
Apoptosis-Inducing Factor Deficiency Induces Tissue-Specific Alterations in Autophagy: Insights from a Preclinical Model of Mitochondrial Disease and Exercise Training Effects


 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Model
2.2. Study Design
2.2.1. Time Course Study
2.2.2. Exercise Training Study
2.3. Cardiomyocyte Cell Culture
2.4. Assessment of Autophagy Flux
2.5. Tissue Processing
2.6. Western Blotting
2.7. Immunocytochemistry
2.8. High Performance Liquid Chromatography
2.9. Statistical Analysis
3. Results
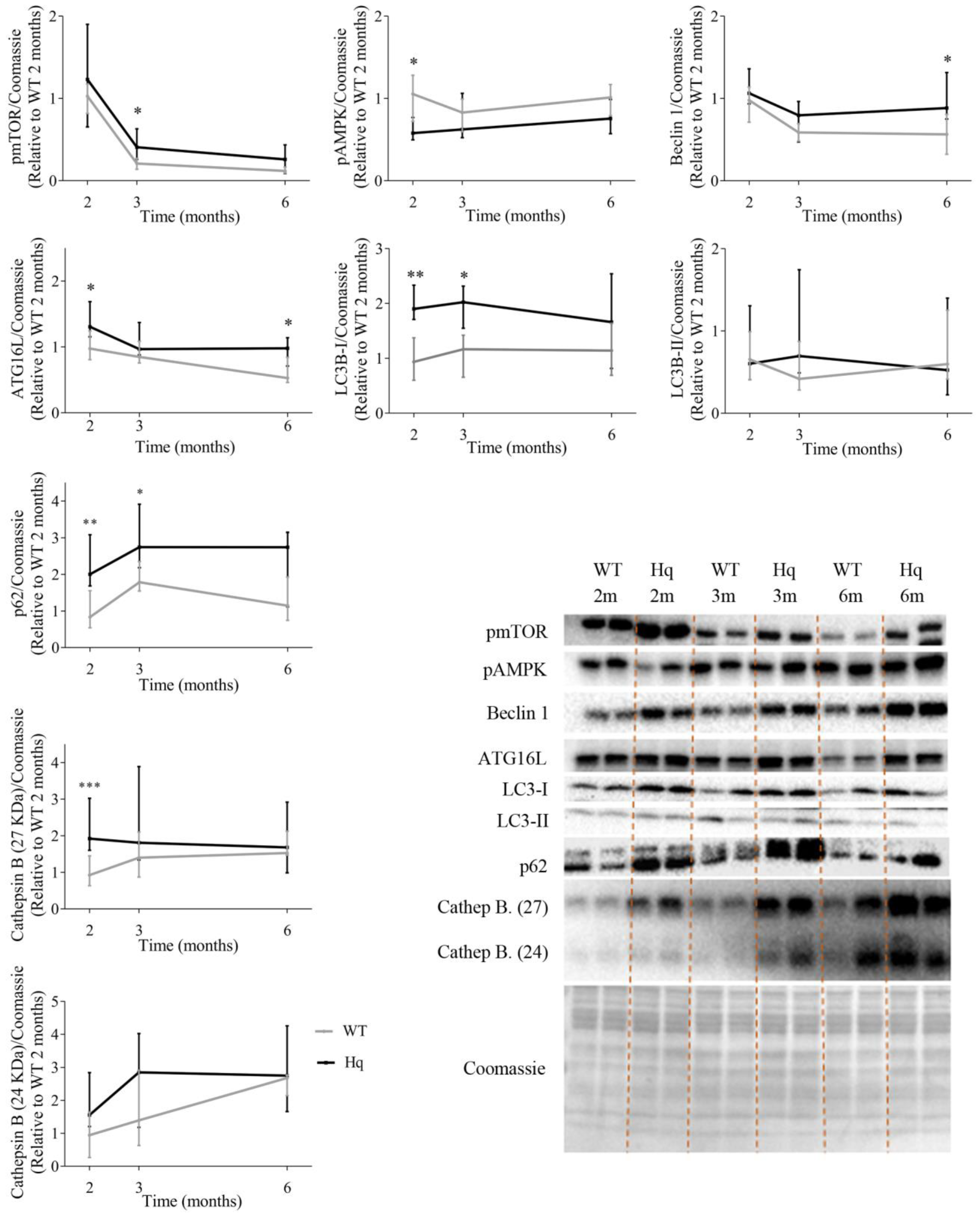
3.1. Autophagy in the Heart: Time Course Study
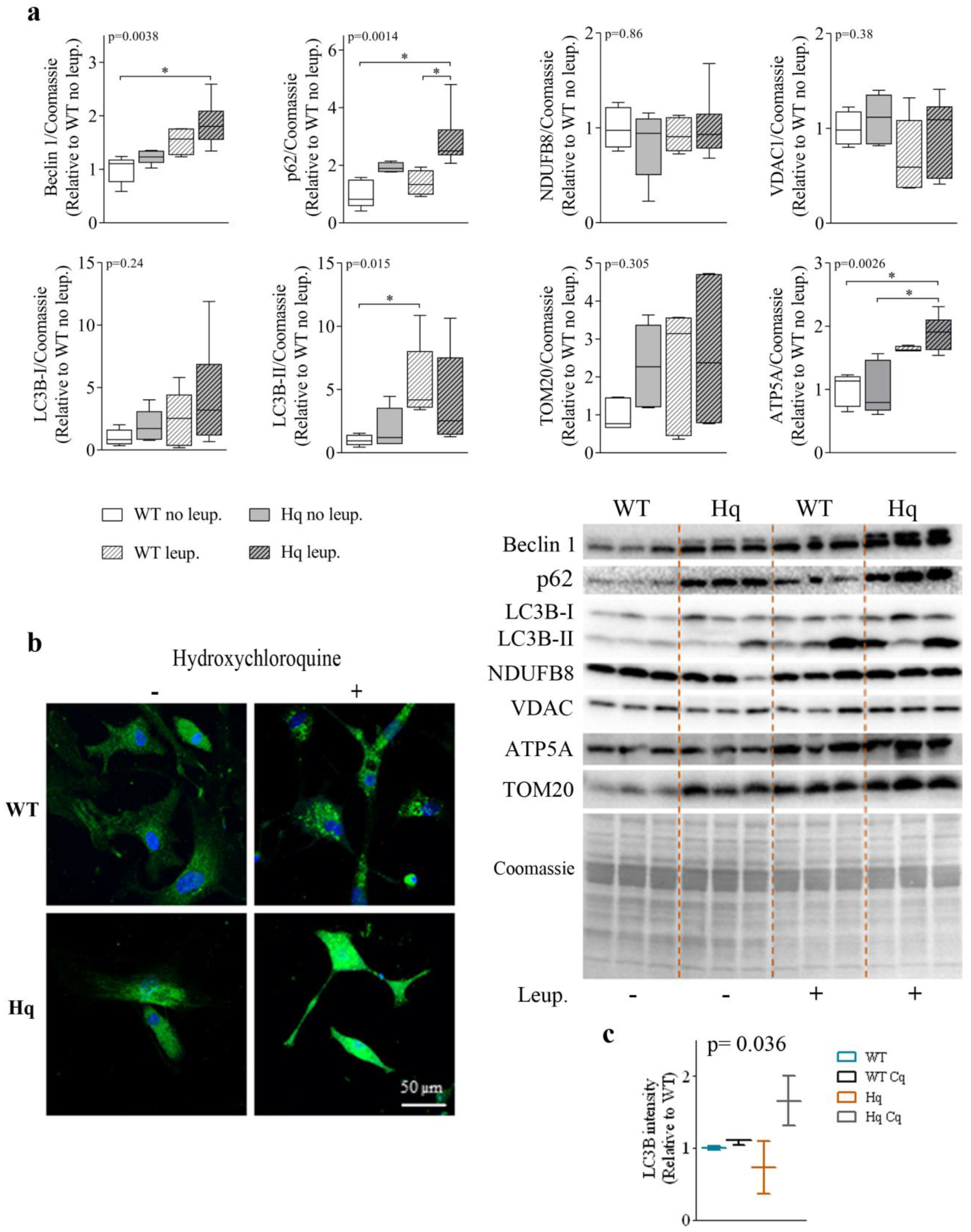
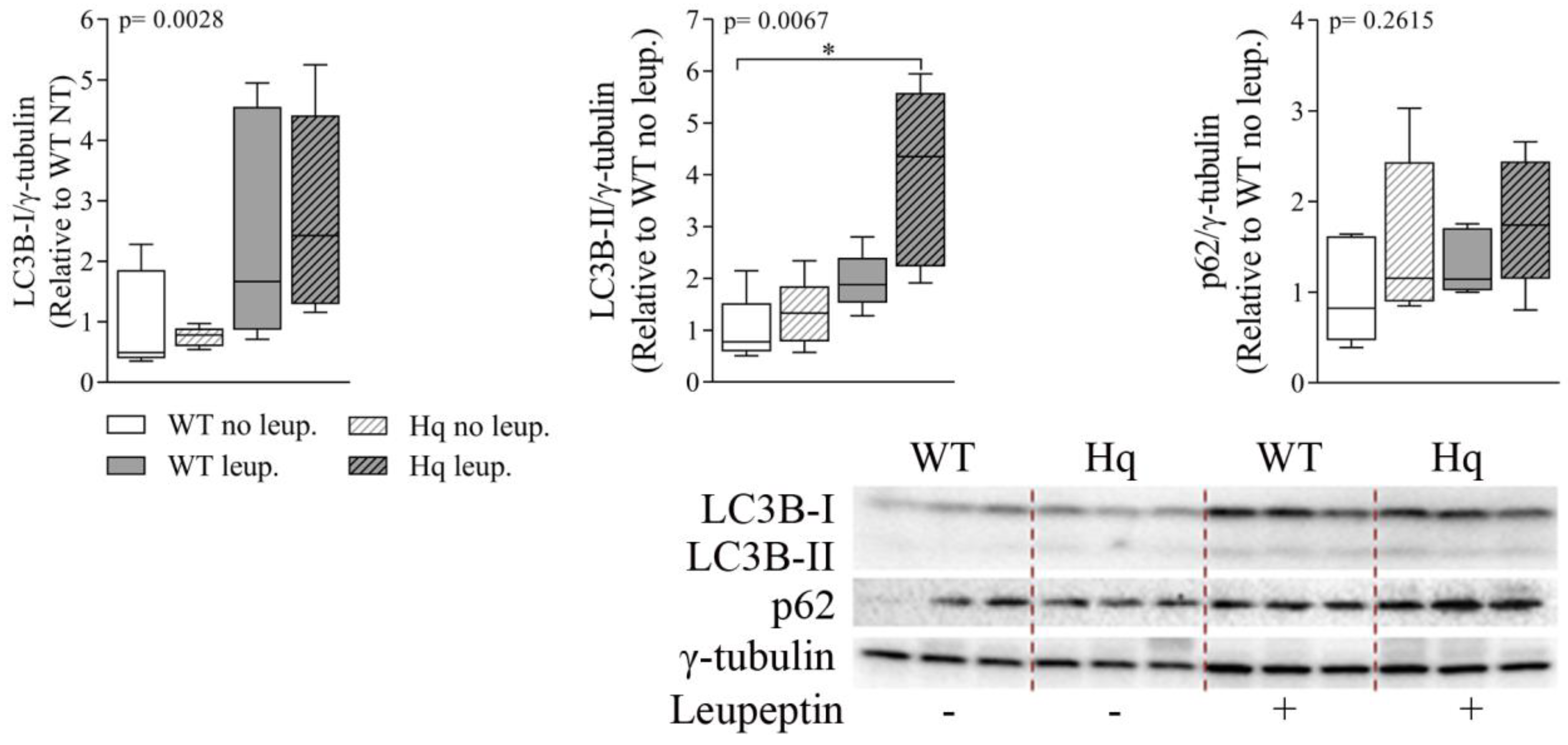
3.2. Autophagic Flux Analysis in the Heart
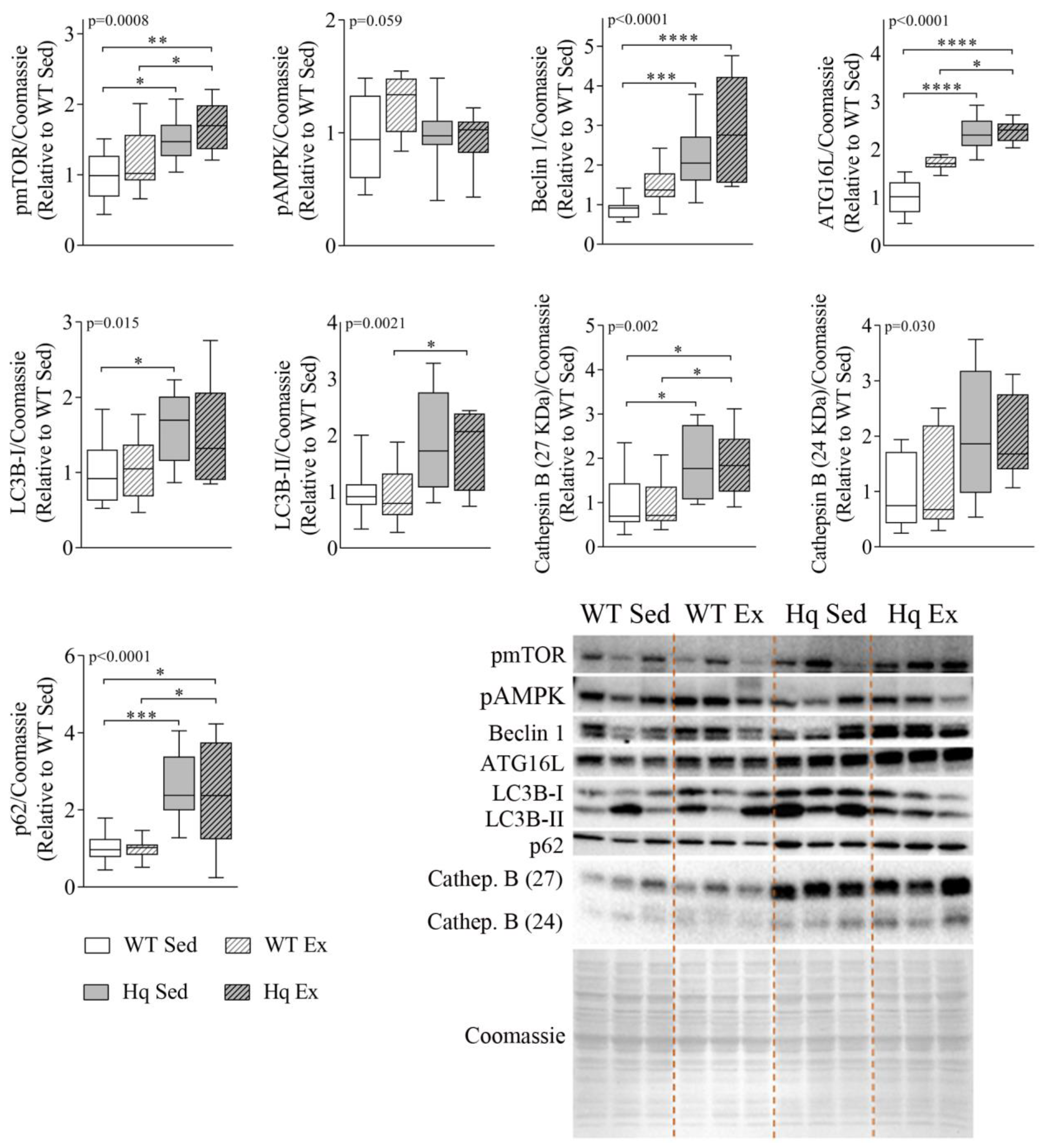
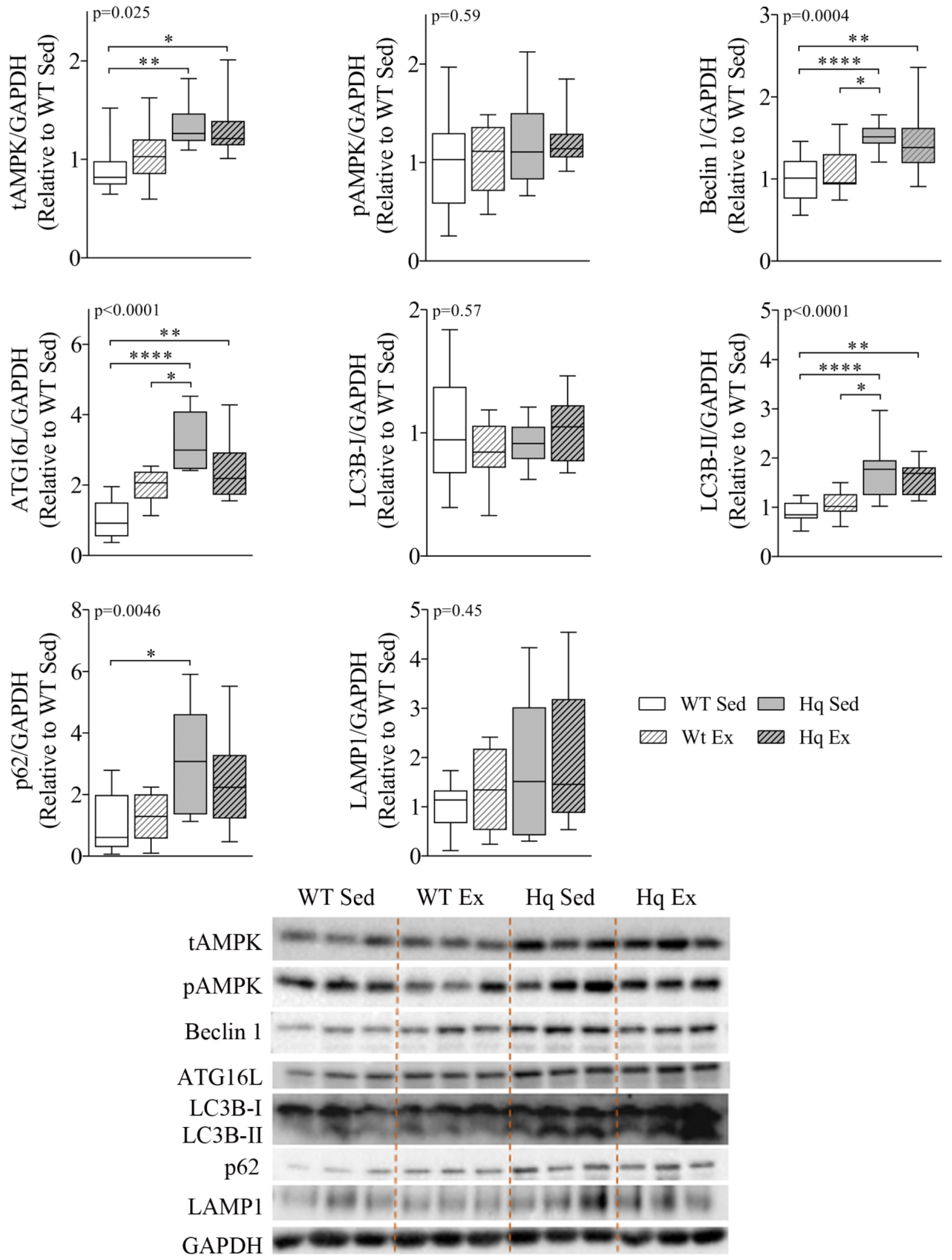
3.3. Effects of Exercise Training on Autophagy in the Heart
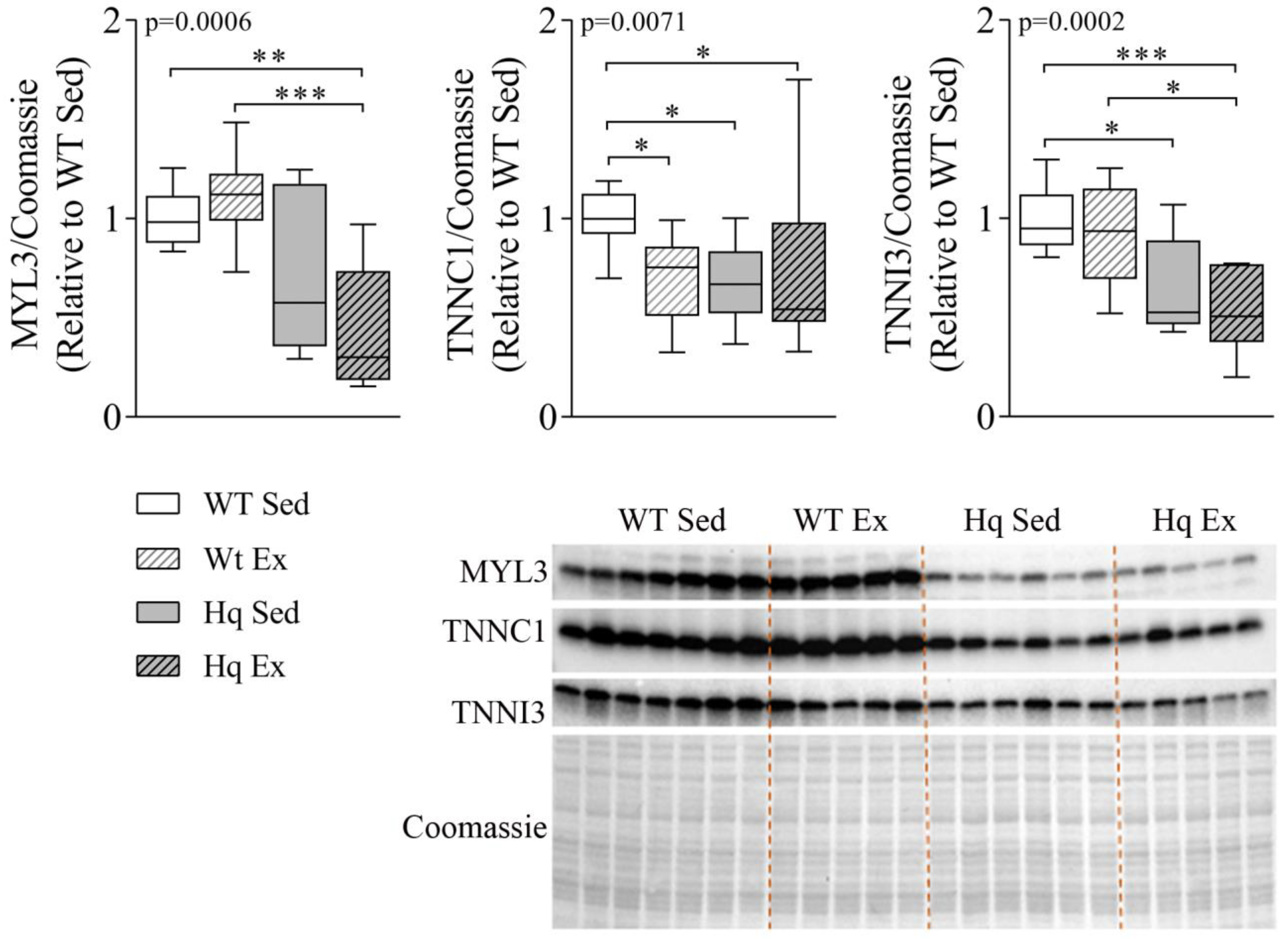
3.4. Sarcomere Proteins Levels in the Heart
3.5. Free Amino Acid and Ammonium Levels in the Heart
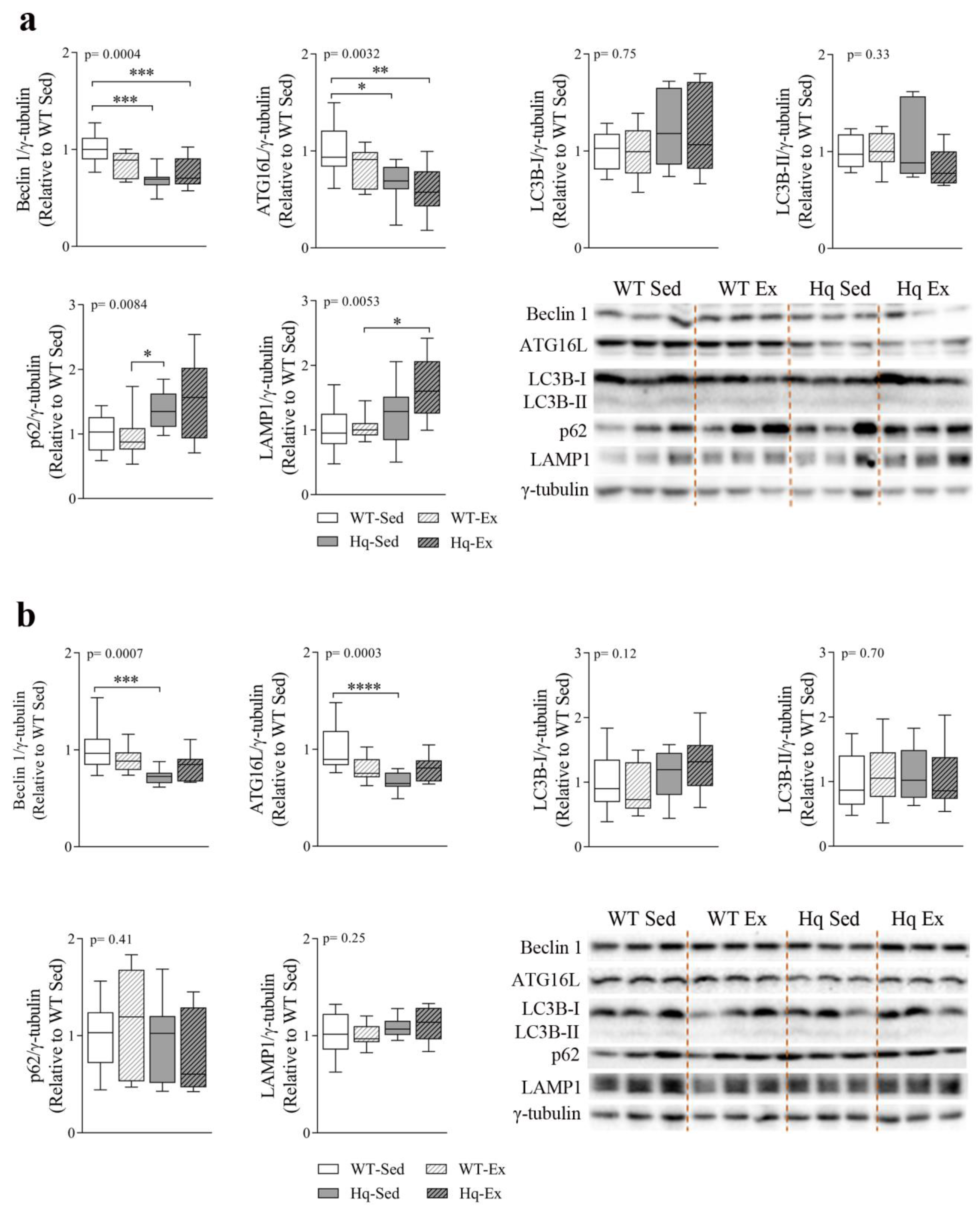
3.6. Autophagy in Skeletal Muscle—Exercise Training Effects
3.7. Free Amino Acids and Ammonium Levels in Skeletal Muscle
3.8. Autophagy Protein Levels and Effects of Exercise Training in Central Nervous System
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schaefer, A.M.; Taylor, R.W.; Turnbull, D.M.; Chinnery, P.F. The epidemiology of mitochondrial disorders—Past, present and future. Biochim. Biophys. Acta (BBA)-Bioenerg. 2004, 1659, 115–120. [Google Scholar] [CrossRef] [Green Version]
- Mcfarland, R.; Turnbull, D.M. Batteries not included: Diagnosis and management of mitochondrial disease. J. Intern. Med. 2009, 265, 210–228. [Google Scholar] [CrossRef]
- Hüttemann, M.; Lee, I.; Pecinova, A.; Pecina, P.; Przyklenk, K.; Doan, J.W. Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. J. Bioenerg. Biomembr. 2008, 40, 445–456. [Google Scholar] [CrossRef]
- Ghezzi, D.; Zeviani, M. Human diseases associated with defects in assembly of OXPHOS complexes. Essays Biochem. 2018, 62, 271–286. [Google Scholar]
- Emperador, S.; Garrido-Pérez, N.; Amezcua-Gil, J.; Gaudó, P.; Andrés-Sanz, J.A.; Yubero, D.; Fernández-Marmiesse, A.; O’Callaghan, M.M.; Escobar, J.D.O.; Iriondo, M.; et al. Molecular Characterization of New FBXL4 Mutations in Patients with mtDNA Depletion Syndrome. Front. Genet. 2019, 10, 1300. [Google Scholar] [CrossRef]
- Kane, M.S.; Alban, J.; Desquiret-Dumas, V.; Gueguen, N.; Ishak, L.; Ferré, M.; Amati-Bonneau, P.; Procaccio, V.; Bonneau, D.; Lenaers, G.; et al. Autophagy controls the pathogenicity of OPA1 mutations in dominant optic atrophy. J. Cell Mol. Med. 2017, 21, 2284–2297. [Google Scholar] [CrossRef] [Green Version]
- De la Mata, M.; Garrido-Maraver, J.; Cotán, D.; Cordero, M.D.; Oropesa-Ávila, M.; Izquierdo, L.G.; de Miguel, M.; Lorite, J.B.; Infante, E.R.; Ybot, P.; et al. Recovery of MERRF fibroblasts and cybrids pathophysiology by coenzyme Q10. Neurotherapeutics 2012, 9, 446–463. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.-H.; Sakiyama, Y.; Higuchi, I.; Inamori, Y.; Higuchi, Y.; Hashiguchi, A.; Higashi, K.; Yoshimura, A.; Takashima, H. Mitochondrial myopathy with autophagic vacuoles in patients with the m.8344A > G mutation. J. Clin. Pathol. 2013, 66, 659–664. [Google Scholar] [CrossRef]
