef]
- Walz, D.T.; DiMartino, M.J.; Griswold, D.E.; Intoccia, A.P.; Flanagan, T.L. Biologic actions and pharmacokinetic studies of auranofin. Am. J. Med. 1983, 75, 90–108. [Google Scholar] [CrossRef]
- Sannella, A.R.; Casini, A.; Gabbiani, C.; Messori, L.; Bilia, A.R.; Vincieri, F.F.; Majori, G.; Severini, C. New uses for old drugs. Auranofin, a clinically established antiarthritic metallodrug, exhibits potent antimalarial effects in vitro: Mechanistic and pharmacological implications. FEBS Lett. 2008, 582, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Newman, Z.L.; Sirianni, N.; Mawhinney, C.; Lee, M.S.; Leppla, S.H.; Moayeri, M.; Johansen, L.M. Auranofin protects against anthrax lethal toxin-induced activation of the Nlrp1b inflammasome. Antimicrob. Agents Chemother. 2011, 55, 1028–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutton, M.L.; Pehlivanoglu, H.; Vidor, C.J.; James, M.L.; Thomson, M.J.; Lyras, D. Repurposing auranofin as a Clostridioides difficile therapeutic. J. Antimicrob. Chemother. 2020, 75, 409–417. [Google Scholar] [CrossRef]
- Sharma, N.; Singh, A.; Sharma, R.; Kumar, A. Repurposing of Auranofin Against Bacterial Infections: An In silico and In vitro Study. Curr. Comput. Aided Drug Des. 2021, 17, 687–701. [Google Scholar] [CrossRef]
- Lewis, M.G.; DaFonseca, S.; Chomont, N.; Palamara, A.T.; Tardugno, M.; Mai, A.; Collins, M.; Wagner, W.L.; Yalley-Ogunro, J.; Greenhouse, J.; et al. Gold drug auranofin restricts the viral reservoir in the monkey AIDS model and induces containment of viral load following ART suspension. AIDS 2011, 25, 1347–1356. [Google Scholar] [CrossRef]
- Ashino, T.; Sugiuchi, J.; Uehara, J.; Naito-Yamamoto, Y.; Kenmotsu, S.; Iwakura, Y.; Shioda, S.; Numazawa, S.; Yoshida, T. Auranofin protects against cocaine-induced hepatic injury through induction of heme oxygenase-1. J. Toxicol. Sci. 2011, 36, 635–643. [Google Scholar] [CrossRef]
- Gromer, S.; Arscott, L.D.; Williams, C.H., Jr.; Schirmer, R.H.; Becker, K. Human placenta thioredoxin reductase. Isolation of the selenoenzyme, steady state kinetics, and inhibition by therapeutic gold compounds. J. Biol. Chem. 1998, 273, 20096–20101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saei, A.A.; Gullberg, H.; Sabatier, P.; Beusch, C.M.; Johansson, K.; Lundgren, B.; Arvidsson, P.I.; Arner, E.S.J.; Zubarev, R.A. Comprehensive chemical proteomics for target deconvolution of the redox active drug auranofin. Redox Biol. 2020, 32, 101491. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.G.; Brown, K.K.; Arner, E.S.; Hampton, M.B. The thioredoxin reductase inhibitor auranofin triggers apoptosis through a Bax/Bak-dependent process that involves peroxiredoxin 3 oxidation. Biochem. Pharmacol. 2008, 76, 1097–1109. [Google Scholar] [CrossRef] [PubMed]
- Olney, K.E.; Du, J.; van’t Erve, T.J.; Witmer, J.R.; Sibenaller, Z.A.; Wagner, B.A.; Buettner, G.R.; Cullen, J.J. Inhibitors of hydroperoxide metabolism enhance ascorbate-induced cytotoxicity. Free Radic. Res. 2013, 47, 154–163. [Google Scholar] [CrossRef]
- Joo, M.K.; Shin, S.; Ye, D.J.; An, H.G.; Kwon, T.U.; Baek, H.S.; Kwon, Y.J.; Chun, Y.J. Combined treatment with auranofin and trametinib induces synergistic apoptosis in breast cancer cells. J. Toxicol. Environ. Health A 2021, 84, 84–94. [Google Scholar] [CrossRef]
