
Intranasal Administration of Nanovectorized Docosahexaenoic Acid (DHA) Improves Cognitive Function in Two Complementary Mouse Models of Alzheimer’s Disease

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. DHA-Loaded Microemulsions
2.3. Assessment of the Antioxidant Capacity of Microemulsions
2.4. In Vivo Evaluation of Microemulsions
2.4.1. Animals
2.4.2. oAβ25–35 Model
2.4.3. J20 Transgenic Model
2.4.4. Behavioral Tests
Y-Maze Test
Splash Test
O-Maze Test
2.4.5. Western Blots
2.4.6. Oxylipins Quantification
2.5. Statistical Analysis
3. Results
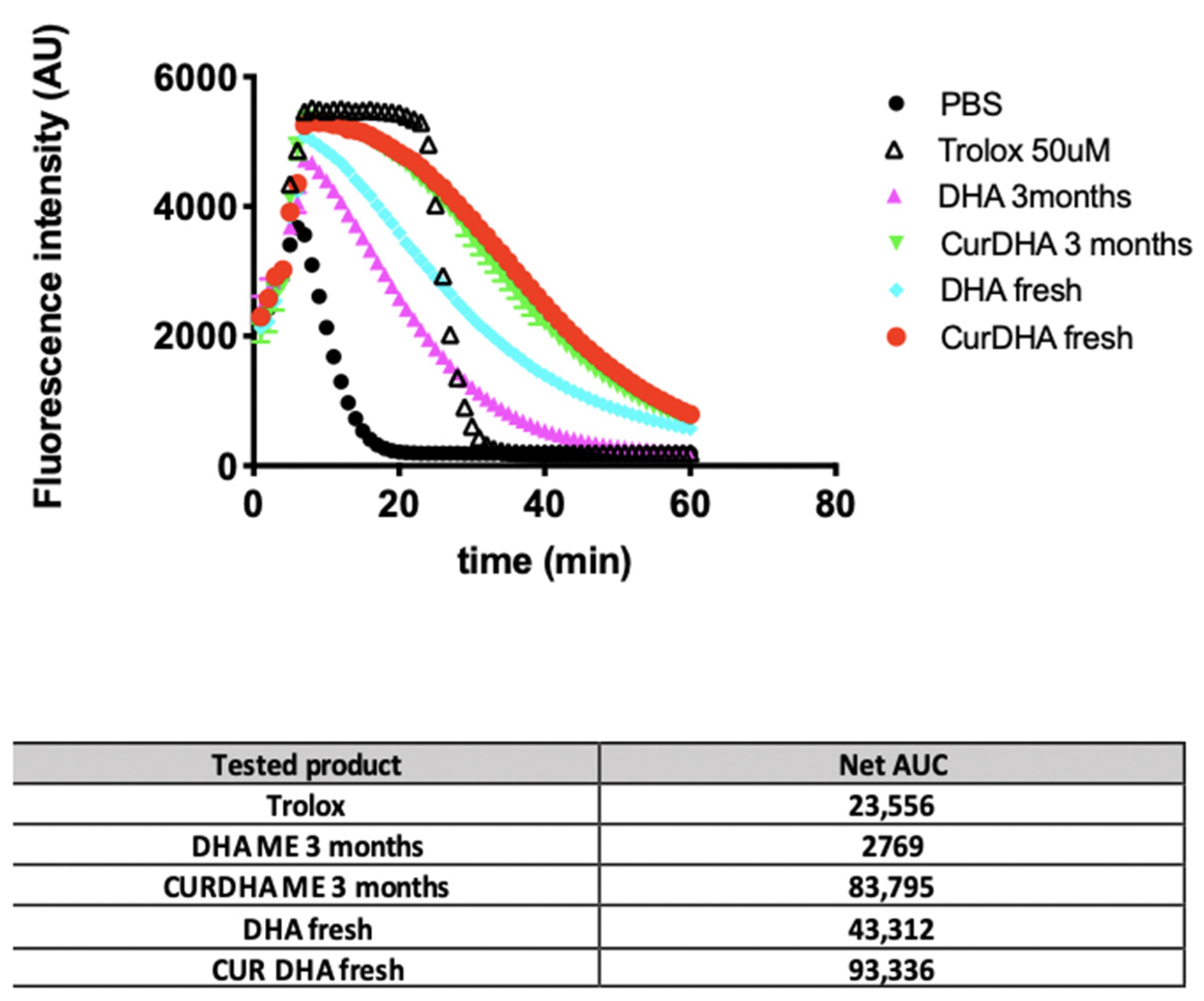
3.1. DHA Stability and Antioxidant Capacity of MEs
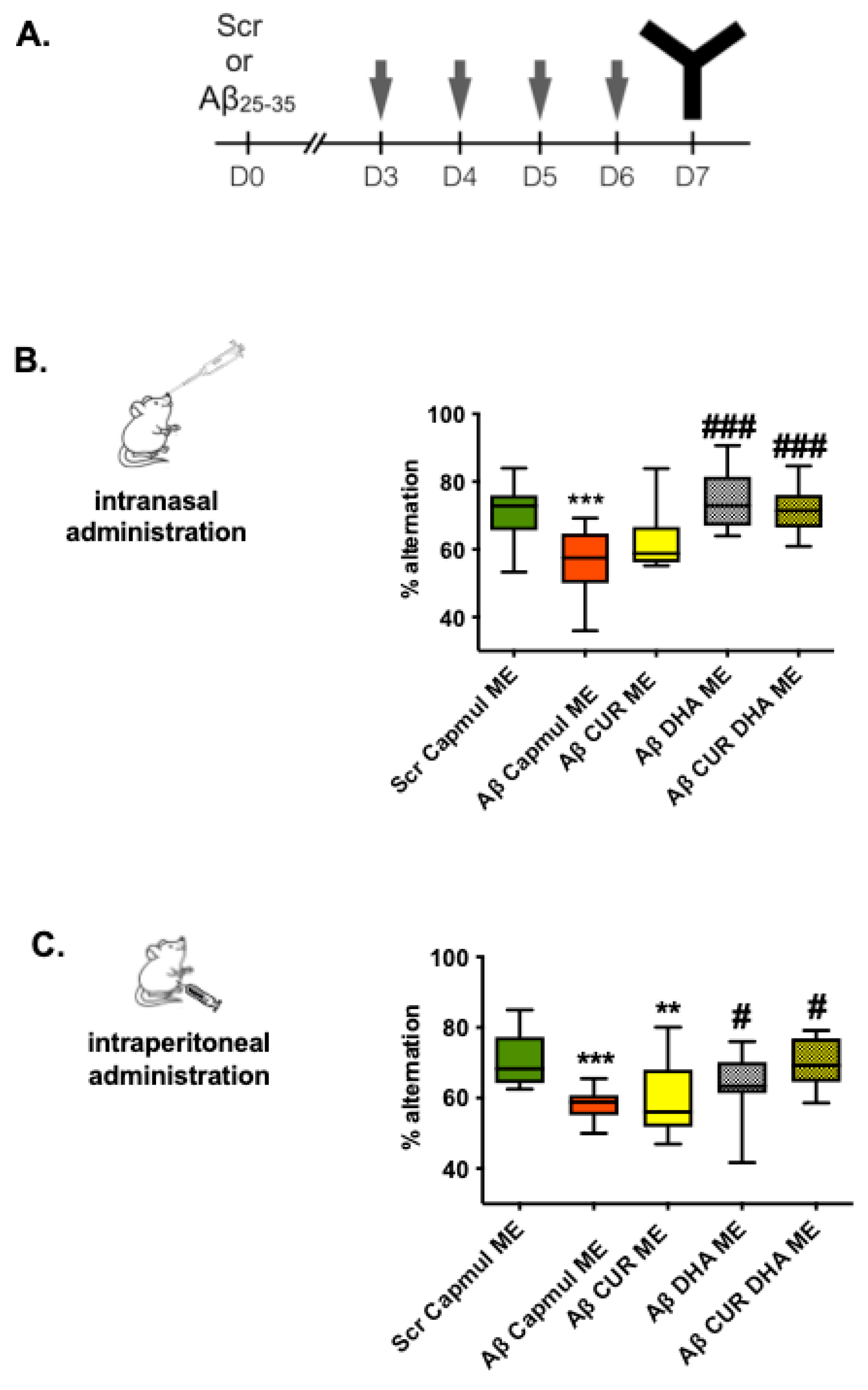
3.2. Effect of MEs on Short-Term Spatial Working Memory in the Aβ25–35 Model
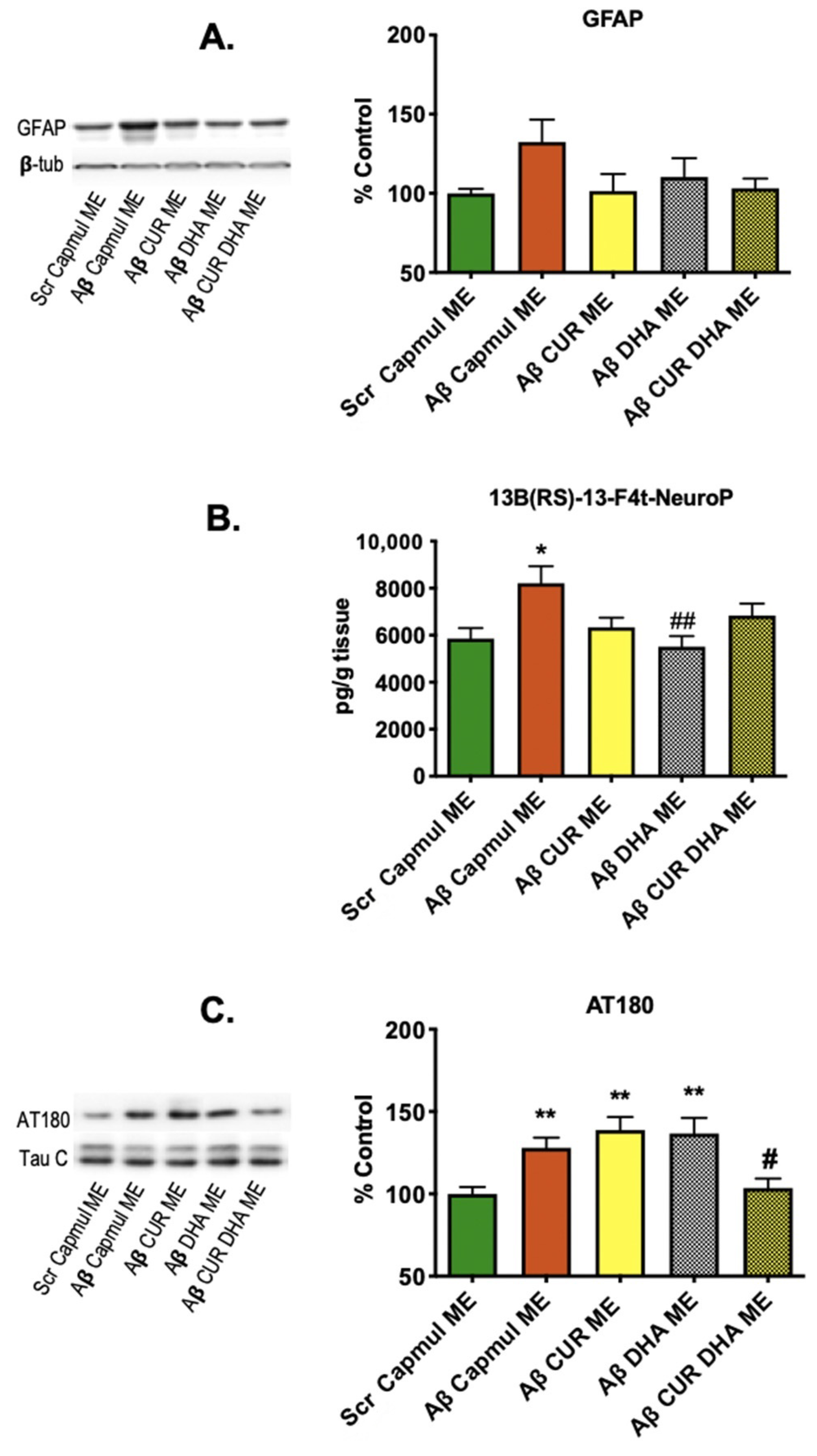
3.3. Effect of MEs on Neuroinflammation, Oxidative Stress, and Tau Phosphorylation Markers in the Aβ25–35 Model
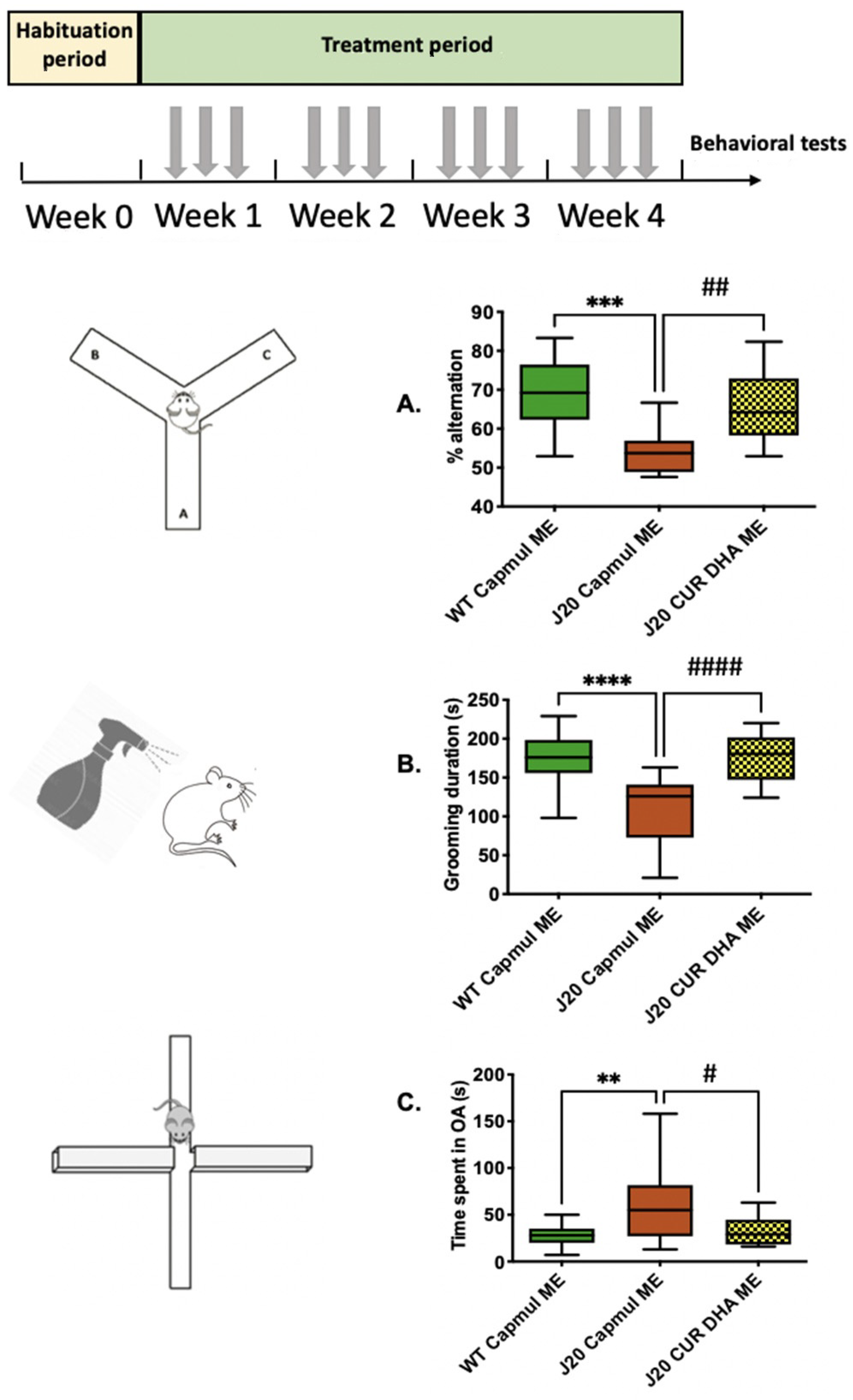
3.4. Effect of CURDHA-MEs on Cognitive Impairment in the J20 Model
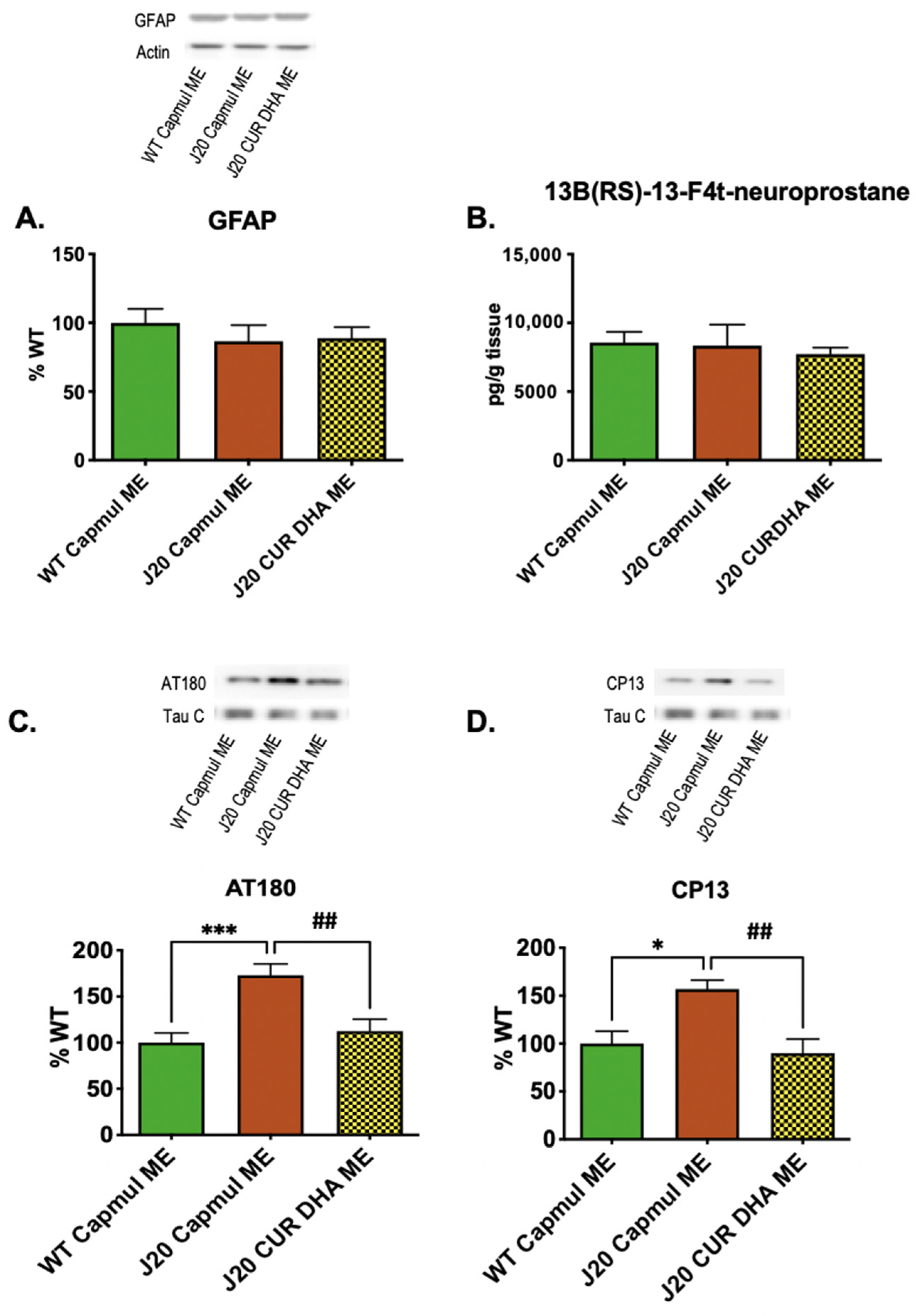
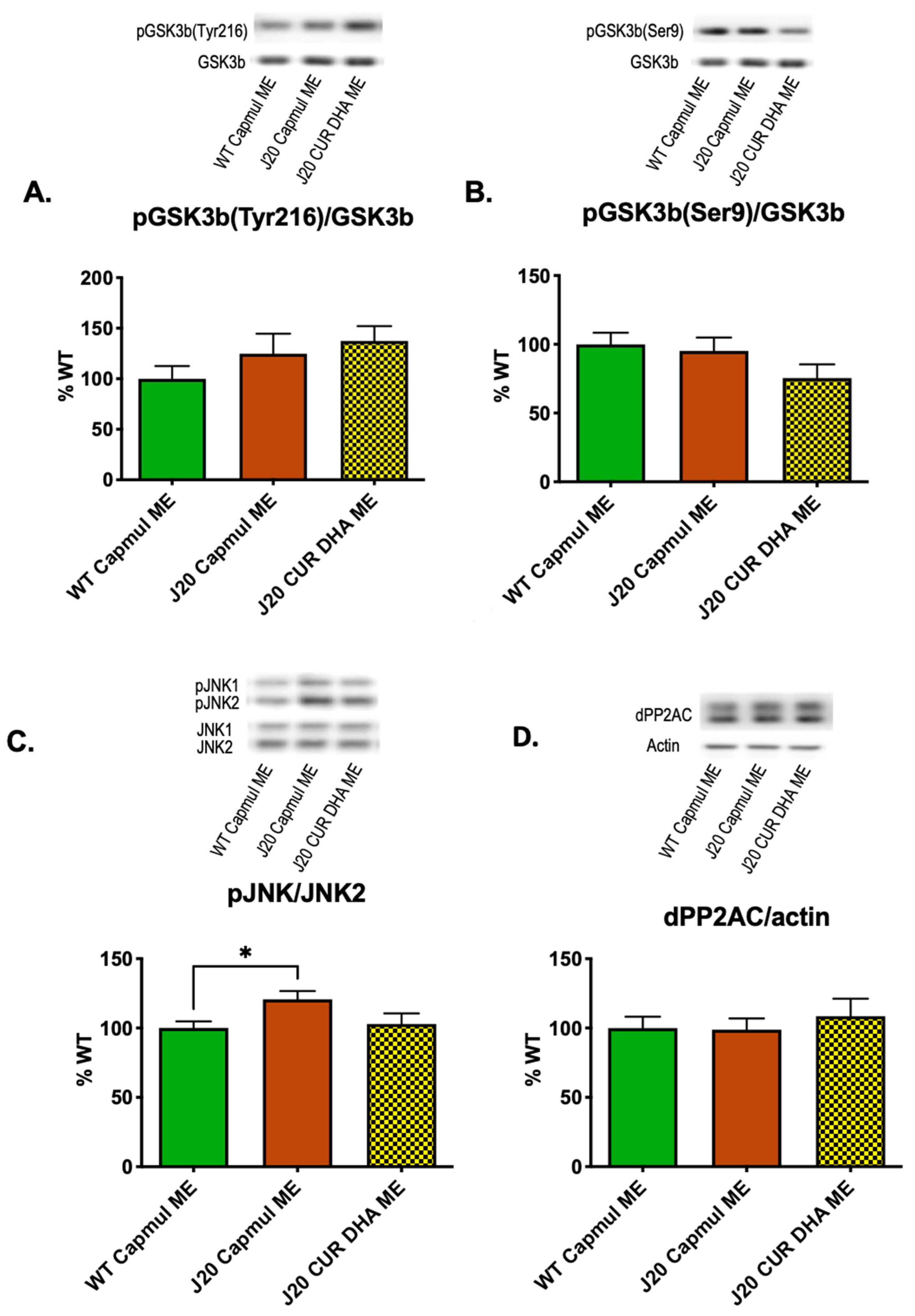
3.5. Effect of CURDHA-MEs on Neuroinflammation, Oxidative Stress, and Tau Phosphorylation Markers in the J20 Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Frisoni, G.B.; Altomare, D.; Thal, D.R.; Ribaldi, F.; van der Kant, R.; Ossenkoppele, R.; Blennow, K.; Cummings, J.; van Duijn, C.; Nilsson, P.M.; et al. The Probabilistic Model of Alzheimer Disease: The Amyloid Hypothesis Revised. Nat. Rev. Neurosci. 2022, 23, 53–66. [Google Scholar] [CrossRef]
- Maclean, C.H.; Issa, A.M.; Newberry, S.J.; Mojica, W.A.; Morton, S.C.; Garland, R.H.; Hilton, L.G.; Traina, S.B.; Shekelle, P.G. Effects of Omega-3 Fatty Acids on Cognitive Function with Aging, Dementia, and Neurological Diseases. Evid. Rep. Technol. Assess. 2005, 114, 1–3. [Google Scholar]
- Mallick, R.; Basak, S.; Duttaroy, A.K. Docosahexaenoic Acid, 22:6n-3: Its Roles in the Structure and Function of the Brain. Int. J. Dev. Neurosci. 2019, 79, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Plourde, M.; Cunnane, S.C. Extremely Limited Synthesis of Long Chain Polyunsaturates in Adults: Implications for Their Dietary Essentiality and Use as Supplements. Appl. Physiol. Nutr. Metab. 2007, 32, 619–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Schacky, C. Importance of EPA and DHA Blood Levels in Brain Structure and Function. Nutrients 2021, 13, 1074. [Google Scholar] [CrossRef]
- Cole, G.M.; Frautschy, S.A. Docosahexaenoic Acid Protects from Amyloid and Dendritic Pathology in an Alzheimer’s Disease Mouse Model. Nutr. Health 2006, 18, 249–259. [Google Scholar] [CrossRef]
- Hooijmans, C.R.; Van der Zee, C.E.E.M.; Dederen, P.J.; Brouwer, K.M.; Reijmer, Y.D.; van Groen, T.; Broersen, L.M.; Lütjohann, D.; Heerschap, A.; Kiliaan, A.J. DHA and Cholesterol Containing Diets Influence Alzheimer-like Pathology, Cognition and Cerebral Vasculature in APPswe/PS1dE9 Mice. Neurobiol. Dis. 2009, 33, 482–498. [Google Scholar] [CrossRef]
