
How Cells Deal with the Fluctuating Environment: Autophagy Regulation under Stress in Yeast and Mammalian Systems

 , ,
, ,
Abstract
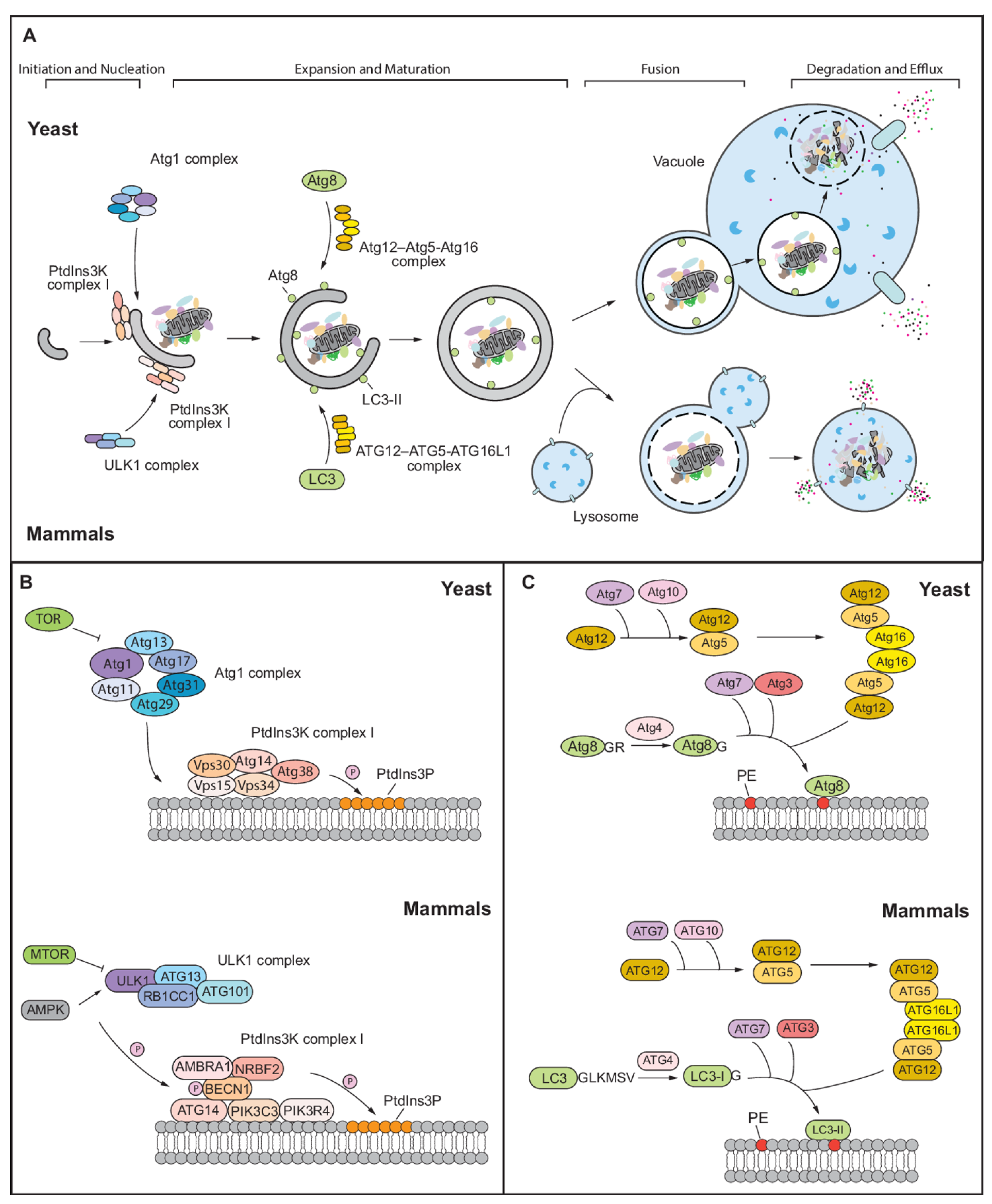
:1. Overview of Autophagy in Yeast and Mammals
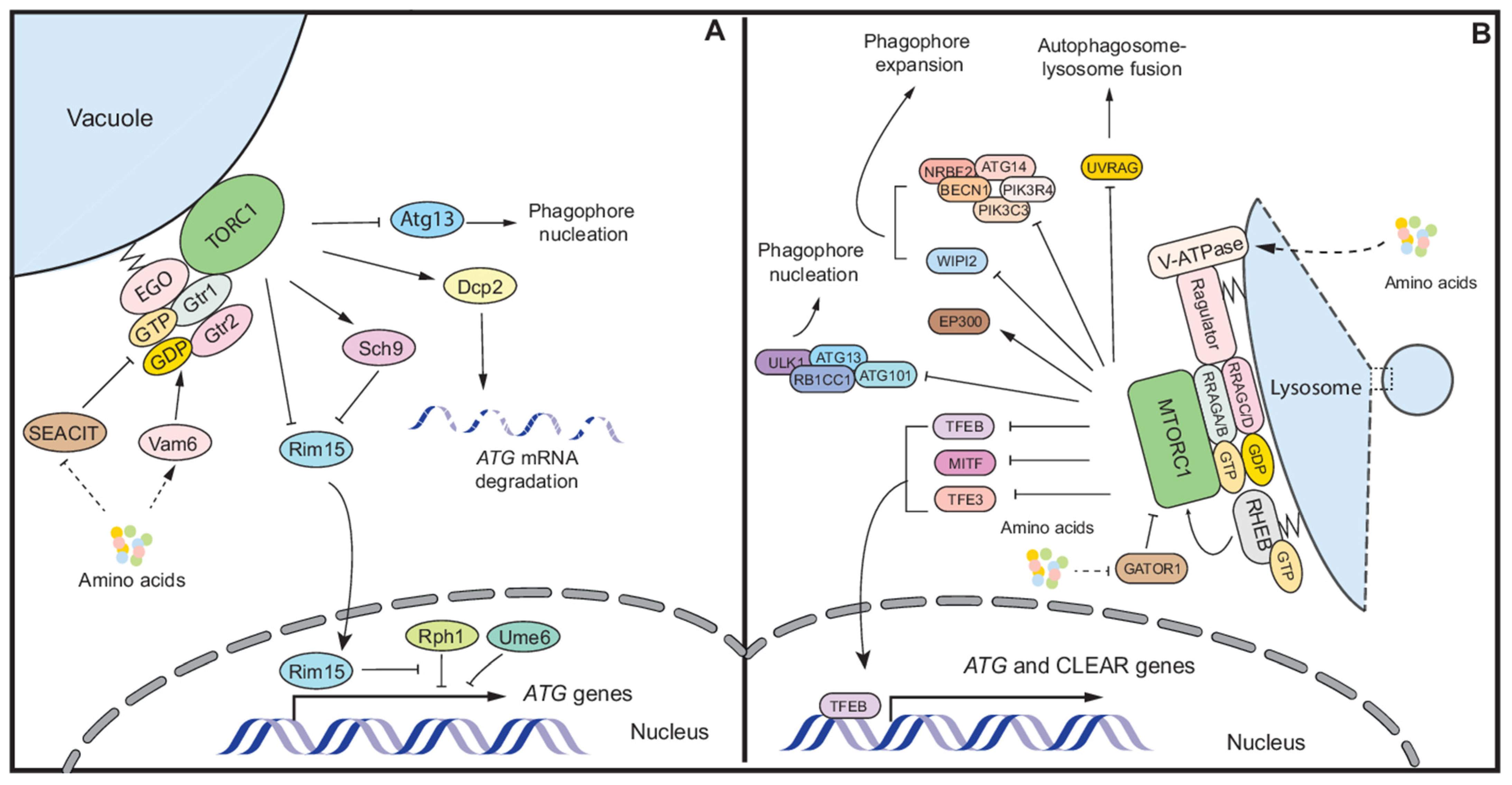
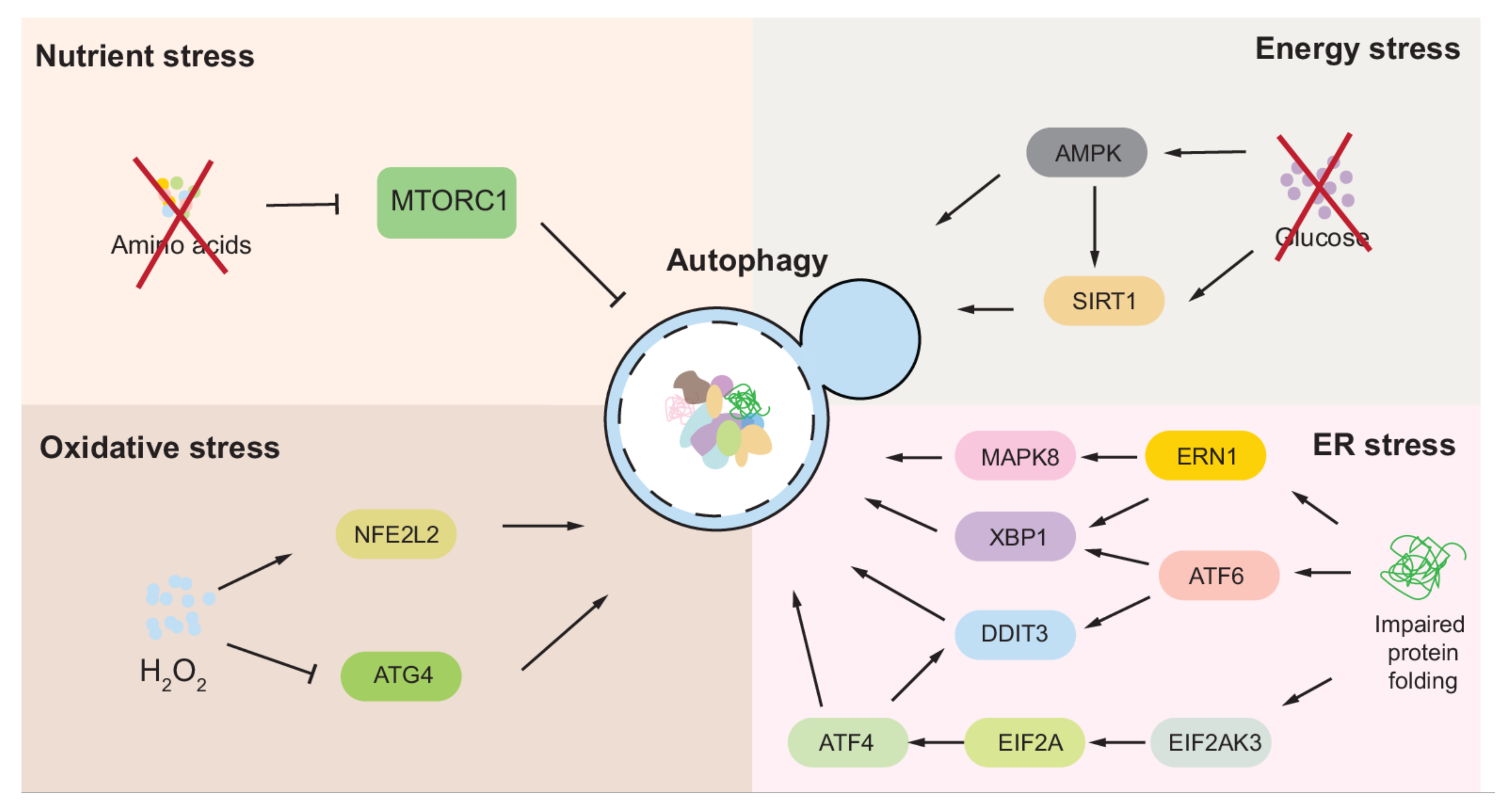
2. Autophagy Regulation under Nutrient Stress
2.1. Mechanisms of Autophagy Regulation by Nutrient Stress in Yeast
2.2. Autophagy Regulation in Mammalian Cells
2.2.1. Autophagy Regulation by MTORC1
2.2.2. Other Autophagy Regulation during Nutrient Stress
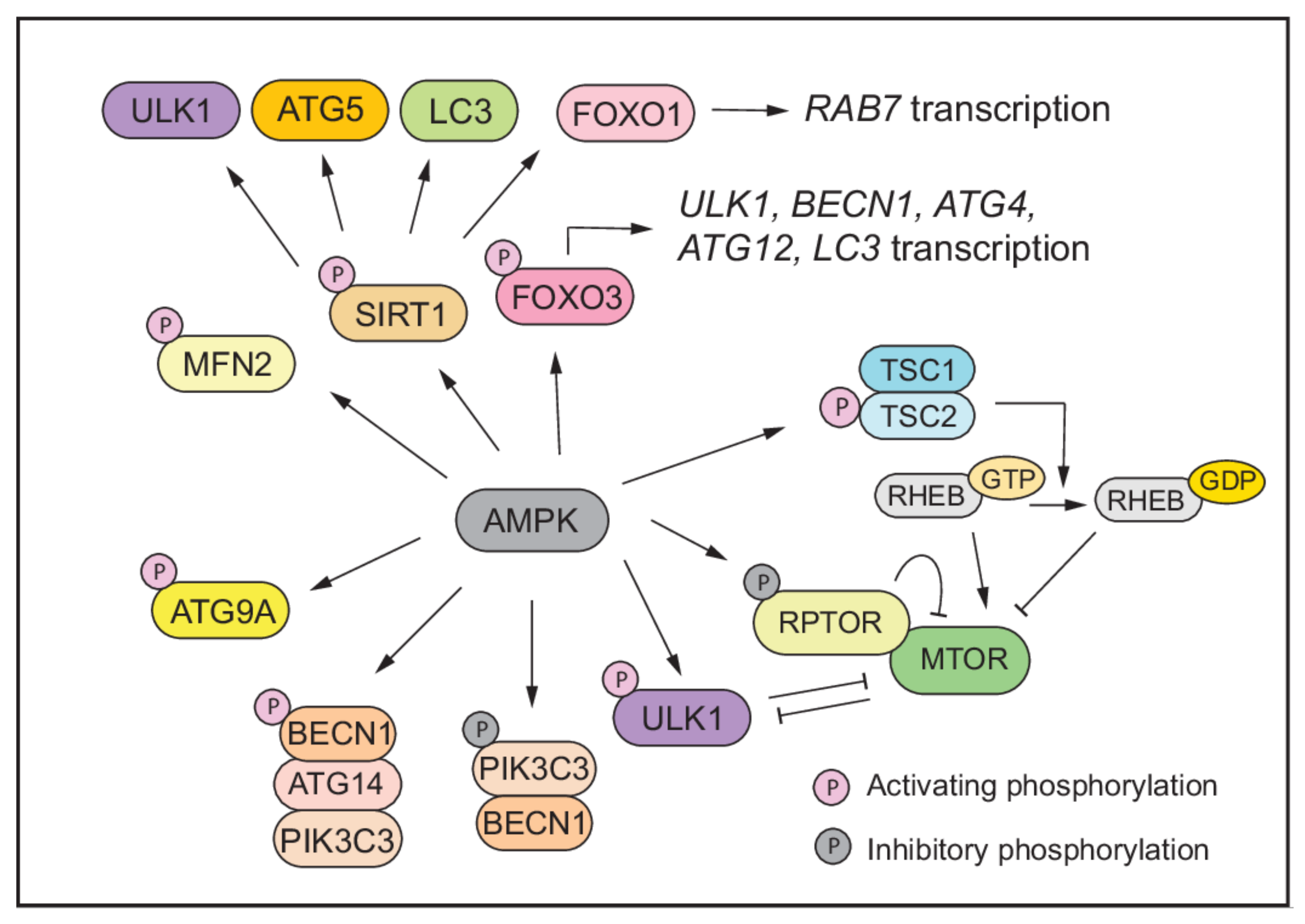
3. Autophagy Regulation under Energy Stress
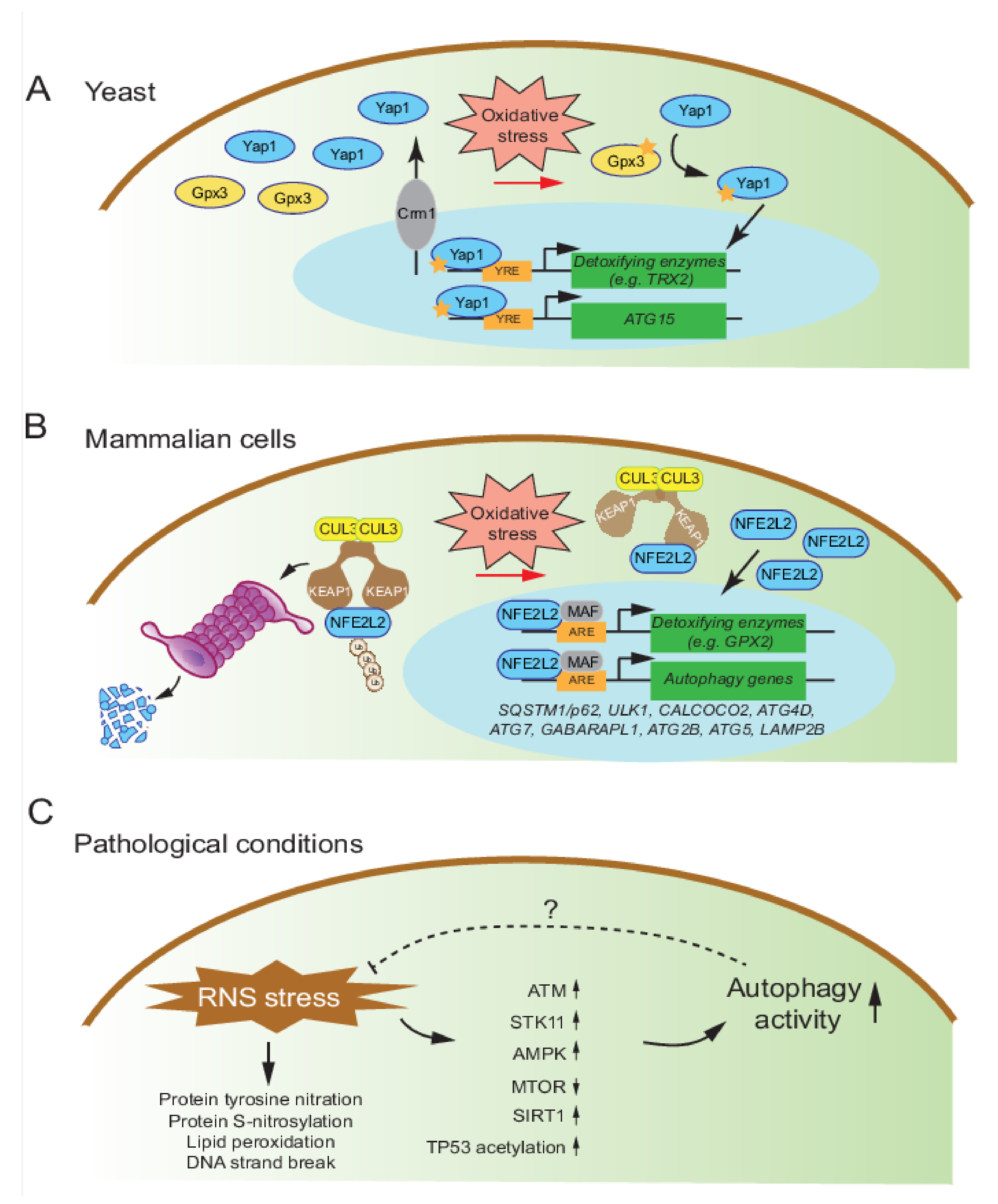
4. Autophagy Regulation under Oxidative/Nitrosative Stress
4.1. Mechanisms of Autophagy Regulation by Oxidative Stress in Yeast
4.2. Mechanisms of Autophagy Regulation by Oxidative Stress in Mammalian Cells
4.3. Mechanisms of Autophagy Regulation by Nitrosative Stress
5. Autophagy Regulation under ER Stress
5.1. Endoplasmic Reticulum Stress and Autophagy
5.2. The Mechanisms of ER Stress-Mediated Autophagy in Yeast
5.3. The Mechanisms of ER Stress-Mediated Autophagy in Mammalian Cells
5.3.1. ERN1
5.3.2. EIF2AK3
5.3.3. ATF6
5.3.4. Calcium
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| ATG | autophagy related |
| ER | endoplasmic reticulum |
| ERAD | ER-associated degradation |
| GAP | GTPase activating protein |
| H2O2 | hydrogen peroxide |
| m6A | N6-methyl-adenosine |
| MTORC1 | mechanistic target of rapamycin kinase complex 1 |
| NO | nitric oxide |
| PAS | phagophore assembly site |
| PE | phosphatidylethanolamine |
| PtdIns3K | class III phosphatidylinositol kinase |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| TFs | transcription factors |
| Ubl | ubiquitin-like |
| UPR | unfolded protein response |
| V-ATPase | vacuolar-type H+-translocating ATPase |
References
- Yorimitsu, T.; Klionsky, D.J. Autophagy: Molecular machinery for self-eating. Cell Death Differ. 2005, 12 (Suppl. 2), 1542–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, X.; Klionsky, D.J. An overview of macroautophagy in yeast. J. Mol. Biol. 2016, 428, 1681–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Klionsky, D.J. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 2009, 335, 1–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Nakatogawa, H. Mechanisms governing autophagosome biogenesis. Nat. Rev. Mol. Cell Biol. 2020, 21, 439–458. [Google Scholar] [CrossRef]
- Yim, W.W.; Mizushima, N. Lysosome biology in autophagy. Cell Discov. 2020, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Ariosa, A.R.; Lahiri, V.; Lei, Y.; Yang, Y.; Yin, Z.; Zhang, Z.; Klionsky, D.J. A perspective on the role of autophagy in cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166262. [Google Scholar] [CrossRef]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Gatica, D.; Chiong, M.; Lavandero, S.; Klionsky, D.J. The role of autophagy in cardiovascular pathology. Cardiovasc. Res. 2022, in press. [Google Scholar] [CrossRef]
- Suzuki, K.; Kirisako, T.; Kamada, Y.; Mizushima, N.; Noda, T.; Ohsumi, Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001, 20, 5971–5981. [Google Scholar] [CrossRef]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Burman, C.; Ktistakis, N.T. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 2010, 584, 1302–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, J.; Klionsky, D.J. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: Beyond the usual suspects’ review series. EMBO Rep. 2008, 9, 859–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shpilka, T.; Mizushima, N.; Elazar, Z. Ubiquitin-like proteins and autophagy at a glance. J. Cell Sci. 2012, 125, 2343–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Z.; Popelka, H.; Lei, Y.; Yang, Y.; Klionsky, D.J. The Roles of Ubiquitin in Mediating Autophagy. Cells 2020, 9, 2025. [Google Scholar] [CrossRef]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef]
- Reggiori, F.; Tucker, K.A.; Stromhaug, P.E.; Klionsky, D.J. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev. Cell 2004, 6, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Klionsky, D.J. The Emerging Roles of Autophagy in Human Diseases. Biomedicines 2021, 9, 1651. [Google Scholar] [CrossRef]
