
Multifaceted Roles of the KEAP1–NRF2 System in Cancer and Inflammatory Disease Milieu


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction—The Physiological Roles of the KEAP1–NRF2 Axis
2. Structural Basis of KEAP1 and NRF2 Functions
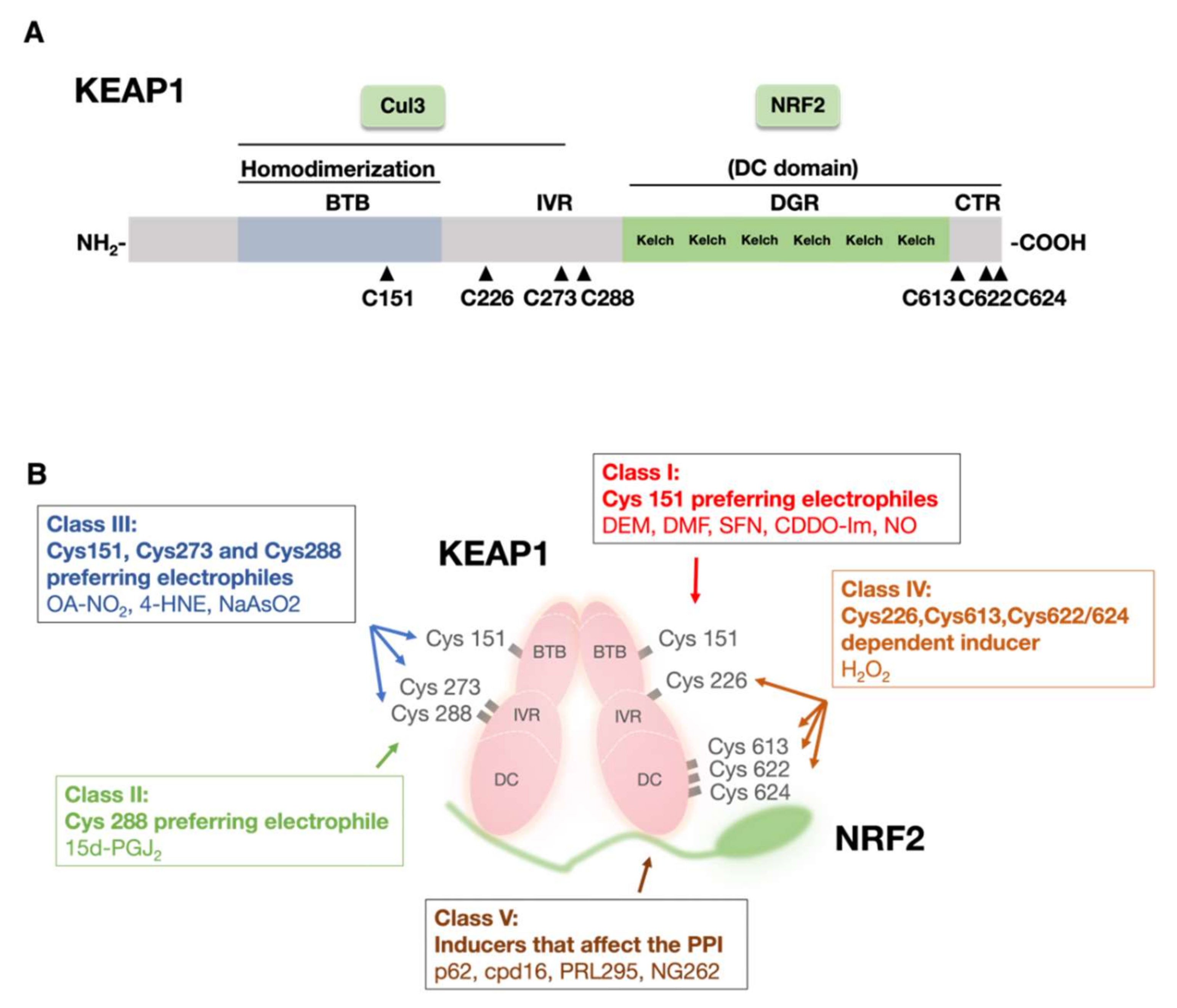
2.1. KEAP1 Senses Electrophiles and ROS
2.2. NRF2 Is Composed of Six Functional Domains
3. The KEAP1–NRF2 Axis in Cancer
3.1. NRF2 Is Constitutively Activated in NRF2-Addicted Cancer
3.2. NRF2 Plays Diverse Roles in Different Cell Fractions in the Tumor Microenvironment
3.2.1. NRF2-Null Host Cells Are Eliminated by Competitive Cell Selection during Chemical Carcinogenesis
3.2.2. NRF2 Activation in Immune Cells Leads to Tumor Suppression
3.3. NRF2 Inhibitors in the Treatment of NRF2-Addicted Cancers
3.4. NRF2 Inducers for the Treatment of NRF2-Activated Cancers
4. KEAP1–NRF2 Axis and Inflammation
4.1. NRF2 Activation Resolves Acute and Chronic Inflammation
4.2. Molecular Basis of NRF2 Anti-Inflammatory Activity
4.3. NRF2 Activation Attenuates SCD Pathology
4.4. Multifarious Roles of NRF2 in Different Types of Cells in SCD
4.5. NRF2 Inducers Ameliorate SCD Pathology
5. Concluding Remarks
Funding
Conflicts of Interest
Glossary
| AREs | Antioxidant responsive elements |
| CDDO-Im | 2-cyano-3,12 dioxooleana-1,9 diene-28-imidazolide |
| ChIP-seq | Chromatin immunoprecipitation-sequencing |
| CsMBEs | CNC–sMaf binding element |
| CUL3 | Cullin 3 |
| DDS | Drug delivery system |
| DMF | Dimethyl fumarate |
| Degron | A portion of protein important for the regulation of the protein degradation |
| EpREs | Electrophile-responsive elements |
| Hmox1 | Heme oxygenase 1 |
| KEAP1 | Kelch-like ECH-associated protein |
| LysM-Cre | Myeloid lineage specific Cre expression |
| MDSCs | Myeloid derived suppressor cells |
| Nqo1 | NADPH: quinone oxidoreductase 1 |
| NRF2 | Nuclear factor erythroid 2-like 2 |
| NSCLC | Non small cell lung cancer |
| RBC | Red blood cell |
| ROS | Reactive oxygen species |
| SCD | Sickle cell disease |
| sMAF | Small MAF |
| SFN | Sulforaphane |
| Tie1-Cre | Endothelial specific Cre expression |
References
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friling, R.S.; Bensimon, A.; Tichauer, Y.; Daniel, V. Xenobiotic-inducible expression of murine glutathione S-transferase Ya subunit gene is controlled by an electrophile-responsive element. Proc. Natl. Acad. Sci. USA 1990, 87, 6258–6262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsuki, A.; Suzuki, M.; Katsuoka, F.; Tsuchida, K.; Suda, H.; Morita, M.; Shimizu, R.; Yamamoto, M. Unique cistrome defined as CsMBE is strictly required for Nrf2-sMaf heterodimer function in cytoprotection. Free Radic. Biol. Med. 2016, 91, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Balla, G.; Vercellotti, G.M.; Muller-Eberhard, U.; Eaton, J.; Jacob, H.S. Exposure of endothelial cells to free heme potentiates damage mediated by granulocytes and toxic oxygen species. Lab. Investig. 1991, 64, 648–655. [Google Scholar]