- Garrido-Maraver, J.; Paz, M.V.; Cordero, M.D.; Bautista-Lorite, J.; Oropesa-Ávila, M.; de la Mata, M.; Pavón, A.D.; Lavera, I.; Alcocer-Gómez, E.; Galán, F. Critical role of AMP-activated protein kinase in the balance between mitophagy and mitochondrial biogenesis in MELAS disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 2535–2553. [Google Scholar] [CrossRef]
- Lin, D.-S.; Huang, Y.-W.; Ho, C.-S.; Hung, P.-L.; Hsu, M.-H.; Wang, T.-J.; Wu, T.-Y.; Lee, T.-H.; Huang, Z.-D.; Chang, P.-C.; et al. Oxidative Insults and Mitochondrial DNA Mutation Promote Enhanced Autophagy and Mitophagy Compromising Cell Viability in Pluripotent Cell Model of Mitochondrial Disease. Cells 2019, 8, 65. [Google Scholar] [CrossRef] [Green Version]
- White, K.E.; Davies, V.J.; Hogan, V.E.; Piechota, M.J.; Nichols, P.P.; Turnbull, D.M.; Votruba, M. OPA1 deficiency associated with increased autophagy in retinal ganglion cells in a murine model of dominant optic atrophy. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2567–2571. [Google Scholar] [CrossRef]
- Villanueva-Paz, M.; Povea-Cabello, S.; Villalón-García, I.; Álvarez-Córdoba, M.; Suárez-Rivero, J.M.; Talaverón-Rey, M.; Jackson, S.; Falcón-Moya, R.; Rodríguez-Moreno, A.; Sánchez-Alcázar, J.A. Parkin-mediated mitophagy and autophagy flux disruption in cellular models of MERRF syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165726. [Google Scholar] [CrossRef]
- Morán, M.; Delmiro, A.; Blázquez, A.; Ugalde, C.; Arenas, J.; Martín, M.A. Bulk autophagy, but not mitophagy, is increased in cellular model of mitochondrial disease. Biochim. Biophys. Acta 2014, 1842, 1059–1070. [Google Scholar] [CrossRef] [Green Version]
- Cotán, D.; Cordero, M.D.; Garrido-Maraver, J.; Oropesa-Avila, M.; Rodríguez-Hernández, A.; Gomez Izquierdo, L.; De la Mata, M.; De Miguel, M.; Lorite, J.B.; Infante, E.R.; et al. Secondary coenzyme Q10 deficiency triggers mitochondria degradation by mitophagy in MELAS fibroblasts. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 2669–2687. [Google Scholar]
- Dombi, E.; Diot, A.; Morten, K.; Carver, J.; Lodge, T.; Fratter, C.; Ng, Y.S.; Liao, C.; Muir, R.; Blakely, E.L. The m.13051G > A mitochondrial DNA mutation results in variable neurology and activated mitophagy. Neurology 2016, 86, 1921–1923. [Google Scholar] [CrossRef] [Green Version]
- Tyynismaa, H.; Mjosund, K.P.; Wanrooij, S.; Lappalainen, I.; Ylikallio, E.; Jalanko, A.; Spelbrink, J.; Paetau, A.; Suomalainen-Wartiovaara, A. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 17687–17692. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Lu, Y.; Xie, Z.; Liu, J.; Yuan, Y.; Wang, Z. RNA-seq profiling, and impaired autophagic process in skeletal muscle of MELAS. Biochem. Biophys. Res Commun. 2020, 523, 91–97. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Z.; Khan, A.; Zheng, H.; Yuan, C.; Jiang, H. Advances in drug therapy for mitochondrial diseases. Ann. Transl. Med. 2020, 8, 17. [Google Scholar] [CrossRef]