- Oster, T.; Pillot, T. Docosahexaenoic Acid and Synaptic Protection in Alzheimer’s Disease Mice. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2010, 1801, 791–798. [Google Scholar] [CrossRef]
- Teng, E.; Taylor, K.; Bilousova, T.; Weiland, D.; Pham, T.; Zuo, X.; Yang, F.; Chen, P.-P.; Glabe, C.G.; Takacs, A.; et al. Dietary DHA Supplementation in an APP/PS1 Transgenic Rat Model of AD Reduces Behavioral and Aβ Pathology and Modulates Aβ Oligomerization. Neurobiol. Dis. 2015, 82, 552–560. [Google Scholar] [CrossRef] [Green Version]
- Gillette Guyonnet, S.; Abellan Van Kan, G.; Andrieu, S.; Barberger Gateau, P.; Berr, C.; Bonnefoy, M.; Dartigues, J.F.; de Groot, L.; Ferry, M.; Galan, P.; et al. IANA Task Force on Nutrition and Cognitive Decline with Aging. J. Nutr. Health Aging 2007, 11, 132–152. [Google Scholar]
- Lagarde, M.; Bernoud, N.; Brossard, N.; Lemaitre-Delaunay, D.; Thiès, F.; Croset, M.; Lecerf, J. Lysophosphatidylcholine as a Preferred Carrier Form of Docosahexaenoic Acid to the Brain. J. Mol. Neurosci. 2001, 16, 201–204. [Google Scholar] [CrossRef]
- Lo Van, A.; Sakayori, N.; Hachem, M.; Belkouch, M.; Picq, M.; Lagarde, M.; Osumi, N.; Bernoud-Hubac, N. Mechanisms of DHA Transport to the Brain and Potential Therapy to Neurodegenerative Diseases. Biochimie 2016, 130, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Witting, L.A.; Horwitt, M.K. Effect of Degree of Fatty Acid Unsaturation in Tocopherol Deficiency-Induced Creatinuria. J. Nutr. 1964, 82, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Erdő, F.; Bors, L.A.; Farkas, D.; Bajza, Á.; Gizurarson, S. Evaluation of Intranasal Delivery Route of Drug Administration for Brain Targeting. Brain Res. Bull. 2018, 143, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Mishra, G.; Sharma, A.K.; Gothwal, A.; Kesharwani, P.; Gupta, U. Intranasal Drug Delivery: A Non-Invasive Approach for the Better Delivery of Neurotherapeutics. Pharm. Nanotechnol. 2018, 5, 203–214. [Google Scholar] [CrossRef]
- Hallschmid, M. Intranasal Insulin for Alzheimer’s Disease. CNS Drugs 2021, 35, 21–37. [Google Scholar] [CrossRef]
- Zhao, Z.Q.; Chen, B.Z.; Zhang, X.P.; Zheng, H.; Guo, X.D. An Update on the Routes for the Delivery of Donepezil. Mol. Pharm. 2021, 18, 2482–2494. [Google Scholar] [CrossRef]
- Shinde, R.L.; Bharkad, G.P.; Devarajan, P.V. Intranasal Microemulsion for Targeted Nose to Brain Delivery in Neurocysticercosis: Role of Docosahexaenoic Acid. Eur. J. Pharm. Biopharm. 2015, 96, 363–379. [Google Scholar] [CrossRef]
- Shinde, R.L.; Devarajan, P.V. Docosahexaenoic Acid–Mediated, Targeted and Sustained Brain Delivery of Curcumin Microemulsion. Drug Deliv. 2017, 24, 152–161. [Google Scholar] [CrossRef] [Green Version]
- Oger, C.; Bultel-Poncé, V.; Guy, A.; Balas, L.; Rossi, J.-C.; Durand, T.; Galano, J.-M. The Handy Use of Brown’s P2-Ni Catalyst for a Skipped Diyne Deuteration: Application to the Synthesis of a [D4]-Labeled F4t-Neuroprostane. Chem. Eur. J. 2010, 16, 13976–13980. [Google Scholar] [CrossRef]