- Lee, J.E.; Kwon, Y.J.; Baek, H.S.; Ye, D.J.; Cho, E.; Choi, H.K.; Oh, K.S.; Chun, Y.J. Synergistic induction of apoptosis by combination treatment with mesupron and auranofin in human breast cancer cells. Arch. Pharm. Res. 2017, 40, 746–759. [Google Scholar] [CrossRef]
- Ye, D.J.; Kwon, Y.J.; Baek, H.S.; Cho, E.; Kwon, T.U.; Chun, Y.J. Combination treatment with auranofin and nutlin-3a induces synergistic cytotoxicity in breast cancer cells. J. Toxicol. Environ. Health A 2019, 82, 626–637. [Google Scholar] [CrossRef]
- Freire Boullosa, L.; Van Loenhout, J.; Flieswasser, T.; De Waele, J.; Hermans, C.; Lambrechts, H.; Cuypers, B.; Laukens, K.; Bartholomeus, E.; Siozopoulou, V.; et al. Auranofin reveals therapeutic anticancer potential by triggering distinct molecular cell death mechanisms and innate immunity in mutant p53 non-small cell lung cancer. Redox Biol. 2021, 42, 101949. [Google Scholar] [CrossRef]
- Lin, Z.; Li, Q.; Zhao, Y.; Lin, Z.; Cheng, N.; Zhang, D.; Liu, G.; Lin, J.; Zhang, H.; Lin, D. Combination of Auranofin and ICG-001 Suppress the Proliferation and Metastasis of Colon Cancer. Front. Oncol. 2021, 11, 738085. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, J.H.; Berek, J.S.; Hu, M.C. Auranofin displays anticancer activity against ovarian cancer cells through FOXO3 activation independent of p53. Int. J. Oncol. 2014, 45, 1691–1698. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, B.; Du, J.; Heer, C.; Van Beek, H.; Sptiz, D.R.; Cullen, J.J. Inhibition of Peroxide Removal Enhances Pharmacological Ascorbate and Ionizing Radiation-Induced Cytotoxicity of Pancreatic Ductal Adenocarcinoma. Free Radical. Bio. Med. 2017, 112, 97. [Google Scholar] [CrossRef]
- Van Beek, H. Inhibition of Peroxide Removal Systems and Ascorbate-Induced Cytotoxicity in Pancreatic Cancer. Master’s Thesis, University of Iowa, Iowa City, IA, USA, 2016. [Google Scholar] [CrossRef] [Green Version]
- Day, A.M.; Brown, J.D.; Taylor, S.R.; Rand, J.D.; Morgan, B.A.; Veal, E.A. Inactivation of a peroxiredoxin by hydrogen peroxide is critical for thioredoxin-mediated repair of oxidized proteins and cell survival. Mol. Cell 2012, 45, 398–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, P.; Karton, A.; Betz, A.; Peskin, A.V.; Pace, P.; O’Reilly, R.J.; Hampton, M.B.; Radom, L.; Winterbourn, C.C. Model for the exceptional reactivity of peroxiredoxins 2 and 3 with hydrogen peroxide: A kinetic and computational study. J. Biol. Chem. 2011, 286, 18048–18055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stocker, S.; Maurer, M.; Ruppert, T.; Dick, T.P. A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat. Chem. Biol. 2018, 14, 148–155. [Google Scholar] [CrossRef]
- Stocker, S.; Van Laer, K.; Mijuskovic, A.; Dick, T.P. The Conundrum of Hydrogen Peroxide Signaling and the Emerging Role of Peroxiredoxins as Redox Relay Hubs. Antioxid. Redox Signal. 2018, 28, 558–573. [Google Scholar] [CrossRef]