- Jeppesen, T.D.; Schwartz, M.; Olsen, D.B.; Wibrand, F.; Krag, T.; Dunø, M.; Hauerslev, S.; Vissing, J. Aerobic training is safe and improves exercise capacity in patients with mitochondrial myopathy. Brain 2006, 129, 3402–3412. [Google Scholar] [CrossRef] [Green Version]
- Porcelli, S.; Marzorati, M.; Morandi, L.; Grassi, B. Home-based aerobic exercise training improves skeletal muscle oxidative metabolism in patients with metabolic myopathies. J. Appl. Physiol. 2016, 121, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Taivassalo, T.; Gardner, J.L.; Taylor, R.W.; Schaefer, A.; Newman, J.; Barron, M.; Haller, R.G.; Turnbull, D.M. Endurance training and detraining in mitochondrial myopathies due to single large-scale mtDNA deletions. Brain 2006, 129, 3391–3401. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.L.; Blakely, E.L.; Schaefer, A.M.; He, L.; Wyrick, P.; Haller, R.G.; Taylor, R.W.; Turnbull, D.M.; Taivassalo, T. Resistance training in patients with single, large-scale deletions of mitochondrial DNA. Brain 2008, 131, 2832–2840. [Google Scholar] [CrossRef] [Green Version]
- Venturelli, M.; Villa, F.; Ruzzante, F.; Tarperi, C.; Rudi, D.; Milanese, C.; Cavedon, V.; Fonte, C.; Picelli, A.; Smania, N. Neuromuscular and Muscle Metabolic Functions in MELAS before and after Resistance Training: A Case Study. Front. Physiol. 2019, 10, 503. [Google Scholar] [CrossRef] [Green Version]
- Cejudo, P.; Bautista, J.; Montemayor, T.; Villagómez, R.; Jiménez, L.; Ortega, F.; Campos, Y.; Sánchez, H.; Arenas, J. Exercise training in mitochondrial myopathy: A randomized controlled trial. Muscle Nerve 2005, 32, 342–350. [Google Scholar] [CrossRef]
- Fiuza-Luces, C.; Díez-Bermejo, J.; Fernández-De La Torre, M.; Rodríguez-Romo, G.; Sanz-Ayán, P.; Delmiro, A.; Munguía-Izquierdo, D.; Rodríguez-Gómez, I.; Ara, I.; Domínguez-González, C.; et al. Health Benefits of an Innovative Exercise Program for Mitochondrial Disorders. Med. Sci. Sports Exerc. 2018, 50, 1142–1151. [Google Scholar] [CrossRef]
- Vainshtein, A.; Hood, D.A. The regulation of autophagy during exercise in skeletal muscle. J. Appl. Physiol. 2016, 120, 664–673. [Google Scholar] [CrossRef] [Green Version]
- Campos, J.C.; Queliconi, B.B.; Bozi, L.H.M.; Bechara, L.R.G.; Dourado, P.M.M.; Andres, A.M.; Jannig, P.R.; Gomes, K.M.S.; Zambelli, V.O.; Rocha-Resende, C.; et al. Exercise reestablishes autophagic flux and mitochondrial quality control in heart failure. Autophagy 2017, 13, 1304–1317. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, J.; Cretoiu, D.; Li, G.; Xiao, J. Exercise-mediated regulation of autophagy in the cardiovascular system. J. Sport Health Sci. 2020, 9, 203–210. [Google Scholar] [CrossRef]
- Andreotti, D.Z.; Silva, J.D.N.; Matumoto, A.M.; Orellana, A.M.; de Mello, P.S.; Kawamoto, E.M. Effects of Physical Exercise on Autophagy and Apoptosis in Aged Brain: Human and Animal Studies. Front Nutr. 2020, 7, 94. [Google Scholar] [CrossRef]
- He, C.; Sumpter, R.J.; Levine, B. Exercise induces autophagy in peripheral tissues and in the brain. Autophagy 2012, 8, 1548–1551. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Liang, J.; Wu, L.; Zhang, H.; Lv, J.; Chen, N. Exercise-Induced Autophagy Suppresses Sarcopenia Through Akt/mTOR and Akt/FoxO3a Signal Pathways and AMPK-Mediated Mitochondrial Quality Control. Front. Physiol. 2020, 11, 583478. [Google Scholar] [CrossRef]