- Yu, X.; Long, Y.C.; Shen, H.M. Differential regulatory functions of three classes of phosphatidylinositol and phosphoinositide 3-kinases in autophagy. Autophagy 2015, 11, 1711–1728. [Google Scholar] [CrossRef] [Green Version]
- Nascimbeni, A.C.; Codogno, P.; Morel, E. Phosphatidylinositol-3-phosphate in the regulation of autophagy membrane dynamics. FEBS J. 2017, 284, 1267–1278. [Google Scholar] [CrossRef]
- Sun, Q.; Fan, W.; Chen, K.; Ding, X.; Chen, S.; Zhong, Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 2008, 105, 19211–19216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; He, L.; Behrends, C.; Araki, M.; Araki, K.; Jun Wang, Q.; Catanzaro, J.M.; Friedman, S.L.; Zong, W.X.; Fiel, M.I.; et al. NRBF2 regulates autophagy and prevents liver injury by modulating Atg14L-linked phosphatidylinositol-3 kinase III activity. Nat. Commun. 2014, 5, 3920. [Google Scholar] [CrossRef] [PubMed]
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polson, H.E.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbe, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Urban, J.; Soulard, A.; Huber, A.; Lippman, S.; Mukhopadhyay, D.; Deloche, O.; Wanke, V.; Anrather, D.; Ammerer, G.; Riezman, H.; et al. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol. Cell 2007, 26, 663–674. [Google Scholar] [CrossRef]
- Tanigawa, M.; Maeda, T. An In Vitro TORC1 Kinase Assay That Recapitulates the Gtr-Independent Glutamine-Responsive TORC1 Activation Mechanism on Yeast Vacuoles. Mol. Cell. Biol. 2017, 37, e00075-17. [Google Scholar] [CrossRef] [Green Version]
- Ikenoue, T.; Hong, S.; Inoki, K. Monitoring mammalian target of rapamycin (mTOR) activity. Methods Enzymol. 2009, 452, 165–180. [Google Scholar] [CrossRef]
- Zhang, C.S.; Hawley, S.A.; Zong, Y.; Li, M.; Wang, Z.; Gray, A.; Ma, T.; Cui, J.; Feng, J.W.; Zhu, M.; et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 2017, 548, 112–116. [Google Scholar] [CrossRef]
- Berg, J.; Hung, Y.P.; Yellen, G. A genetically encoded fluorescent reporter of ATP:ADP ratio. Nat. Methods 2009, 6, 161–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tantama, M.; Martinez-Francois, J.R.; Mongeon, R.; Yellen, G. Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat. Commun. 2013, 4, 2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zegzouti, H.; Zdanovskaia, M.; Hsiao, K.; Goueli, S.A. ADP-Glo: A Bioluminescent and homogeneous ADP monitoring assay for kinases. Assay Drug Dev. Technol. 2009, 7, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Munday, M.R.; Scott, J.; Yang, X.; Carlson, M.; Carling, D. Yeast SNF1 is functionally related to mammalian AMP-activated protein kinase and regulates acetyl-CoA carboxylase in vivo. J. Biol. Chem. 1994, 269, 19509–19515. [Google Scholar] [CrossRef]
- Lim, C.T.; Lolli, F.; Thomas, J.D.; Kola, B.; Korbonits, M. Measurement of AMP-activated protein kinase activity and expression in response to ghrelin. Methods Enzymol. 2012, 514, 271–287. [Google Scholar] [CrossRef]
- Hempel, S.L.; Buettner, G.R.; O’Malley, Y.Q.; Wessels, D.A.; Flaherty, D.M. Dihydrofluorescein diacetate is superior for detecting intracellular oxidants: Comparison with 2′,7′-dichlorodihydrofluorescein diacetate, 5(and 6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate, and dihydrorhodamine 123. Free Radic. Biol. Med. 1999, 27, 146–159. [Google Scholar] [CrossRef]
- Miller, E.W.; Tulyathan, O.; Isacoff, E.Y.; Chang, C.J. Molecular imaging of hydrogen peroxide produced for cell signaling. Nat. Chem. Biol. 2007, 3, 263–267. [Google Scholar] [CrossRef]
- Uggeri, J.; Gatti, R.; Belletti, S.; Scandroglio, R.; Corradini, R.; Rotoli, B.M.; Orlandini, G. Calcein-AM is a detector of intracellular oxidative activity. Histochem. Cell Biol. 2004, 122, 499–505. [Google Scholar] [CrossRef]
- Tian, R.; Abarientos, A.; Hong, J.; Hashemi, S.H.; Yan, R.; Drager, N.; Leng, K.; Nalls, M.A.; Singleton, A.B.; Xu, K.; et al. Genome-wide CRISPRi/a screens in human neurons link lysosomal failure to ferroptosis. Nat. Neurosci. 2021, 24, 1020–1034. [Google Scholar] [CrossRef]
- Camera, E.; Picardo, M. Analytical methods to investigate glutathione and related compounds in biological and pathological processes. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2002, 781, 181–206. [Google Scholar] [CrossRef]