- Galano, J.-M.; Mas, E.; Barden, A.; Mori, T.A.; Signorini, C.; De Felice, C.; Barrett, A.; Opere, C.; Pinot, E.; Schwedhelm, E.; et al. Isoprostanes and Neuroprostanes: Total Synthesis, Biological Activity and Biomarkers of Oxidative Stress in Humans. Prostaglandins Other Lipid Mediat. 2013, 107, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Acar, N.; Berdeaux, O.; Grégoire, S.; Cabaret, S.; Martine, L.; Gain, P.; Thuret, G.; Creuzot-Garcher, C.P.; Bron, A.M.; Bretillon, L. Lipid Composition of the Human Eye: Are Red Blood Cells a Good Mirror of Retinal and Optic Nerve Fatty Acids? PLoS ONE 2012, 7, e35102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acar, N.; Merle, B.M.J.; Ajana, S.; He, Z.; Grégoire, S.; Hejblum, B.P.; Martine, L.; Buaud, B.; Bron, A.M.; Creuzot-Garcher, C.P.; et al. Predicting the Retinal Content in Omega-3 Fatty Acids for Age-related Macular-degeneration. Clin. Transl. Med. 2021, 11, e404. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A Simple Method for the Isolation and Purification of Total Lipides from Animal Tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Morrison, W.R.; Smith, L.M. Preparation of fatty acid methyl esters and dimethylacetals from lipids with boron fluoride–methanol. J. Lipid Res. 1964, 5, 600–608. [Google Scholar] [CrossRef]
- Huang, D.; Ou, B.; Hampsch-Woodill, M.; Flanagan, J.A.; Prior, R.L. High-Throughput Assay of Oxygen Radical Absorbance Capacity (ORAC) Using a Multichannel Liquid Handling System Coupled with a Microplate Fluorescence Reader in 96-Well Format. J. Agric. Food Chem. 2002, 50, 4437–4444. [Google Scholar] [CrossRef]
- Zussy, C.; Brureau, A.; Keller, E.; Marchal, S.; Blayo, C.; Delair, B.; Ixart, G.; Maurice, T.; Givalois, L. Alzheimer’s Disease Related Markers, Cellular Toxicity and Behavioral Deficits Induced Six Weeks after Oligomeric Amyloid-β Peptide Injection in Rats. PLoS ONE 2013, 8, e53117. [Google Scholar] [CrossRef]
- Mansuy, M.; Baille, S.; Canet, G.; Borie, A.; Cohen-Solal, C.; Vignes, M.; Perrier, V.; Chevallier, N.; Le Guern, N.; Deckert, V.; et al. Deletion of Plasma Phospholipid Transfer Protein (PLTP) Increases Microglial Phagocytosis and Reduces Cerebral Amyloid-β Deposition in the J20 Mouse Model of Alzheimer’s Disease. Oncotarget 2018, 9, 19688–19703. [Google Scholar] [CrossRef] [Green Version]
- Franklin, K.B.J.; Paxinos, G. Paxinos and Franklin’s The Mouse Brain in Stereotaxic Coordinates, 4th ed.; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2013; ISBN 978-0-12-391057-8. [Google Scholar]
- Mucke, L.; Masliah, E.; Yu, G.-Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-Level Neuronal Expression of Aβ 1–42 in Wild-Type Human Amyloid Protein Precursor Transgenic Mice: Synaptotoxicity without Plaque Formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef] [Green Version]