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of novel NRF2-regulated genes by ChIP-Seq: Influence on retinoid X receptor alpha. Nucleic Acids Res. 2012, 40, 7416–7429. [Google Scholar] [CrossRef] [Green Version]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.D.; Yamamoto, M. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Motohashi, H.; Yamamoto, M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol. Sci. 2013, 34, 340–346. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Takaya, K.; Suzuki, T.; Motohashi, H.; Onodera, K.; Satomi, S.; Kensler, T.W.; Yamamoto, M. Validation of the multiple sensor mechanism of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2012, 53, 817–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Jedlicka, A.E.; Reddy, S.P.; Kensler, T.W.; Yamamoto, M.; Zhang, L.Y.; Kleeberger, S.R. Role of NRF2 in protection against hyperoxic lung injury in mice. Am. J. Respir. Cell Mol. Biol. 2002, 26, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Reddy, S.P.; Yamamoto, M.; Kleeberger, S.R. The transcription factor NRF2 protects against pulmonary fibrosis. FASEB J. 2004, 18, 1258–1260. [Google Scholar] [CrossRef] [PubMed]
- Okawa, H.; Motohashi, H.; Kobayashi, A.; Aburatani, H.; Kensler, T.W.; Yamamoto, M. Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem. Biophys. Res. Commun. 2006, 339, 79–88. [Google Scholar] [CrossRef]
- Reisman, S.A.; Buckley, D.B.; Tanaka, Y.; Klaassen, C.D. CDDO-Im protects from acetaminophen hepatotoxicity through induction of Nrf2-dependent genes. Toxicol. Appl. Pharmacol. 2009, 236, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Sussan, T.E.; Rangasamy, T.; Blake, D.J.; Malhotra, D.; El-Haddad, H.; Bedja, D.; Yates, M.S.; Kombairaju, P.; Yamamoto, M.; Liby, K.T.; et al. Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 250–255. [Google Scholar] [CrossRef] [Green Version]
- Higashi, C.; Kawaji, A.; Tsuda, N.; Hayashi, M.; Saito, R.; Yagishita, Y.; Suzuki, T.; Uruno, A.; Nakamura, M.; Nakao, K.; et al. The novel Nrf2 inducer TFM-735 ameliorates experimental autoimmune encephalomyelitis in mice. Eur. J. Pharmacol. 2017, 802, 76–84. [Google Scholar] [CrossRef]
- Ishii, Y.; Itoh, K.; Morishima, Y.; Kimura, T.; Kiwamoto, T.; Iizuka, T.; Hegab, A.E.; Hosoya, T.; Nomura, A.; Sakamoto, T.; et al. Transcription factor Nrf2 plays a pivotal role in protection against elastase-induced pulmonary inflammation and emphysema. J. Immunol. 2005, 175, 6968–6975. [Google Scholar] [CrossRef]