- Owen, J.B.; Butterfield, D.A. Measurement of oxidized/reduced glutathione ratio. Methods Mol. Biol. 2010, 648, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Dooley, C.T.; Dore, T.M.; Hanson, G.T.; Jackson, W.C.; Remington, S.J.; Tsien, R.Y. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J. Biol. Chem. 2004, 279, 22284–22293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, A.J.; Dick, T.P. Fluorescent protein-based redox probes. Antioxid. Redox Signal. 2010, 13, 621–650. [Google Scholar] [CrossRef] [PubMed]
- Pap, E.H.; Drummen, G.P.; Winter, V.J.; Kooij, T.W.; Rijken, P.; Wirtz, K.W.; Op den Kamp, J.A.; Hage, W.J.; Post, J.A. Ratio-fluorescence microscopy of lipid oxidation in living cells using C11-BODIPY(581/591). FEBS Lett. 1999, 453, 278–282. [Google Scholar] [CrossRef] [Green Version]
- Dahle, L.K.; Hill, E.G.; Holman, R.T. The thiobarbituric acid reaction and the autoxidations of polyunsaturated fatty acid methyl esters. Arch. Biochem. Biophys. 1962, 98, 253–261. [Google Scholar] [CrossRef]
- Tang, Q.; Su, Y.W.; Xian, C.J. Determining Oxidative Damage by Lipid Peroxidation Assay in Rat Serum. Bio Protoc. 2019, 9, e3263. [Google Scholar] [CrossRef]
- Lajoie, P.; Fazio, E.N.; Snapp, E.L. Approaches to imaging unfolded secretory protein stress in living cells. Endoplasmic Reticulum Stress Dis. 2014, 1, 27–39. [Google Scholar] [CrossRef]
- Beriault, D.R.; Werstuck, G.H. Detection and quantification of endoplasmic reticulum stress in living cells using the fluorescent compound, Thioflavin T. Biochim. Biophys. Acta 2013, 1833, 2293–2301. [Google Scholar] [CrossRef] [Green Version]
- Iwawaki, T.; Akai, R.; Kohno, K.; Miura, M. A transgenic mouse model for monitoring endoplasmic reticulum stress. Nat. Med. 2004, 10, 98–102. [Google Scholar] [CrossRef]
- Samali, A.; Fitzgerald, U.; Deegan, S.; Gupta, S. Methods for monitoring endoplasmic reticulum stress and the unfolded protein response. Int. J. Cell Biol. 2010, 2010, 830307. [Google Scholar] [CrossRef]
- Nadanaka, S.; Yoshida, H.; Kano, F.; Murata, M.; Mori, K. Activation of mammalian unfolded protein response is compatible with the quality control system operating in the endoplasmic reticulum. Mol. Biol. Cell 2004, 15, 2537–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, A.; Hall, M.N. Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. 2017, 36, 397–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, A.; Hall, M.N.; Lin, S.C.; Hardie, D.G. AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef]
- Vlahakis, A.; Graef, M.; Nunnari, J.; Powers, T. TOR complex 2-Ypk1 signaling is an essential positive regulator of the general amino acid control response and autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 10586–10591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stracka, D.; Jozefczuk, S.; Rudroff, F.; Sauer, U.; Hall, M.N. Nitrogen source activates TOR (target of rapamycin) complex 1 via glutamine and independently of Gtr/Rag proteins. J. Biol. Chem. 2014, 289, 25010–25020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binda, M.; Peli-Gulli, M.P.; Bonfils, G.; Panchaud, N.; Urban, J.; Sturgill, T.W.; Loewith, R.; De Virgilio, C. The Vam6 GEF controls TORC1 by activating the EGO complex. Mol. Cell 2009, 35, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Panchaud, N.; Peli-Gulli, M.P.; De Virgilio, C. Amino acid deprivation inhibits TORC1 through a GTPase-activating protein complex for the Rag family GTPase Gtr1. Sci. Signal. 2013, 6, ra42. [Google Scholar] [CrossRef] [Green Version]
- Kamada, Y.; Funakoshi, T.; Shintani, T.; Nagano, K.; Ohsumi, M.; Ohsumi, Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 2000, 150, 1507–1513. [Google Scholar] [CrossRef] [Green Version]
- Kamada, Y.; Yoshino, K.; Kondo, C.; Kawamata, T.; Oshiro, N.; Yonezawa, K.; Ohsumi, Y. Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol. Cell. Biol. 2010, 30, 1049–1058. [Google Scholar] [CrossRef] [Green Version]