- Willner, P. Chronic Mild Stress (CMS) Revisited: Consistency and Behavioural-Neurobiological Concordance in the Effects of CMS. Neuropsychobiology 2005, 52, 90–110. [Google Scholar] [CrossRef]
- Moretti, M.; Colla, A.; de Oliveira Balen, G.; dos Santos, D.B.; Budni, J.; de Freitas, A.E.; Farina, M.; Severo Rodrigues, A.L. Ascorbic Acid Treatment, Similarly to Fluoxetine, Reverses Depressive-like Behavior and Brain Oxidative Damage Induced by Chronic Unpredictable Stress. J. Psychiatr. Res. 2012, 46, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Coutellier, L.; Friedrich, A.-C.; Failing, K.; Marashi, V.; Würbel, H. Effects of Foraging Demand on Maternal Behaviour and Adult Offspring Anxiety and Stress Response in C57BL/6 Mice. Behav. Brain Res. 2009, 196, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Zussy, C.; Brureau, A.; Delair, B.; Marchal, S.; Keller, E.; Ixart, G.; Naert, G.; Meunier, J.; Chevallier, N.; Maurice, T.; et al. Time-Course and Regional Analyses of the Physiopathological Changes Induced after Cerebral Injection of an Amyloid β Fragment in Rats. Am. J. Pathol. 2011, 179, 315–334. [Google Scholar] [CrossRef]
- Dupuy, A.; Le Faouder, P.; Vigor, C.; Oger, C.; Galano, J.-M.; Dray, C.; Lee, J.C.-Y.; Valet, P.; Gladine, C.; Durand, T.; et al. Simultaneous Quantitative Profiling of 20 Isoprostanoids from Omega-3 and Omega-6 Polyunsaturated Fatty Acids by LC–MS/MS in Various Biological Samples. Anal. Chim. Acta 2016, 921, 46–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigor, C.; Oger, C.; Reversat, G.; Rocher, A.; Zhou, B.; Linares-Maurizi, A.; Guy, A.; Bultel-Poncé, V.; Galano, J.-M.; Vercauteren, J.; et al. Isoprostanoid Profiling of Marine Microalgae. Biomolecules 2020, 10, 1073. [Google Scholar] [CrossRef]
- Balachandar, R.; Soundararajan, S.; Bagepally, B.S. Docosahexaenoic Acid Supplementation in Age-Related Cognitive Decline: A Systematic Review and Meta-Analysis. Eur. J. Clin. Pharmacol. 2020, 76, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Troesch, B.; Eggersdorfer, M.; Laviano, A.; Rolland, Y.; Smith, A.D.; Warnke, I.; Weimann, A.; Calder, P.C. Expert Opinion on Benefits of Long-Chain Omega-3 Fatty Acids (DHA and EPA) in Aging and Clinical Nutrition. Nutrients 2020, 12, 2555. [Google Scholar] [CrossRef]
- Arellanes, I.C.; Choe, N.; Solomon, V.; He, X.; Kavin, B.; Martinez, A.E.; Kono, N.; Buennagel, D.P.; Hazra, N.; Kim, G.; et al. Brain Delivery of Supplemental Docosahexaenoic Acid (DHA): A Randomized Placebo-Controlled Clinical Trial. EBioMedicine 2020, 59, 102883. [Google Scholar] [CrossRef]
- He, Z.; Zeng, W.; Zhu, X.; Zhao, H.; Lu, Y.; Lu, Z. Influence of Surfactin on Physical and Oxidative Stability of Microemulsions with Docosahexaenoic Acid. Colloids Surf. Biointerf. 2017, 151, 232–239. [Google Scholar] [CrossRef]