- Uruno, A.; Matsumaru, D.; Ryoke, R.; Saito, R.; Kadoguchi, S.; Saigusa, D.; Saito, T.; Saido, T.C.; Kawashima, R.; Yamamoto, M. Nrf2 Suppresses Oxidative Stress and Inflammation in Alzheimer’s disease model mice. Mol. Cell. Biol. 2020, 40, e00467-19. [Google Scholar] [CrossRef]
- Uruno, A.; Furusawa, Y.; Yagishita, Y.; Fukutomi, T.; Muramatsu, H.; Negishi, T.; Sugawara, A.; Kensler, T.W.; Yamamoto, M. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol. Cell. Biol. 2013, 33, 2996–3010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keleku-Lukwete, N.; Suzuki, M.; Otsuki, A.; Tsuchida, K.; Katayama, S.; Hayashi, M.; Naganuma, E.; Moriguchi, T.; Tanabe, O.; Engel, J.D.; et al. Amelioration of inflammation and tissue damage in sickle cell model mice by Nrf2 activation. Proc. Natl. Acad. Sci. USA 2015, 112, 12169–12174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, H.; Keleku-Lukwete, N.; Kuga, A.; Fuke, N.; Suganuma, H.; Suzuki, M.; Yamamoto, M. Dietary supplementation with sulforaphane attenuates liver damage and heme overload in a sickle cell disease murine model. Exp. Hematol. 2019, 77, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooi, A.; Wong, J.C.; Petillo, D.; Roossien, D.; Perrier-Trudova, V.; Whitten, D.; Min, B.W.; Tan, M.H.; Zhang, Z.; Yang, X.J.; et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 2011, 20, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Adam, J.; Hatipoglu, E.; O’Flaherty, L.; Ternette, N.; Sahgal, N.; Lockstone, H.; Baban, D.; Nye, E.; Stamp, G.W.; Wolhuter, K.; et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: Roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 2011, 20, 524–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, M.; Xiong, Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell. Biol. 2005, 25, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [Green Version]
- Hong, F.; Freeman, M.L.; Liebler, D.C. Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chem. Res. Toxicol. 2005, 18, 1917–1926. [Google Scholar] [CrossRef]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fourquet, S.; Guerois, R.; Biard, D.; Toledano, M.B. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J. Biol. Chem. 2010, 285, 8463–8471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, M.; Li, L.; Iwamoto, N.; Nakajima-Takagi, Y.; Kaneko, H.; Nakayama, Y.; Eguchi, M.; Wada, Y.; Kumagai, Y.; Yamamoto, M. The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol. Cell. Biol. 2009, 29, 493–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, M.; Lamont, D.J.; Beattie, K.A.; Hayes, J.D. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc. Natl. Acad. Sci. USA 2010, 107, 18838–18843. [Google Scholar] [CrossRef] [Green Version]