- Tate, J.J.; Georis, I.; Dubois, E.; Cooper, T.G. Distinct phosphatase requirements and GATA factor responses to nitrogen catabolite repression and rapamycin treatment in Saccharomyces cerevisiae. J. Biol. Chem. 2010, 285, 17880–17895. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela, L.; Aranda, C.; Gonzalez, A. TOR modulates GCN4-dependent expression of genes turned on by nitrogen limitation. J. Bacteriol. 2001, 183, 2331–2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, A.; Jin, M.; Xu, Z.; Klionsky, D.J. A large-scale analysis of autophagy-related gene expression identifies new regulators of autophagy. Autophagy 2015, 11, 2114–2122. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.F.; Bertram, P.G.; Ai, W.; Zheng, X.F. Regulation of APG14 expression by the GATA-type transcription factor Gln3p. J. Biol. Chem. 2001, 276, 6463–6467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.; Delorme-Axford, E.; Backues, S.K.; Klionsky, D.J. Atg41/Icy2 regulates autophagosome formation. Autophagy 2015, 11, 2288–2299. [Google Scholar] [CrossRef] [Green Version]
- Swinnen, E.; Ghillebert, R.; Wilms, T.; Winderickx, J. Molecular mechanisms linking the evolutionary conserved TORC1-Sch9 nutrient signalling branch to lifespan regulation in Saccharomyces cerevisiae. FEMS Yeast Res. 2014, 14, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Bernard, A.; Jin, M.; Gonzalez-Rodriguez, P.; Fullgrabe, J.; Delorme-Axford, E.; Backues, S.K.; Joseph, B.; Klionsky, D.J. Rph1/KDM4 mediates nutrient-limitation signaling that leads to the transcriptional induction of autophagy. Curr. Biol. 2015, 25, 546–555. [Google Scholar] [CrossRef] [Green Version]
- Bartholomew, C.R.; Suzuki, T.; Du, Z.; Backues, S.K.; Jin, M.; Lynch-Day, M.A.; Umekawa, M.; Kamath, A.; Zhao, M.; Xie, Z.; et al. Ume6 transcription factor is part of a signaling cascade that regulates autophagy. Proc. Natl. Acad. Sci. USA 2012, 109, 11206–11210. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; McQuiston, T.; Bernard, A.; Park, Y.D.; Qiu, J.; Vural, A.; Zhang, N.; Waterman, S.R.; Blewett, N.H.; Myers, T.G.; et al. A conserved mechanism of TOR-dependent RCK-mediated mRNA degradation regulates autophagy. Nat. Cell Biol. 2015, 17, 930–942. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.; He, D.; Backues, S.K.; Freeberg, M.A.; Liu, X.; Kim, J.K.; Klionsky, D.J. Transcriptional regulation by Pho23 modulates the frequency of autophagosome formation. Curr. Biol. 2014, 24, 1314–1322. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Imamura, Y.; Ueno, M.; Suzuki, S.W.; Ohsumi, Y.; Yukawa, M.; Tsuchiya, E. The yeast chromatin remodeler Rsc1-RSC complex is required for transcriptional activation of autophagy-related genes and inhibition of the TORC1 pathway in response to nitrogen starvation. Biochem. Biophys. Res. Commun. 2015, 464, 1248–1253. [Google Scholar] [CrossRef]
- Wen, X.; Gatica, D.; Yin, Z.; Hu, Z.; Dengjel, J.; Klionsky, D.J. The transcription factor Spt4-Spt5 complex regulates the expression of ATG8 and ATG41. Autophagy 2020, 16, 1172–1185. [Google Scholar] [CrossRef]
- Delorme-Axford, E.; Abernathy, E.; Lennemann, N.J.; Bernard, A.; Ariosa, A.; Coyne, C.B.; Kirkegaard, K.; Klionsky, D.J. The exoribonuclease Xrn1 is a post-transcriptional negative regulator of autophagy. Autophagy 2018, 14, 898–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Yao, Z.; Jin, M.; Namkoong, S.; Yin, Z.; Lee, J.H.; Klionsky, D.J. Dhh1 promotes autophagy-related protein translation during nitrogen starvation. PLoS Biol. 2019, 17, e3000219. [Google Scholar] [CrossRef]
- Gatica, D.; Hu, G.; Liu, X.; Zhang, N.; Williamson, P.R.; Klionsky, D.J. The Pat1-Lsm Complex Stabilizes ATG mRNA during Nitrogen Starvation-Induced Autophagy. Mol. Cell 2019, 73, 314–324.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Z.; Liu, X.; Ariosa, A.; Huang, H.; Jin, M.; Karbstein, K.; Klionsky, D.J. Psp2, a novel regulator of autophagy that promotes autophagy-related protein translation. Cell Res. 2019, 29, 994–1008. [Google Scholar] [CrossRef]
- Lahiri, V.; Metur, S.P.; Hu, Z.; Song, X.; Mari, M.; Hawkins, W.D.; Bhattarai, J.; Delorme-Axford, E.; Reggiori, F.; Tang, D.; et al. Post-transcriptional regulation of ATG1 is a critical node that modulates autophagy during distinct nutrient stresses. Autophagy 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, C.; Tan, L.J.; Mochida, K.; Kirisako, H.; Koizumi, M.; Asai, E.; Sakoh-Nakatogawa, M.; Ohsumi, Y.; Nakatogawa, H. Hrr25 triggers selective autophagy-related pathways by phosphorylating receptor proteins. J. Cell Biol. 2014, 207, 91–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Ariosa, A.R.; Yang, Y.; Hu, Z.; Dengjel, J.; Klionsky, D.J. Downregulation of autophagy by Met30-mediated Atg9 ubiquitination. Proc. Natl. Acad. Sci. USA 2021, 118, e2005539118. [Google Scholar] [CrossRef]