- Shehzad, Q.; Rehman, A.; Jafari, S.M.; Zuo, M.; Khan, M.A.; Ali, A.; Khan, S.; Karim, A.; Usman, M.; Hussain, A.; et al. Improving the Oxidative Stability of Fish Oil Nanoemulsions by Co-Encapsulation with Curcumin and Resveratrol. Colloids Surf. Biointerf. 2021, 199, 111481. [Google Scholar] [CrossRef]
- Véricel, E.; Polette, A.; Bacot, S.; Calzada, C.; Lagarde, M. Pro- and Antioxidant Activities of Docosahexaenoic Acid on Human Blood Platelets: DHA and Platelet Redox Status. J. Thromb. Haemost. 2003, 1, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, J.; Qiu, J.; Li, Y.; Wang, J.; Jiao, J. Intakes of Fish and Polyunsaturated Fatty Acids and Mild-to-Severe Cognitive Impairment Risks: A Dose-Response Meta-Analysis of 21 Cohort Studies1–3. Am. J. Clin. Nutr. 2015, 103, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Han, X.; Zhang, X.; Chen, Z.; Mi, Y.; Gou, X. Dietary Fatty Acid Factors in Alzheimer’s Disease: A Review. J. Alzheimers Dis. 2020, 78, 887–904. [Google Scholar] [CrossRef]
- Burckhardt, M.; Herke, M.; Wustmann, T.; Watzke, S.; Langer, G.; Fink, A. Omega-3 Fatty Acids for the Treatment of Dementia. Cochrane Database Syst. Rev. 2016, 4, CD009002. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Khalil, H.; Nicolazzo, J. The Impact of Docosahexaenoic Acid on Alzheimer’s Disease: Is There a Role of the Blood-Brain Barrier? Curr. Clin. Pharmacol. 2015, 10, 222–241. [Google Scholar] [CrossRef]
- Ochiai, Y.; Uchida, Y.; Ohtsuki, S.; Tachikawa, M.; Aizawa, S.; Terasaki, T. The Blood-Brain Barrier Fatty Acid Transport Protein 1 (FATP1/SLC27A1) Supplies Docosahexaenoic Acid to the Brain, and Insulin Facilitates Transport. J. Neurochem. 2017, 141, 400–412. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.N.; Ma, D.; Shui, G.; Wong, P.; Cazenave-Gassiot, A.; Zhang, X.; Wenk, M.R.; Goh, E.L.K.; Silver, D.L. Mfsd2a Is a Transporter for the Essential Omega-3 Fatty Acid Docosahexaenoic Acid. Nature 2014, 509, 503–506. [Google Scholar] [CrossRef]
- Tomaszewski, N.; He, X.; Solomon, V.; Lee, M.; Mack, W.J.; Quinn, J.F.; Braskie, M.N.; Yassine, H.N. Effect of APOE Genotype on Plasma Docosahexaenoic Acid (DHA), Eicosapentaenoic Acid, Arachidonic Acid, and Hippocampal Volume in the Alzheimer’s Disease Cooperative Study-Sponsored DHA Clinical Trial. J. Alzheimers Dis. 2020, 74, 975–990. [Google Scholar] [CrossRef]
- Joumard-Cubizolles, L.; Lee, J.C.-Y.; Vigor, C.; Leung, H.H.; Bertrand-Michel, J.; Galano, J.-M.; Mazur, A.; Durand, T.; Gladine, C. Insight into the Contribution of Isoprostanoids to the Health Effects of Omega 3 PUFAs. Prostaglandins Other Lipid Mediat. 2017, 133, 111–122. [Google Scholar] [CrossRef]
- Roy, J.; Le Guennec, J.-Y.; Galano, J.-M.; Thireau, J.; Bultel-Poncé, V.; Demion, M.; Oger, C.; Lee, J.C.-Y.; Durand, T. Non-Enzymatic Cyclic Oxygenated Metabolites of Omega-3 Polyunsaturated Fatty Acid: Bioactive Drugs? Biochimie 2016, 120, 56–61. [Google Scholar] [CrossRef]