- Saito, R.; Suzuki, T.; Hiramoto, K.; Asami, S.; Naganuma, E.; Suda, H.; Iso, T.; Yamamoto, H.; Morita, M.; Baird, L.; et al. Characterizations of Three Major Cysteine Sensors of Keap1 in Stress Response. Mol. Cell. Biol. 2016, 36, 271–284. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Maher, J.; Yamamoto, M. Select heterozygous Keap1 mutations have a dominant-negative effect on wild-type Keap1 in vivo. Cancer Res. 2011, 71, 1700–1709. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Suzuki, T.; Kobayashi, A.; Wakabayashi, J.; Maher, J.; Motohashi, H.; Yamamoto, M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol. Cell. Biol. 2008, 28, 2758–2770. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Muramatsu, A.; Saito, R.; Iso, T.; Shibata, T.; Kuwata, K.; Kawaguchi, S.I.; Iwawaki, T.; Adachi, S.; Suda, H.; et al. Molecular Mechanism of Cellular Oxidative Stress Sensing by Keap1. Cell Rep. 2019, 28, 746–758.e4. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Lazzara, P.R.; David, B.P.; Ankireddy, A.; Richardson, B.G.; Dye, K.; Ratia, K.M.; Reddy, S.P.; Moore, T.W. Isoquinoline Kelch-like ECH-Associated Protein 1-Nuclear Factor (Erythroid-Derived 2)-like 2 (KEAP1-NRF2) Inhibitors with High Metabolic Stability. J. Med. Chem. 2020, 63, 6547–6560. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Lu, M.C.; Xu, L.L.; Yang, T.T.; Xi, M.Y.; Xu, X.L.; Guo, X.K.; Zhang, X.J.; You, Q.D.; Sun, H.P. Discovery of potent Keap1-Nrf2 protein-protein interaction inhibitor based on molecular binding determinants analysis. J. Med. Chem. 2014, 57, 2736–2745. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell. Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Sekine, H.; Okazaki, K.; Ota, N.; Shima, H.; Katoh, Y.; Suzuki, N.; Igarashi, K.; Ito, M.; Motohashi, H.; Yamamoto, M. The Mediator Subunit MED16 Transduces NRF2-Activating Signals into Antioxidant Gene Expression. Mol. Cell. Biol. 2016, 36, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Tong, K.I.; Padmanabhan, B.; Kobayashi, A.; Shang, C.; Hirotsu, Y.; Yokoyama, S.; Yamamoto, M. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol. Cell. Biol. 2007, 27, 7511–7521. [Google Scholar] [CrossRef] [Green Version]
- Fukutomi, T.; Takagi, K.; Mizushima, T.; Ohuchi, N.; Yamamoto, M. Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex degron and Keap1. Mol. Cell. Biol. 2014, 34, 832–846. [Google Scholar] [CrossRef] [Green Version]
- Horie, Y.; Suzuki, T.; Inoue, J.; Iso, T.; Wells, G.; Moore, T.W.; Mizushima, T.; Dinkova-Kostova, A.T.; Kasai, T.; Kamei, T.; et al. Molecular basis for the disruption of Keap1-Nrf2 interaction via Hinge & Latch mechanism. Commun. Biol. 2021, 4, 576. [Google Scholar] [CrossRef]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef] [Green Version]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobón-Velasco, J.C.; Devijver, H.; García-Mayoral, M.F.; Van Leuven, F.; et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/β-TrCP axis. Mol. Cell. Biol. 2012, 32, 3486–3499. [Google