- Yi, C.; Ma, M.; Ran, L.; Zheng, J.; Tong, J.; Zhu, J.; Ma, C.; Sun, Y.; Zhang, S.; Feng, W.; et al. Function and molecular mechanism of acetylation in autophagy regulation. Science 2012, 336, 474–477. [Google Scholar] [CrossRef]
- Fullgrabe, J.; Lynch-Day, M.A.; Heldring, N.; Li, W.; Struijk, R.B.; Ma, Q.; Hermanson, O.; Rosenfeld, M.G.; Klionsky, D.J.; Joseph, B. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature 2013, 500, 468–471. [Google Scholar] [CrossRef]
- Liu, X.; Jin, M.; Yao, Z.; Bernard, A.; Klionsky, D.J. Bidirectional roles of Dhh1 in regulating autophagy. Autophagy 2019, 15, 1838–1839. [Google Scholar] [CrossRef] [PubMed]
- Noda, T. Regulation of Autophagy through TORC1 and mTORC1. Biomolecules 2017, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, A.; Wallace, R.L.; Wellington, R.; Luo, X.; Capaldi, A.P. Multilayered regulation of TORC1-body formation in budding yeast. Mol. Biol. Cell 2019, 30, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Ukai, H.; Araki, Y.; Kira, S.; Oikawa, Y.; May, A.I.; Noda, T. Gtr/Ego-independent TORC1 activation is achieved through a glutamine-sensitive interaction with Pib2 on the vacuolar membrane. PLoS Genet. 2018, 14, e1007334. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, R.; Peli-Gulli, M.P.; Hu, Z.; Jaquenoud, M.; Garcia Osuna, G.M.; Sardu, A.; Dengjel, J.; De Virgilio, C. Spatially Distinct Pools of TORC1 Balance Protein Homeostasis. Mol. Cell 2019, 73, 325–338.e8. [Google Scholar] [CrossRef] [Green Version]
- McDaniel, S.L.; Hepperla, A.J.; Huang, J.; Dronamraju, R.; Adams, A.T.; Kulkarni, V.G.; Davis, I.J.; Strahl, B.D. H3K36 Methylation Regulates Nutrient Stress Response in Saccharomyces cerevisiae by Enforcing Transcriptional Fidelity. Cell Rep. 2017, 19, 2371–2382. [Google Scholar] [CrossRef] [Green Version]
- Bar-Peled, L.; Sabatini, D.M. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014, 24, 400–406. [Google Scholar] [CrossRef]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [Green Version]
- Powis, K.; De Virgilio, C. Conserved regulators of Rag GTPases orchestrate amino acid-dependent TORC1 signaling. Cell Discov. 2016, 2, 15049. [Google Scholar] [CrossRef] [Green Version]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Peled, L.; Schweitzer, L.D.; Zoncu, R.; Sabatini, D.M. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 2012, 150, 1196–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, K.; Choe, A.; Sabatini, D.M. Intersubunit Crosstalk in the Rag GTPase Heterodimer Enables mTORC1 to Respond Rapidly to Amino Acid Availability. Mol. Cell 2017, 68, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef] [Green Version]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Demetriades, C.; Plescher, M.; Teleman, A.A. Lysosomal recruitment of TSC2 is a universal response to cellular stress. Nat. Commun. 2016, 7, 10662. [Google Scholar] [CrossRef]
- Yao, Y.; Hong, S.; Ikeda, T.; Mori, H.; MacDougald, O.A.; Nada, S.; Okada, M.; Inoki, K. Amino Acids Enhance Polyubiquitination of Rheb and Its Binding to mTORC1 by Blocking Lysosomal ATXN3 Deubiquitinase Activity. Mol. Cell 2020, 80, 437–451.e6. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Chan, E.Y. mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci. Signal. 2009, 2, pe51. [Google Scholar] [CrossRef]