- Grimm, M.O.W.; Haupenthal, V.J.; Mett, J.; Stahlmann, C.P.; Blümel, T.; Mylonas, N.T.; Endres, K.; Grimm, H.S.; Hartmann, T. Oxidized Docosahexaenoic Acid Species and Lipid Peroxidation Products Increase Amyloidogenic Amyloid Precursor Protein Processing. Neurodegener. Dis. 2016, 16, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.A.; Devidze, N.; Halabisky, B.; Lo, I.; Thwin, M.T.; Yu, G.-Q.; Bredesen, D.E.; Masliah, E.; Mucke, L. Many Neuronal and Behavioral Impairments in Transgenic Mouse Models of Alzheimer’s Disease Are Independent of Caspase Cleavage of the Amyloid Precursor Protein. J. Neurosci. 2010, 30, 372–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quartey, M.O.; Nyarko, J.N.K.; Pennington, P.R.; Heistad, R.M.; Chaharyn, B.M.; Wei, Z.; Bainbridge, D.; Baker, G.B.; Mousseau, D.D. Age- and Sex-Dependent Profiles of APP Fragments and Key Secretases Align with Changes in Despair-like Behavior and Cognition in Young APPSwe/Ind Mice. Biochem. Biophys. Res. Commun. 2019, 511, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.D.C.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s Disease Hyperphosphorylated Tau Sequesters Normal Tau into Tangles of Filaments and Disassembles Microtubules. Nat. Med. 1996, 2, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.M.; Yee, C.; May, T.; Dhanala, A.; Mitchell, C.S. Cognitive Decline in Preclinical Alzheimer’s Disease: Amyloid-Beta versus Tauopathy. J. Alzheimers Dis. 2017, 61, 265–281. [Google Scholar] [CrossRef] [Green Version]
- Ossenkoppele, R.; Smith, R.; Mattsson-Carlgren, N.; Groot, C.; Leuzy, A.; Strandberg, O.; Palmqvist, S.; Olsson, T.; Jögi, J.; Stormrud, E.; et al. Accuracy of Tau Positron Emission Tomography as a Prognostic Marker in Preclinical and Prodromal Alzheimer Disease: A Head-to-Head Comparison Against Amyloid Positron Emission Tomography and Magnetic Resonance Imaging. JAMA Neurol. 2021, 78, 961. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Datta, D.; Del Tredici, K.; Braak, H. Hypothesis: Tau Pathology Is an Initiating Factor in Sporadic Alzheimer’s Disease. Alzheimers Dement. 2021, 17, 115–124. [Google Scholar] [CrossRef]
- Ma, Q.-L.; Yang, F.; Rosario, E.R.; Ubeda, O.J.; Beech, W.; Gant, D.J.; Chen, P.P.; Hudspeth, B.; Chen, C.; Zhao, Y.; et al. β-Amyloid Oligomers Induce Phosphorylation of Tau and Inactivation of Insulin Receptor Substrate via c-Jun N-Terminal Kinase Signaling: Suppression by Omega-3 Fatty Acids and Curcumin. J. Neurosci. 2009, 29, 9078–9089. [Google Scholar] [CrossRef]
- Luna-Muñoz, J.; García-Sierra, F.; Falcón, V.; Menéndez, I.; Chávez-Macías, L.; Mena, R. Regional Conformational Change Involving Phosphorylation of Tau Protein at the Thr231, Precedes the Structural Change Detected by Alz-50 Antibody in Alzheimer’s Disease. J. Alzheimers Dis. 2005, 8, 29–41. [Google Scholar] [CrossRef]
- Kimura, T.; Ono, T.; Takamatsu, J.; Yamamoto, H.; Ikegami, K.; Kondo, A.; Hasegawa, M.; Ihara, Y.; Miyamoto, E.; Miyakawa, T. Sequential Changes of Tau-Site-Specific Phosphorylation during Development of Paired Helical Filaments. Dement. Geriatr. Cogn. Disord. 1996, 7, 177–181. [Google Scholar] [CrossRef]