- McMillan, E.M.; Paré, M.F.; Baechler, B.L.; Graham, D.A.; Rush, J.W.E.; Quadrilatero, J. Autophagic signaling and proteolytic enzyme activity in cardiac and skeletal muscle of spontaneously hypertensive rats following chronic aerobic exercise. PLoS ONE 2015, 10, e0119382. [Google Scholar] [CrossRef] [Green Version]
- Almeida, M.F.; Silva, C.M.; Chaves, R.S.; Lima, N.C.R.; Almeida, R.S.; Melo, K.P.; Demasi, M.; Fernandes, T.; Oliveira, E.M.; Netto, L.E.S.; et al. Effects of mild running on substantia nigra during early neurodegeneration. J. Sports Sci. 2018, 36, 1363–1370. [Google Scholar] [CrossRef]
- Ng, S.Y.; Mikhail, A.; Ljubicic, V. Chronic physical activity is beneficial for SMA patients, however the cellular and molecular mechanisms of exercise biology are largely undefined in SMA. J. Physiol. 2019, 597, 4757–4778. [Google Scholar] [CrossRef]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Constantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef]
- Otera, H.; Ohsakaya, S.; Nagaura, Z.I.; Lorenzo, H.K.; Zamzami, N. Export of mitochondrial AIF in response to proapoptotic stimuli depends on processing at the intermembrane space. EMBO J. 2005, 24, 1375–1386. [Google Scholar] [CrossRef]
- Chinta, S.K.; Rane, A.; Yadava, N.; Andersen, J.K.; Nicholls, D.G.; Polster, B. Reactive oxygen species regulation by AIF- and complex I- depleted brain mitochondria. Free Radic. Biol. Med. 2009, 2346, 939–947. [Google Scholar] [CrossRef] [Green Version]
- Hangen, E.; Féraud, O.; Lachkar, S.; Mou, H.; Doti, N.; Fimia, G.M.; Lam, N.-v.; Zhu, C.; Godin, I.; Muller, K.; et al. Interaction between AIF and CHCHD4 Regulates Respiratory Chain Biogenesis. Mol. Cell 2015, 58, 1001–1014. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.; Buettner, S.; Ghezzi, D.; Zeviani, M.; Bano, D.; Nicotera, P. Loss of apoptosis-inducing factor critically affects MIA40 function. Cell Death Dis. 2015, 6, e1814. [Google Scholar] [CrossRef]
- Bano, D.; Prehn, J.H.M. Apoptosis-Inducing Factor (AIF) in Physiology and Disease: The Tale of a Repented Natural Born Killer. EBioMedicine 2018, 30, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Fiuza-Luces, C.; Valenzuela, P.L.; Laine-Menéndez, S.; Fernández-de la Torre, M.; Bermejo-Gómez, V.; Rufián-Vázquez, L.; Arenas, J.; Martín, M.A.; Lucia, A.; Morán, M. Physical Exercise and Mitochondrial Disease: Insights from a Mouse Model. Front. Neurol. 2019, 10, 790. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.A.; Longo-Guess, C.M.; Rossmann, M.P.; Seburn, K.L.; Hurd, R.E.; Frankel, W.N.; Bronson, R.T.; Ackerman, S.L. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature 2002, 419, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Bénit, P.; Goncalves, S.; Dassa, E.P.; Brière, J.-J.; Rustin, P. The variability of the harlequin mouse phenotype resembles that of human mitochondrial-complex I-deficiency syndromes. PLoS ONE 2008, 3, e3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vahsen, N.; Candé, C.; Brière, J.J.; Bénit, P.; Joza, N.; Larochette, N.; Mastroberardino, P.G.; Pequignot, M.O.; Casares, N.; Lazar, V.; et al. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004, 23, 4679–4689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Empel, V.P.M.; Bertrand, A.T.; van der Nagel, R.; Kostin, S.; Doevendans, P.A.; Crijns, H.J.; de Wit, E.; Sluiter, W.; Ackerman, S.L.; De Windt, L.J. Downregulation of apoptosis-inducing factor in harlequin mutant mice sensitizes the myocardium to oxidative stress-related cell death and pressure overload-induced decompensation. Circ. Res. 2005, 96, e92–e101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welinder, C.; Ekblad, L. Coomassie staining as loading control in Western blot analysis. J. Proteome Res. 2011, 10, 1416–1419. [Google Scholar] [CrossRef]
- Haspel, J.; Shaik, R.S.; Ifedigbo, E.; Nakahira, K.; Dolinay, T.; Englert, J.A.; Choi, A.M.K. Characterization of macroautophagic flux in vivo using a leupeptin-based assay. Characterization of macroautophagic flux in vivo using a leupeptin-based assay. Autophagy 2011, 7, 629–642. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.A.; Nikkanen, J.; Yatsuga, S.; Jackson, C.; Wang, L.; Pradhan, S.; Kivelä, R.; Pessia, A.; Velagapudi, V.; Suomalainen, A. mTORC1 Regulates Mitochondrial Integrated Stress Response and Mitochondrial Myopathy Progression. Cell Metab. 2017, 26, 419–428.e5. [Google Scholar] [CrossRef]
- McKnight, N.C.; Zhong, Y.; Wold, M.S.; Gong, S.; Phillips, G.R.; Dou, Z.; Zhao, Y.; Heintz, N.; Zong, W.-X.; Yue, Z. Beclin 1 is required for neuron viability and regulates endosome pathways via the UVRAG-VPS34 complex. PLoS Genet. 2014, 10, e1004626. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Fernández-de la Torre, M.; Fiuza-Luces, C.; Valenzuela, P.L.; Laine-Menéndez, S.; Arenas, J.; Martín, M.A.; Turnbull, D.M.; Lucia, A.; Morán, M. Exercise Training and Neurodegeneration in Mitochondrial Disorders: Insights from the Harlequin Mouse. Front. Physiol. 2020, 11, 594223. [Google Scholar] [CrossRef] [PubMed]
- Danieli, A.; Martens, S. p62-mediated phase separation at the intersection of the ubiquitin-proteasome system and autophagy. J. Cell Sci. 2018, 131, jcs214304. [Google Scholar] [CrossRef] [Green Version]
- Shin, W.H.; Park, J.H.; Chung, K.C. The central regulator p62 between ubiquitin proteasome system and autophagy and its role in the mitophagy and Parkinson’s disease. BMB Rep. 2020, 53, 56–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, R.; Enaka, M.; Muragaki, Y. Activation of KEAP1/NRF2/P62 signaling alleviates high phosphate-induced calcification of vascular smooth muscle cells by suppressing reactive oxygen species production. Sci. Rep. 2019, 9, 10366. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Li, Y.; Pitti, R.; Lawrence, D.; Pham, V.C.; Lill, J.R.; Ashkenazi, A. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell 2009, 137, 721–735. [Google Scholar] [CrossRef] [Green Version]
- Lucas, S.; Chen, G.; Aras, S.; Wang, J. Serine catabolism is essential to maintain mitochondrial respiration in mammalian cells. Life Sci. Alliance 2018, 1, 1–10. [Google Scholar] [CrossRef]
- Al-Bari, M.A.A.; Xu, P. Molecular regulation of autophagy machinery by mTOR-dependent and -independent pathways. Ann. N. Y. Acad. Sci. 2020, 1467, 3–20. [Google Scholar] [CrossRef]