- Dunlop, E.A.; Hunt, D.K.; Acosta-Jaquez, H.A.; Fingar, D.C.; Tee, A.R. ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy 2011, 7, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Seo, M.; Otto, N.M.; Kim, D.H. ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy 2011, 7, 1212–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.X.; Russell, R.C.; Guan, K.L. Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy 2013, 9, 1983–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Zhang, S.; He, L.; Rong, Y.; Brier, L.W.; Sun, Q.; Liu, R.; Fan, W.; Chen, S.; Yue, Z.; et al. MTORC1-mediated NRBF2 phosphorylation functions as a switch for the class III PtdIns3K and autophagy. Autophagy 2017, 13, 592–607. [Google Scholar] [CrossRef] [Green Version]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Jung, C.H.; Seo, M.; Kim, E.K.; Park, J.M.; Bae, S.S.; Kim, D.H. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol. Cell 2015, 57, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Wan, W.; You, Z.; Zhou, L.; Xu, Y.; Peng, C.; Zhou, T.; Yi, C.; Shi, Y.; Liu, W. mTORC1-Regulated and HUWE1-Mediated WIPI2 Degradation Controls Autophagy Flux. Mol. Cell 2018, 72, 303–315.e6. [Google Scholar] [CrossRef] [Green Version]
- Wan, W.; You, Z.; Xu, Y.; Zhou, L.; Guan, Z.; Peng, C.; Wong, C.C.L.; Su, H.; Zhou, T.; Xia, H.; et al. mTORC1 Phosphorylates Acetyltransferase p300 to Regulate Autophagy and Lipogenesis. Mol. Cell 2017, 68, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.H.; Finkel, T. Regulation of autophagy by the p300 acetyltransferase. J. Biol. Chem. 2009, 284, 6322–6328. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [Green Version]
- Martina, J.A.; Puertollano, R. Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J. Cell Biol. 2013, 200, 475–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega-Rubin-de-Celis, S.; Pena-Llopis, S.; Konda, M.; Brugarolas, J. Multistep regulation of TFEB by MTORC1. Autophagy 2017, 13, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, G.; Esposito, A.; Choi, H.; Matarese, M.; Benedetti, V.; Di Malta, C.; Monfregola, J.; Medina, D.L.; Lippincott-Schwartz, J.; Ballabio, A. mTOR-dependent phosphorylation controls TFEB nuclear export. Nat. Commun. 2018, 9, 3312. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, Y.; Liu, J.; Zhang, J.; Xu, M.; You, Z.; Peng, C.; Gong, Z.; Liu, W. Acetyltransferase GCN5 regulates autophagy and lysosome biogenesis by targeting TFEB. EMBO Rep. 2020, 21, e48335. [Google Scholar] [CrossRef] [PubMed]
- Martina, J.A.; Diab, H.I.; Lishu, L.; Jeong, A.L.; Patange, S.; Raben, N.; Puertollano, R. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci. Signal. 2014, 7, ra9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ploper, D.; Taelman, V.F.; Robert, L.; Perez, B.S.; Titz, B.; Chen, H.W.; Graeber, T.G.; von Euw, E.; Ribas, A.; De Robertis, E.M. MITF drives endolysosomal biogenesis and potentiates Wnt signaling in melanoma cells. Proc. Natl. Acad. Sci. USA 2015, 112, E420–E429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller, K.; Sigurbjornsdottir, S.; Arnthorsson, A.O.; Pogenberg, V.; Dilshat, R.; Fock, V.; Brynjolfsdottir, S.H.; Bindesboll, C.; Bessadottir, M.; Ogmundsdottir, H.M.; et al. MITF has a central role in regulating starvation-induced autophagy in melanoma. Sci. Rep. 2019, 9, 1055. [Google Scholar] [CrossRef] [Green Version]
- Di Malta, C.; Siciliano, D.; Calcagni, A.; Monfregola, J.; Punzi, S.; Pastore, N.; Eastes, A.N.; Davis, O.; De Cegli, R.; Zampelli, A.; et al. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science 2017, 356, 1188–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wengrod, J.; Wang, D.; Weiss, S.; Zhong, H.; Osman, I.; Gardner, L.B. Phosphorylation of eIF2alpha triggered by mTORC1 inhibition and PP6C activation is required for autophagy and is aberrant in PP6C-mutated melanoma. Sci. Signal. 2015, 8, ra27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.W.; Weng, J.H.; Lee, W.X.; Hu, Y.; Gu, L.; Cho, S.; Lee, G.; Binari, R.; Li, C.; Cheng, M.E.; et al. mTORC1-chaperonin CCT signaling regulates m(6)A RNA methylation to suppress autophagy. Proc. Natl. Acad. Sci. USA 2021, 118, e2021945118. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Kim, K.I. Epigenetic Control of Autophagy: Nuclear Events Gain More Attention. Mol. Cell 2017, 65, 781–785. [Google Scholar] [CrossRef] [Green Version]
- Bowman, C.J.; Ayer, D.E.; Dynlacht, B.D. Foxk proteins repress the initiation of starvation-induced atrophy and autophagy programs. Nat. Cell Biol. 2014, 16, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; An, Z.; Zou, Z.; Sumpter, R.; Su, M.; Zang, X.; Sinha, S.; Gaestel, M.; Levine, B. The stress-responsive kinases MAPKAPK2/MAPKAPK3 activate starvation-induced autophagy through Beclin 1 phosphorylation. Elife 2015, 4, e05289. [Google Scholar] [CrossRef]