Scholar] [CrossRef] [Green Version]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.H.; Cha, Y.N.; Surh, Y.J. Peroxynitrite induces HO-1 expression via PI3K/Akt-dependent activation of NF-E2-related factor 2 in PC12 cells. Free Radic. Biol. Med. 2006, 41, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Hayes, J.D.; Wolf, C.R. Generation of a stable antioxidant response element-driven reporter gene cell line and its use to show redox-dependent activation of nrf2 by cancer chemotherapeutic agents. Cancer Res. 2006, 66, 10983–10994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padmanabhan, B.; Tong, K.I.; Ohta, T.; Nakamura, Y.; Scharlock, M.; Ohtsuji, M.; Kang, M.I.; Kobayashi, A.; Yokoyama, S.; Yamamoto, M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol. Cell 2006, 21, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Shibata, T.; Ohta, T.; Tong, K.I.; Kokubu, A.; Odogawa, R.; Tsuta, K.; Asamura, H.; Yamamoto, M.; Hirohashi, S. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc. Natl. Acad. Sci. USA 2008, 105, 13568–13573. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Lee, H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Baylin, S.B.; Sidransky, D.; Gabrielson, E.; et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006, 3, e420. [Google Scholar] [CrossRef] [Green Version]
- Nioi, P.; Nguyen, T. A mutation of Keap1 found in breast cancer impairs its ability to repress Nrf2 activity. Biochem. Biophys. Res. Commun. 2007, 362, 816–821. [Google Scholar] [CrossRef]
- Shibata, T.; Kokubu, A.; Gotoh, M.; Ojima, H.; Ohta, T.; Yamamoto, M.; Hirohashi, S. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology 2008, 135, 1358–1368. [Google Scholar] [CrossRef]
- Kim, Y.R.; Oh, J.E.; Kim, M.S.; Kang, M.R.; Park, S.W.; Han, J.Y.; Eom, H.S.; Yoo, N.J.; Lee, S.H. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J. Pathol. 2010, 220, 446–451. [Google Scholar] [CrossRef]
- Martinez, V.D.; Vucic, E.A.; Thu, K.L.; Pikor, L.A.; Lam, S.; Lam, W.L. Disruption of KEAP1/CUL3/RBX1 E3-ubiquitin ligase complex components by multiple genetic mechanisms: Association with poor prognosis in head and neck cancer. Head Neck 2015, 37, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Ooi, A.; Dykema, K.; Ansari, A.; Petillo, D.; Snider, J.; Kahnoski, R.; Anema, J.; Craig, D.; Carpten, J.; Teh, B.T.; et al. CUL3 and NRF2 mutations confer an NRF2 activation phenotype in a sporadic form of papillary renal cell carcinoma. Cancer Res. 2013, 73, 2044–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
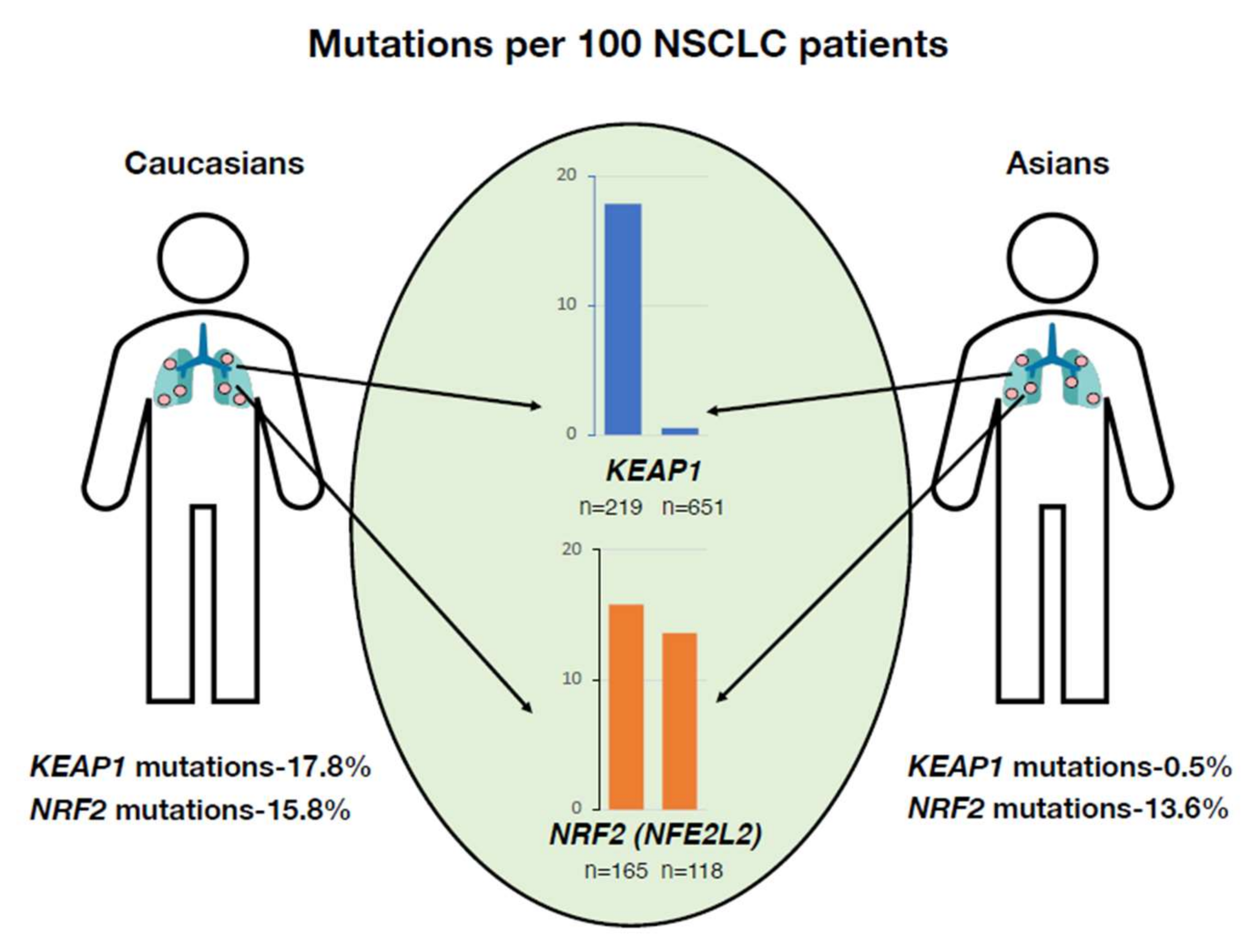