- Vincow, E.S.; Thomas, R.E.; Merrihew, G.E.; Shulman, N.J.; Bammler, T.K.; MacDonald, J.W.; MacCoss, M.J.; Pallanck, L.J. Autophagy accounts for approximately one-third of mitochondrial protein turnover and is protein selective. Autophagy 2019, 15, 1592–1605. [Google Scholar] [CrossRef]
- Bayod, S.; Del Valle, J.; Pelegri, C.; Vilaplana, J.; Canudas, A.M.; Camins, A.; Jimenez, A.; Sanchez-Roige, S.; Lalanza, J.F.; Escorihuela, R.M.; et al. Macroautophagic process was differentially modulated by long-term moderate exercise in rat brain and peripheral tissues. J. Physiol. Pharmacol. Off. J. Polish Physiol. Soc. 2014, 65, 229–239. [Google Scholar]
- Marques-Aleixo, I.; Santos-Alves, E.; Balça, M.M.; Rizo-Roca, D.; Moreira, P.I.; Oliveira, P.J.; Magalhães, J.; Ascensão, A. Physical exercise improves brain cortex and cerebellum mitochondrial bioenergetics and alters apoptotic, dynamic and auto(mito)phagy markers. Neuroscience 2015, 301, 480–495. [Google Scholar] [CrossRef] [PubMed]
- Schwalm, C.; Jamart, C.; Benoit, N.; Naslain, D.; Prémont, C.; Prevet, J.; Van Thienen, R.; Deldicque, L.; Francaux, M. Activation of autophagy in human skeletal muscle is dependent on exercise intensity and AMPK activation. FASEB J. 2015, 29, 3515–3526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocchi, A.; He, C. Regulation of Exercise-Induced Autophagy in Skeletal Muscle. Curr. Pathobiol. Rep. 2017, 5, 177–186. [Google Scholar] [CrossRef]
- Pan, G.; Jin, L.; Shen, W.; Zhang, J.; Pan, J.; Cheng, J.; Xie, Q.; Hu, Q.; Wu, S.; Zhang, H.; et al. Treadmill exercise improves neurological function by inhibiting autophagy and the binding of HMGB1 to Beclin1 in MCAO juvenile rats. Life Sci. 2020, 243, 117279. [Google Scholar] [CrossRef]
- Herring, A.; Münster, Y.; Metzdorf, J.; Bolczek, B.; Krüssel, S.; Krieter, D.; Yavuz, I.; Karim, F.; Roggendorf, C.; Stang, A.; et al. Late running is not too late against Alzheimer’s pathology. Neurobiol. Dis. 2016, 94, 44–54. [Google Scholar] [CrossRef]
- Fucà, E.; Guglielmotto, M.; Boda, E.; Rossi, F.; Leto, K.; Buffo, A. Preventive motor training but not progenitor grafting ameliorates cerebellar ataxia and deregulated autophagy in tambaleante mice. Neurobiol. Dis. 2017, 102, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.; Kwon, I.; Song, W.; Cosio-Lima, L.M.; Lee, Y. Endurance Exercise Mediates Neuroprotection Against MPTP-mediated Parkinson’s Disease via Enhanced Neurogenesis, Antioxidant Capacity, and Autophagy. Neuroscience 2018, 379, 292–301. [Google Scholar] [CrossRef]
- Koo, J.-H.; Cho, J.-Y. Treadmill Exercise Attenuates α-Synuclein Levels by Promoting Mitochondrial Function and Autophagy Possibly via SIRT1 in the Chronic MPTP/P-Induced Mouse Model of Parkinson’s Disease. Neurotox. Res. 2017, 32, 473–486. [Google Scholar] [CrossRef]
- Fritzen, A.M.; Frøsig, C.; Jeppesen, J.; Jensen, T.E.; Lundsgaard, A.-M.; Serup, A.K.; Schjerling, P.; Proud, C.G.; Richter, E.A.; Kiens, B. Role of AMPK in regulation of LC3 lipidation as a marker of autophagy in skeletal muscle. Cell. Signal. 2016, 28, 663–674. [Google Scholar] [CrossRef]