- Comb, W.C.; Cogswell, P.; Sitcheran, R.; Baldwin, A.S. IKK-dependent, NF-kappaB-independent control of autophagic gene expression. Oncogene 2011, 30, 1727–1732. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Carling, D.; Halford, N. Roles of the Snf1/Rkin1/AMP-activated protein kinase family in the response to environmental and nutritional stress. Semin. Cell Biol. 1994, 5, 409–416. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawley, S.A.; Davison, M.; Woods, A.; Davies, S.P.; Beri, R.K.; Carling, D.; Hardie, D.G. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 1996, 271, 27879–27887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, B.; Heath, R.; Saiu, P.; Leiper, F.C.; Leone, P.; Jing, C.; Walker, P.A.; Haire, L.; Eccleston, J.F.; Davis, C.T.; et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 2007, 449, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Scott, J.W.; Pan, D.A.; Hudson, E.R. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003, 546, 113–120. [Google Scholar] [CrossRef]
- Hawley, S.A.; Selbert, M.A.; Goldstein, E.G.; Edelman, A.M.; Carling, D.; Hardie, D.G. 5′-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J. Biol. Chem. 1995, 270, 27186–27191. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.P.; Helps, N.R.; Cohen, P.T.; Hardie, D.G. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett. 1995, 377, 421–425. [Google Scholar] [CrossRef] [Green Version]
- Gowans, G.J.; Hawley, S.A.; Ross, F.A.; Hardie, D.G. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 2013, 18, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weerasekara, V.K.; Panek, D.J.; Broadbent, D.G.; Mortenson, J.B.; Mathis, A.D.; Logan, G.N.; Prince, J.T.; Thomson, D.M.; Thompson, J.W.; Andersen, J.L. Metabolic-stress-induced rearrangement of the 14-3-3zeta interactome promotes autophagy via a ULK1- and AMPK-regulated 14-3-3zeta interaction with phosphorylated Atg9. Mol. Cell. Biol. 2014, 34, 4379–4388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, E.L.; Oskoui, P.R.; Banko, M.R.; Maniar, J.M.; Gygi, M.P.; Gygi, S.P.; Brunet, A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 2007, 282, 30107–30119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamargo-Gomez, I.; Marino, G. AMPK: Regulation of Metabolic Dynamics in the Context of Autophagy. Int. J. Mol. Sci. 2018, 19, 3812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.; Su, H.; Zhang, D.; Wang, Y.; Shen, Q.; Liu, B.; Huang, R.; Zhou, T.; Peng, C.; Wong, C.C.; et al. AMPK-Dependent Phosphorylation of GAPDH Triggers Sirt1 Activation and Is Necessary for Autophagy upon Glucose Starvation. Mol. Cell 2015, 60, 930–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [Green Version]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; Depinho, R.A.; Sadoshima, J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Chen, H.; Zhang, L.; Lin, X.; Li, X.; Zhuang, H.; Fan, H.; Meng, T.; He, Z.; Huang, H.; et al. The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy 2021, 17, 1142–1156. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Q. Oxidative Stress and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 179–198. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiffin, R.; Bandyopadhyay, U.; Cuervo, A.M. Oxidative stress and autophagy. Antioxid. Redox Signal. 2006, 8, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008, 15, 171–182. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccafico, S.; Mammucari, C.; Boncompagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; Del Prete, Z.; et al. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 2008, 8, 425–436. [Google Scholar] [CrossRef]
- Park, E.; Chung, S.W. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019, 10, 822. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Elazar, Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007, 17, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef] [Green Version]
- Hayyan, M.; Hashim, M.A.; AlNashef, I.M. Superoxide Ion: Generation and Chemical Implications. Chem. Rev. 2016, 116, 3029–3085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groeger, G.; Quiney, C.; Cotter, T.G. Hydrogen peroxide as a cell-survival signaling molecule. Antioxid. Redox Signal. 2009, 11, 2655–2671. [Google Scholar] [CrossRef]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, R.V.; Hanna, P.M.; Mason, R.P. The origin of the hydroxyl radical oxygen in the Fenton reaction. Free Radic. Biol. Med. 1997, 22, 885–888. [Google Scholar] [CrossRef]