- Izumi, M.; Suzumura, T.; Ogawa, K.; Matsumoto, Y.; Sawa, K.; Yoshimoto, N.; Tani, Y.; Watanabe, T.; Kaneda, H.; Mitsuoka, S.; et al. Differences in molecular epidemiology of lung cancer among ethnicities (Asian vs. Caucasian). J. Thorac. Dis. 2020, 12, 3776–3784. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R.; et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 2004, 91, 355–358. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Leiserson, M.D.; Vandin, F.; Wu, H.T.; Dobson, J.R.; Eldridge, J.V.; Thomas, J.L.; Papoutsaki, A.; Kim, Y.; Niu, B.; McLellan, M.; et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet. 2015, 47, 106–114. [Google Scholar] [CrossRef]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef]
- Muscarella, L.A.; Barbano, R.; D’Angelo, V.; Copetti, M.; Coco, M.; Balsamo, T.; La Torre, A.; Notarangelo, A.; Troiano, M.; Parisi, S.; et al. Regulation of KEAP1 expression by promoter methylation in malignant gliomas and association with patient’s outcome. Epigenetics 2011, 6, 317–325. [Google Scholar] [CrossRef] [Green Version]
- Muscarella, L.A.; Parrella, P.; D’Alessandro, V.; la Torre, A.; Barbano, R.; Fontana, A.; Tancredi, A.; Guarnieri, V.; Balsamo, T.; Coco, M.; et al. Frequent epigenetics inactivation of KEAP1 gene in non-small cell lung cancer. Epigenetics 2011, 6, 710–719. [Google Scholar] [CrossRef] [Green Version]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Nakamura, R.M.; Dent, E.D.; Zhang, J.Y.; Nielsen, F.C.; Christiansen, J.; Chan, E.K.; Tan, E.M. Aberrant expression of fetal RNA-binding protein p62 in liver cancer and liver cirrhosis. Am. J. Pathol. 2001, 159, 945–953. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, I.P.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002, 30, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.D.; Lee, J.; Gnad, F.; Klijn, C.; Schaub, A.; Reeder, J.; Daemen, A.; Bakalarski, C.E.; Holcomb, T.; Shames, D.S.; et al. Recurrent Loss of NFE2L2 Exon 2 Is a Mechanism for Nrf2 Pathway Activation in Human Cancers. Cell Rep. 2016, 16, 2605–2617. [Google Scholar] [CrossRef] [Green Version]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [Green Version]
- DeNicola, G.M.; Chen, P.H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E.; et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 2015, 47, 1475–1481. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, M.; Taguchi, K.; Hirose, W.; Tsuchida, K.; Suzuki, M.; Taniyama, Y.; Kamei, T.; Yamamoto, M. Cellular Nrf2 Levels Determine Cell Fate during Chemical Carcinogenesis in Esophageal Epithelium. Mol. Cell. Biol. 2021, 41, e00536-20. [Google Scholar] [CrossRef]
- Satoh, H.; Moriguchi, T.; Taguchi, K.; Takai, J.; Maher, J.M.; Suzuki, T.; Winnard, P.T.; Raman, V.; Ebina, M.; Nukiwa, T.; et al. Nrf2-deficiency creates a responsive microenvironment for metastasis to the lung. Carcinogenesis 2010, 31, 1833–1843. [Google Scholar] [CrossRef] [Green Version]