- Kim, Y.A.; Kim, Y.S.; Song, W. Autophagic response to a single bout of moderate exercise in murine skeletal muscle. J. Physiol. Biochem. 2012, 68, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.M.; Coppotelli, G.; Branca, R.M.; Kim, K.M.; Lehtiö, J.; Sinclair, D.A.; Olson, L. Voluntary exercise normalizes the proteomic landscape in muscle and brain and improves the phenotype of progeroid mice. Aging Cell 2019, 18, e13029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenz, T.; Diaz, F.; Hernandez, D.; Moraes, C.T. Endurance exercise is protective for mice with mitochondrial myopathy. J. Appl. Physiol. 2009, 106, 1712–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, N.N.; Tian, H.; Chen, P.; Wang, D.; Ren, J.; Zhang, Y. Physical Exercise and Selective Autophagy: Benefit and Risk on Cardiovascular Health. Cells 2019, 8, 1436. [Google Scholar] [CrossRef] [Green Version]
- Codina-Martínez, H.; Fernández-García, B.; Díez-Planelles, C.; Fernández, Á.F.; Higarza, S.G.; Fernández-Sanjurjo, M.; Díez-Robles, S.; Iglesias-Gutiérrez, E.; Tomás-Zapico, C. Autophagy is required for performance adaptive response to resistance training and exercise-induced adult neurogenesis. Scand. J. Med. Sci. Sports 2020, 30, 238–253. [Google Scholar] [CrossRef]
- Wischhof, L.; Gioran, A.; Sonntag-Bensch, D.; Piazzesi, A.; Stork, M.; Nicotera, P.; Bano, D. A disease-associated Aifm1 variant induces severe myopathy in knockin mice. Mol. Metab. 2018, 13, 10–23. [Google Scholar] [CrossRef]
- Nikkanen, J.; Forsström, S.; Euro, L.; Paetau, I.; Kohnz, R.A.; Wang, L.; Chilov, D.; Viinamäki, J.; Roivainen, A.; Marjamäki, P.; et al. Mitochondrial DNA Replication Defects Disturb Cellular dNTP Pools and Remodel One-Carbon Metabolism. Cell Metab. 2016, 23, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Tyynismaa, H.; Carroll, C.J.; Raimundo, N.; Ahola-Erkkila, S.; Wenz, T.; Ruhanen, H.; Guse, K.; Hemminki, A.; Peltola-Mjosund, K.E.; Tulkki, V.; et al. Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet. 2010, 19, 3948–3958. [Google Scholar] [CrossRef] [Green Version]
- Morscher, R.J.; Ducker, G.S.; Li, S.H.-J.; Mayer, J.A.; Gitai, Z.; Sperl, W.; Rabinowitz, J.D. Mitochondrial translation requires folate-dependent tRNA methylation. Nature 2018, 554, 128–132. [Google Scholar] [CrossRef]
- Zieliński, Ł.P.; Smith, A.C.; Smith, A.G.; Robinson, A.J. Metabolic flexibility of mitochondrial respiratory chain disorders predicted by computer modelling. Mitochondrion 2016, 31, 45–55. [Google Scholar] [CrossRef]
- Chen, Q.; Kirk, K.; Shurubor, Y.I.; Zhao, D.; Arreguin, A.J.; Shahi, I.; Valsecchi, F.; Primiano, G.; Calder, E.L.; Carelli, V.; et al. Rewiring of Glutamine Metabolism Is a Bioenergetic Adaptation of Human Cells with Mitochondrial DNA Mutations. Cell Metab. 2018, 27, 1007–1025.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino Acid | WT µmol/L | Hq µmol/L | p-Value |
|---|---|---|---|
| Threonine | 14.3 [12.9–15.6] | 22.3 [19.5–25.5] | 0.008 |
| Serine | 17.0 [15.5–17.8] | 22.4 [20.1–24.9] | 0.016 |
| Asparagine | 7.9 [3.3–9.4] | 9.2 [6.9–11.4] | 0.309 |
| Glutamate | 192.6 [159.1–227.5] | 211.4 [191.5–252.9] | 0.309 |
| Glutamine | 206.7 [171.4–310.3] | 294.9 [234.9–303.3] | 0.309 |
| Glycine | 24.4 [20.6–26.2] | 41.2 [36.6–42.8] | 0.007 |
| Alanine | 131.5 [97.9–158.7] | 170.5 [135.3–209.2] | 0.095 |
| Citrulline | 7.0 [5.5–8.9] | 7.1 [6.3–8.0] | 0.690 |