- Neddens, J.; Temmel, M.; Flunkert, S.; Kerschbaumer, B.; Hoeller, C.; Loeffler, T.; Niederkofler, V.; Daum, G.; Attems, J.; Hutter-Paier, B. Phosphorylation of Different Tau Sites during Progression of Alzheimer’s Disease. Acta Neuropathol. Commun. 2018, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- Prvulovic, D.; Hampel, H. Amyloid β (Aβ) and Phospho-Tau (p-Tau) as Diagnostic Biomarkers in Alzheimer’s Disease. Clin. Chem. Lab. Med. 2011, 49, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Su, J.H.; Cummings, B.J.; Cotman, C.W. Early Phosphorylation of Tau in Alzheimer’s Disease Occurs at Ser-202 and Is Preferentially Located within Neurites. Neuroreport 1994, 5, 2358–2362. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Qureshi, H.Y.; Lu, Y.; Paudel, H.K. Familial FTDP-17 Missense Mutations Inhibit Microtubule Assembly-Promoting Activity of Tau by Increasing Phosphorylation at Ser202 in Vitro. J. Biol. Chem. 2009, 284, 13422–13433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, H.; Hastie, C.J.; McLauchlan, H.; Cohen, P.; Goedert, M. Phosphorylation of Microtubule-Associated Protein Tau by Isoforms of c-Jun N-Terminal Kinase (JNK). J. Neurochem. 2004, 90, 352–358. [Google Scholar] [CrossRef]
- Yarza, R.; Vela, S.; Solas, M.; Ramirez, M.J. C-Jun N-Terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Front. Pharmacol. 2016, 6, 321. [Google Scholar] [CrossRef] [Green Version]
- Ploia, C.; Antoniou, X.; Sclip, A.; Grande, V.; Cardinetti, D.; Colombo, A.; Canu, N.; Benussi, L.; Ghidoni, R.; Forloni, G.; et al. JNK Plays a Key Role in Tau Hyperphosphorylation in Alzheimer’s Disease Models. J. Alzheimers Dis. 2011, 26, 315–329. [Google Scholar] [CrossRef]
- Zhu, W.; Zhao, L.; Li, T.; Xu, H.; Ding, Y.; Cui, G. Docosahexaenoic Acid Ameliorates Traumatic Brain Injury Involving JNK-Mediated Tau Phosphorylation Signaling. Neurosci. Res. 2020, 157, 44–50. [Google Scholar] [CrossRef]
- Goedert, M.; Hasegawa, M.; Jakes, R.; Lawler, S.; Cuenda, A.; Cohen, P. Phosphorylation of Microtubule-Associated Protein Tau by Stress-Activated Protein Kinases. FEBS Lett. 1997, 409, 57–62. [Google Scholar] [CrossRef]
- Reynolds, C.H.; Betts, J.C.; Blackstock, W.P.; Nebreda, A.R.; Anderton, B.H. Phosphorylation Sites on Tau Identified by Nanoelectrospray Mass Spectrometry: Differences In Vitro Between the Mitogen-Activated Protein Kinases ERK2, c-Jun N-Terminal Kinase and P38, and Glycogen Synthase Kinase-3β. J. Neurochem. 2002, 74, 1587–1595. [Google Scholar] [CrossRef]
- Shen, H.-M.; Liu, Z. JNK Signaling Pathway Is a Key Modulator in Cell Death Mediated by Reactive Oxygen and Nitrogen Species. Free Radic. Biol. Med. 2006, 40, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Iwakami, N.; Takeuchi, S.; Waragai, M.; Suzuki, M.; Kanazawa, I.; Lippa, C.F.; Ono, S.; Okazawa, H. JNK Activation Is Associated with Intracellular β-Amyloid Accumulation. Mol. Brain Res. 2000, 85, 221–233. [Google Scholar] [CrossRef]