- Chang, T.S.; Cho, C.S.; Park, S.; Yu, S.; Kang, S.W.; Rhee, S.G. Peroxiredoxin III, a mitochondrion-specific peroxidase, regulates apoptotic signaling by mitochondria. J. Biol. Chem. 2004, 279, 41975–41984. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.W.; Chang, T.S.; Lee, T.H.; Kim, E.S.; Yu, D.Y.; Rhee, S.G. Cytosolic peroxiredoxin attenuates the activation of Jnk and p38 but potentiates that of Erk in Hela cells stimulated with tumor necrosis factor-alpha. J. Biol. Chem. 2004, 279, 2535–2543. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Lee, T.H.; Park, E.S.; Suh, J.M.; Park, S.J.; Chung, H.K.; Kwon, O.Y.; Kim, Y.K.; Ro, H.K.; Shong, M. Role of peroxiredoxins in regulating intracellular hydrogen peroxide and hydrogen peroxide-induced apoptosis in thyroid cells. J. Biol. Chem. 2000, 275, 18266–18270. [Google Scholar] [CrossRef] [Green Version]
- Collins, J.A.; Wood, S.T.; Nelson, K.J.; Rowe, M.A.; Carlson, C.S.; Chubinskaya, S.; Poole, L.B.; Furdui, C.M.; Loeser, R.F. Oxidative Stress Promotes Peroxiredoxin Hyperoxidation and Attenuates Pro-survival Signaling in Aging Chondrocytes. J. Biol. Chem. 2016, 291, 6641–6654. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Kim, T.J.; Lee, K.Y. A novel function of peroxiredoxin 1 (Prx-1) in apoptosis signal-regulating kinase 1 (ASK1)-mediated signaling pathway. FEBS Lett. 2008, 582, 1913–1918. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Niu, W.; Zhang, J.; Ge, L.; Yang, J.; Sun, Z.; Tang, X. Peroxiredoxin 1 suppresses apoptosis via regulation of the apoptosis signal-regulating kinase 1 signaling pathway in human oral leukoplakia. Oncol. Lett. 2015, 10, 1841–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Zhang, H.; Cao, M.; Wang, L.; Wu, S.; Fang, B. Auranofin Enhances Ibrutinib’s Anticancer Activity in EGFR-Mutant Lung Adenocarcinoma. Mol. Cancer Ther. 2018, 17, 2156–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobotta, M.C.; Liou, W.; Stocker, S.; Talwar, D.; Oehler, M.; Ruppert, T.; Scharf, A.N.; Dick, T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015, 11, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Hatem, E.; Azzi, S.; El Banna, N.; He, T.; Heneman-Masurel, A.; Vernis, L.; Baille, D.; Masson, V.; Dingli, F.; Loew, D.; et al. Auranofin/Vitamin C: A Novel Drug Combination Targeting Triple-Negative Breast Cancer. J. Natl. Cancer Inst. 2019, 111, 597–608. [Google Scholar] [CrossRef]
- Graczyk-Jarzynka, A.; Goral, A.; Muchowicz, A.; Zagozdzon, R.; Winiarska, M.; Bajor, M.; Trzeciecka, A.; Fidyt, K.; Krupka, J.A.; Cyran, J.; et al. Inhibition of thioredoxin-dependent H2O2 removal sensitizes malignant B-cells to pharmacological ascorbate. Redox Biol. 2019, 21, 101062. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steers, G.J.; Chen, G.Y.; O’Leary, B.R.; Du, J.; Van Beek, H.; Cullen, J.J. Auranofin and Pharmacologic Ascorbate as Radiomodulators in the Treatment of Pancreatic Cancer. Antioxidants 2022, 11, 971. https://doi.org/10.3390/antiox11050971
Steers GJ, Chen GY, O’Leary BR, Du J, Van Beek H, Cullen JJ. Auranofin and Pharmacologic Ascorbate as Radiomodulators in the Treatment of Pancreatic Cancer. Antioxidants. 2022; 11(5):971. https://doi.org/10.3390/antiox11050971
Chicago/Turabian StyleSteers, Garett J., Gloria Y. Chen, Brianne R. O’Leary, Juan Du, Hannah Van Beek, and Joseph J. Cullen. 2022. "Auranofin and Pharmacologic Ascorbate as Radiomodulators in the Treatment of Pancreatic Cancer" Antioxidants 11, no. 5: 971. https://doi.org/10.3390/antiox11050971