| Valine | 4.8 [4.2–6.0] | 10.4 [7.4–10.5] | 0.031 |
| Methionine | 4.1 [2.9–4.6] | 5.2 [3.7–5.5] | 0.222 |
| Isoleucine | 9.0 [6.8–10.2] | 11.6 [9.6–12.2] | 0.151 |
| Leucine | 13.6 [8.2–13.9] | 18.5 [14.1–19.1] | 0.056 |
| Tyrosine | 4.9 [4.2–6.3] | 6.5 [5.3–7.0] | 0.309 |
| Phenylalanine | 5.8 [4.8–6.2] | 5.2 [4.5–6.3] | 0.690 |
| Ammonium | 313.9 [253.2–345.7] | 336.9 [288.9–339.2] | >0.99 |
| Lysine | 24.4 [20.6–25.0] | 27.9 [23.4–33.3] | 0.095 |
| Histidine | 7.4 [6.7–10.2] | 10.0 [8.6–11.3] | 0.222 |
| Arginine | 16.2 [13.1–18.0] | 17.8 [16.5–22.0] | 0.309 |
| Amino Acid | WT (µM) | Hq (µM) | p-Value |
|---|---|---|---|
| Threonine | 38.2 [36.5–44.7] | 44.7 [44.1–62.5] | 0.095 |
| Serine | 47.6 [34.9–65.5] | 58.7 [47.3–82.4] | 0.420 |
| Asparagine | 5.6 [4.8–7.3] | 6.6 [5.8–13.1] | 0.548 |
| Glutamate | 56.5 [48.4–72.9] | 66.2 [57.3–131.2] | 0.151 |
| Glutamine | 165.3 [143.4–202.1] | 204.3 [181.8–235.5] | 0.095 |
| Glycine | 205.3 [170.3–311.2] | 282.1 [235.1–334.4] | 0.222 |
| Alanine | 348.7 [277.4–371.1] | 379.4 [354.5–475.1] | 0.095 |
| Citrulline | 13.6 [12.3–16.2] | 15.6 [10.1–18.2] | >0.999 |
| Valine | 24.7 [23.1–25.9] | 31.1 [29.3–39.1] | 0.008 |
| Methionine | 9.9 [9.4–10.4] | 12.1 [8.2–13.8] | 0.690 |
| Cysteine | 8.3 [7.9–8.4] | 8.6 [7.8–9.4] | 0.151 |
| Isoleucine | 17.9 [15.8–18.8] | 21.3 [18.7–23.3] | 0.055 |
| Leucine | 23.6 [22.9–25.5] | 33.7 [25.7–38.3] | 0.095 |
| Tyrosine | 16.2 [13.2–16.3] | 16.6 [14.5–18.5] | 0.222 |
| Phenylalanine | 11.5 [11.2–13.1] | 15.7 [11.5–17.3] | 0.095 |
| Ammonium | 575.3 [573.5–600.0] | 569.0 [555.1–598.5] | 0.421 |
| Ornithine | 4.4 ± [4.1–5.1] | 8.3 [6.2–12.8] | 0.008 |
| Lysine | 53.6 [44.9–58.3] | 66.8 [63.4–84.5] | 0.008 |
| Histidine | 14.8 [12.9–18.7] | 18.0 [14.9–21.5] | 0.309 |
| Arginine | 25.0 [18.7–25.7] | 34.1 [27.9–46.4] | 0.008 |
| Hydroxyproline | 11.7 [10.5–14.2] | 14.6 [12.3–16.1] | 0.222 |
| Proline | 34.4 [16.7–37.3] | 36.7 [28.9–57.8] | 0.309 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laine-Menéndez, S.; Fernández-de la Torre, M.; Fiuza-Luces, C.; Delmiro, A.; Arenas, J.; Martín, M.Á.; Boya, P.; Lucia, A.; Morán, M. Apoptosis-Inducing Factor Deficiency Induces Tissue-Specific Alterations in Autophagy: Insights from a Preclinical Model of Mitochondrial Disease and Exercise Training Effects. Antioxidants 2022, 11, 510. https://doi.org/10.3390/antiox11030510
Laine-Menéndez S, Fernández-de la Torre M, Fiuza-Luces C, Delmiro A, Arenas J, Martín MÁ, Boya P, Lucia A, Morán M. Apoptosis-Inducing Factor Deficiency Induces Tissue-Specific Alterations in Autophagy: Insights from a Preclinical Model of Mitochondrial Disease and Exercise Training Effects. Antioxidants. 2022; 11(3):510. https://doi.org/10.3390/antiox11030510
Chicago/Turabian StyleLaine-Menéndez, Sara, Miguel Fernández-de la Torre, Carmen Fiuza-Luces, Aitor Delmiro, Joaquín Arenas, Miguel Ángel Martín, Patricia Boya, Alejandro Lucia, and María Morán. 2022. "Apoptosis-Inducing Factor Deficiency Induces Tissue-Specific Alterations in Autophagy: Insights from a Preclinical Model of Mitochondrial Disease and Exercise Training Effects" Antioxidants 11, no. 3: 510. https://doi.org/10.3390/antiox11030510