- Zhu, X.; Raina, A.K.; Rottkamp, C.A.; Aliev, G.; Perry, G.; Boux, H.; Smith, M.A. Activation and Redistribution of C-Jun N-Terminal Kinase/Stress Activated Protein Kinase in Degenerating Neurons in Alzheimer’s Disease. J. Neurochem. 2001, 76, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Porte, B.; Marguerit, G.; Thomasseau, S.; Paquet, C.; Hugon, J. Dose-Dependent Neuroprotective Effect of the JNK Inhibitor Brimapitide in 5xFAD Transgenic Mice. Brain Res. 2020, 1727, 146587. [Google Scholar] [CrossRef] [PubMed]
- Vela, S.; Sainz, N.; Moreno-Aliaga, M.J.; Solas, M.; Ramirez, M.J. DHA Selectively Protects SAMP-8-Associated Cognitive Deficits Through Inhibition of JNK. Mol. Neurobiol. 2019, 56, 1618–1627. [Google Scholar] [CrossRef]
- Yahfoufi, N.; Alsadi, N.; Jambi, M.; Matar, C. The Immunomodulatory and Anti-Inflammatory Role of Polyphenols. Nutrients 2018, 10, 1618. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.S.; Muhammad, T.; Ikram, M.; Kim, M.O. Dietary Supplementation of the Antioxidant Curcumin Halts Systemic LPS-Induced Neuroinflammation-Associated Neurodegeneration and Memory/Synaptic Impairment via the JNK/NF- κ B/Akt Signaling Pathway in Adult Rats. Oxid. Med. Cell. Longev. 2019, 2019, 7860650. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Months After Preparation | DHA ME | CURDHA ME |
|---|---|---|
| 0 | 20.00 | 21.81 |
| 1 | 18.23 | 21.49 |
| 2 | 10.90 | 20.13 |
| 3 | 4.10 | 18.87 |
| DHA ME | CURDHA ME | |
|---|---|---|
| 15-A2t-IsoP | 1367 ± 82 | 304 ± 20 ** |
| 5(S)-5-F3t-IsoP | 1504 ± 134 | 1015 ± 189 ** |
| 4(RS)-4-F4t-NeuroP | 11,276 ± 1464 | 6986 ± 755 ** |
| 13B(RS)-13-F4t-NeuroP | 4282 ± 252 | 2658 ± 295 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zussy, C.; John, R.; Urgin, T.; Otaegui, L.; Vigor, C.; Acar, N.; Canet, G.; Vitalis, M.; Morin, F.; Planel, E.; et al. Intranasal Administration of Nanovectorized Docosahexaenoic Acid (DHA) Improves Cognitive Function in Two Complementary Mouse Models of Alzheimer’s Disease. Antioxidants 2022, 11, 838. https://doi.org/10.3390/antiox11050838
Zussy C, John R, Urgin T, Otaegui L, Vigor C, Acar N, Canet G, Vitalis M, Morin F, Planel E, et al. Intranasal Administration of Nanovectorized Docosahexaenoic Acid (DHA) Improves Cognitive Function in Two Complementary Mouse Models of Alzheimer’s Disease. Antioxidants. 2022; 11(5):838. https://doi.org/10.3390/antiox11050838
Chicago/Turabian StyleZussy, Charleine, Rijo John, Théo Urgin, Léa Otaegui, Claire Vigor, Niyazi Acar, Geoffrey Canet, Mathieu Vitalis, Françoise Morin, Emmanuel Planel, and et al. 2022. "Intranasal Administration of Nanovectorized Docosahexaenoic Acid (DHA) Improves Cognitive Function in Two Complementary Mouse Models of Alzheimer’s Disease" Antioxidants 11, no. 5: 838. https://doi.org/10.3390/antiox11050838