- Hiramoto, K.; Satoh, H.; Suzuki, T.; Moriguchi, T.; Pi, J.; Shimosegawa, T.; Yamamoto, M. Myeloid lineage-specific deletion of antioxidant system enhances tumor metastasis. Cancer Prev. Res. 2014, 7, 835–844. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, M.; Kuga, A.; Suzuki, M.; Panda, H.; Kitamura, H.; Motohashi, H.; Yamamoto, M. Microenvironmental Activation of Nrf2 Restricts the Progression of Nrf2-Activated Malignant Tumors. Cancer Res. 2020, 80, 3331–3344. [Google Scholar] [CrossRef]
- Tesi, R.J. MDSC; the Most Important Cell You Have Never Heard of. Trends Pharmacol. Sci. 2019, 40, 4–7. [Google Scholar] [CrossRef]
- Wang, P.F.; Song, S.Y.; Wang, T.J.; Ji, W.J.; Li, S.W.; Liu, N.; Yan, C.X. Prognostic role of pretreatment circulating MDSCs in patients with solid malignancies: A meta-analysis of 40 studies. Oncoimmunology 2018, 7, e1494113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [Green Version]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, K.; Tsujita, T.; Hayashi, M.; Ojima, A.; Keleku-Lukwete, N.; Katsuoka, F.; Otsuki, A.; Kikuchi, H.; Oshima, Y.; Suzuki, M.; et al. Halofuginone enhances the chemo-sensitivity of cancer cells by suppressing NRF2 accumulation. Free Radic. Biol. Med. 2017, 103, 236–247. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. [Google Scholar] [CrossRef]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Kemper, E.K.; Suciu, R.M.; Vinogradova, E.V.; Backus, K.M.; Horning, B.D.; Paul, T.A.; Ichu, T.A.; Svensson, R.U.; Olucha, J.; et al. Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell 2017, 171, 696–709. [Google Scholar] [CrossRef] [Green Version]
- Baird, L.; Suzuki, T.; Takahashi, Y.; Hishinuma, E.; Saigusa, D.; Yamamoto, M. Geldanamycin-Derived HSP90 Inhibitors Are Synthetic Lethal with NRF2. Mol. Cell. Biol. 2020, 40, e00377-20. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Yamamoto, M. NRF2-Dependent Bioactivation of Mitomycin C as a Novel Strategy To Target KEAP1-NRF2 Pathway Activation in Human Cancer. Mol. Cell. Biol. 2021, 41, e00473-20. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Scollick, C.; Traore, K.; Yates, M.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem. Biophys. Res. Commun. 2006, 351, 883–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Mochizuki, M.; Ishii, Y.; Ishii, T.; Shibata, T.; Kawamoto, Y.; Kelly, V.; Sekizawa, K.; Uchida, K.; Yamamoto, M. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Delta(12,14)-prostaglandin j(2). Mol. Cell. Biol. 2004, 24, 36–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef] [Green Version]
- INGRAM, V.M. Gene mutations in human haemoglobin: The chemical difference between normal and sickle cell haemoglobin. Nature 1957, 180, 326–328. [Google Scholar] [CrossRef]
- Bunn, H.F. Pathogenesis and treatment of sickle cell disease. N. Engl. J. Med. 1997, 337, 762–769. [Google Scholar] [CrossRef]
- Li, C.; Wright, M.M.; Jackson, R.M. Reactive species mediated injury of human lung epithelial cells after hypoxia-reoxygenation. Exp. Lung Res. 2002, 28, 373–389. [Google Scholar] [CrossRef]
- Camus, S.M.; De Moraes, J.A.; Bonnin, P.; Abbyad, P.; Le Jeune, S.; Lionnet, F.; Loufrani, L.; Grimaud, L.; Lambry, J.C.; Charue, D.; et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood 2015, 125, 3805–3814. [Google Scholar] [CrossRef] [Green Version]
- Repka, T.; Hebbel, R.P. Hydroxyl radical formation by sickle erythrocyte membranes: Role of pathologic iron deposits and cytoplasmic reducing agents. Blood 1991, 78, 2753–2758. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.M.; Ciavatta, D.J.; Townes, T.M. Knockout-transgenic mouse model of sickle cell disease. Science 1997, 278, 873–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Xi, C.; Thomas, B.; Pace, B.S. Loss of NRF2 function exacerbates the pathophysiology of sickle cell disease in a transgenic mouse model. Blood 2018, 131, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Keleku-Lukwete, N.; Suzuki, M.; Panda, H.; Otsuki, A.; Katsuoka, F.; Saito, R.; Saigusa, D.; Uruno, A.; Yamamoto, M. Nrf2 activation in myeloid cells and endothelial cells differentially mitigates sickle cell disease pathology in mice. Blood Adv. 2019, 3, 1285–1297. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Ihunnah, C.A.; Hazra, R.; Walker, A.L.; Hansen, J.M.; Archer, D.R.; Owusu-Ansah, A.T.; Ofori-Acquah, S.F. Nonhematopoietic Nrf2 dominantly impedes adult progression of sickle cell anemia in mice. JCI Insight 2016, 1, e81090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Zhang, P.; Abdulla, F.; Nguyen, P.; Killeen, T.; Xu, P.; O’Sullivan, G.; Nath, K.A.; et al. Control of Oxidative Stress and Inflammation in Sickle Cell Disease with the Nrf2 Activator Dimethyl Fumarate. Antioxid. Redox Signal. 2017, 26, 748–762. [Google Scholar] [CrossRef]
- Doss, J.F.; Jonassaint, J.C.; Garrett, M.E.; Ashley-Koch, A.E.; Telen, M.J.; Chi, J.T. Phase 1 Study of a Sulforaphane-Containing Broccoli Sprout Homogenate for Sickle Cell Disease. PLoS ONE 2016, 11, e0152895. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panda, H.; Wen, H.; Suzuki, M.; Yamamoto, M. Multifaceted Roles of the KEAP1–NRF2 System in Cancer and Inflammatory Disease Milieu. Antioxidants 2022, 11, 538. https://doi.org/10.3390/antiox11030538
Panda H, Wen H, Suzuki M, Yamamoto M. Multifaceted Roles of the KEAP1–NRF2 System in Cancer and Inflammatory Disease Milieu. Antioxidants. 2022; 11(3):538. https://doi.org/10.3390/antiox11030538
Chicago/Turabian StylePanda, Harit, Huaichun Wen, Mikiko Suzuki, and Masayuki Yamamoto. 2022. "Multifaceted Roles of the KEAP1–NRF2 System in Cancer and Inflammatory Disease Milieu" Antioxidants 11, no. 3: 538. https://doi.org/10.3390/antiox11030538