- Halliwell, B.; Chirico, S. Lipid peroxidation: Its mechanism, measurement, and significance. Am. J. Clin. Nutr. 1993, 57, 715S–724S, discussion 724S—725S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Kong, X.; Kang, J.; Su, J.; Li, Y.; Zhong, J.; Sun, L. Oxidative stress induces parallel autophagy and mitochondria dysfunction in human glioma U251 cells. Toxicol. Sci. 2009, 110, 376–388. [Google Scholar] [CrossRef]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.H.; Sohn, S.; Kwon, H.J.; Kim, S.U.; Kim, M.J.; Lee, S.J.; Choi, K.S. Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res. 2007, 67, 6314–6324. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.W.; Ni, B.B.; Li, B.; Yang, Y.H.; Jiang, S.D.; Jiang, L.S. The responses of autophagy and apoptosis to oxidative stress in nucleus pulposus cells: Implications for disc degeneration. Cell. Physiol. Biochem. 2014, 34, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Fan, S.M.; Song, J.K.; Tashiro, S.; Onodera, S.; Ikejima, T. Hydroxyl radical (·OH) played a pivotal role in oridonin-induced apoptosis and autophagy in human epidermoid carcinoma A431 cells. Biol. Pharm. Bull. 2012, 35, 2148–2159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barsoum, M.J.; Yuan, H.; Gerencser, A.A.; Liot, G.; Kushnareva, Y.; Gräber, S.; Kovacs, I.; Lee, W.D.; Waggoner, J.; Cui, J.; et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006, 25, 3900–3911. [Google Scholar] [CrossRef] [PubMed]
- Liot, G.; Bossy, B.; Lubitz, S.; Kushnareva, Y.; Sejbuk, N.; Bossy-Wetzel, E. Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA- and ROS-dependent pathway. Cell Death Differ. 2009, 16, 899–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, N.; Vacek, J.C.; Givvimani, S.; Sen, U.; Tyagi, S.C. Cardiac specific deletion of N-methyl-d-aspartate receptor 1 ameliorates mtMMP-9 mediated autophagy/mitophagy in hyperhomocysteinemia. J. Recept. Signal Transduct. Res. 2010, 30, 78–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, M.; Wani, W.Y.; Redmann, M.; Benavides, G.A.; Johnson, M.S.; Ouyang, X.; Cofield, S.S.; Mitra, K.; Darley-Usmar, V.; Zhang, J. Regulation of autophagy, mitochondrial dynamics, and cellular bioenergetics by 4-hydroxynonenal in primary neurons. Autophagy 2017, 13, 1828–1840. [Google Scholar] [CrossRef] [Green Version]
- De Gaetano, A.; Gibellini, L.; Zanini, G.; Nasi, M.; Cossarizza, A.; Pinti, M. Mitophagy and Oxidative Stress: The Role of Aging. Antioxidants 2021, 10, 794. [Google Scholar] [CrossRef]
- Deffieu, M.; Bhatia-Kissová, I.; Salin, B.; Galinier, A.; Manon, S.; Camougrand, N. Glutathione participates in the regulation of mitophagy in yeast. J. Biol. Chem. 2009, 284, 14828–14837. [Google Scholar] [CrossRef] [Green Version]
- Jing, H.; Liu, H.; Zhang, L.; Gao, J.; Song, H.; Tan, X. Ethanol Induces Autophagy Regulated by Mitochondrial ROS in Saccharomyces cerevisiae. J. Microbiol. Biotechnol. 2018, 28, 1982–1991. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues-Pousada, C.; Devaux, F.; Caetano, S.M.; Pimentel, C.; da Silva, S.; Cordeiro, A.C.; Amaral, C. Yeast AP-1 like transcription factors (Yap) and stress response: A current overview. Microb. Cell 2019, 6, 267–285. [Google Scholar] [CrossRef]
- Toledano, M.B.; Delaunay, A.; Monceau, L.; Tacnet, F. Microbial H2O2 sensors as archetypical redox signaling modules. Trends Biochem. Sci. 2004, 29, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Harshman, K.D.; Moye-Rowley, W.S.; Parker, C.S. Transcriptional activation by the SV40 AP-1 recognition element in yeast is mediated by a factor similar to AP-1 that is distinct from GCN4. Cell 1988, 53, 321–330. [Google Scholar] [CrossRef]
- Fernandes, L.; Rodrigues-Pousada, C.; Struhl, K. Yap, a novel family of eight bZIP proteins in Saccharomyces cerevisiae with distinct biological functions. Mol. Cell. Biol. 1997, 17, 6982–6993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuge, S.; Jones, N. YAP1 dependent activation of TRX2 is essential for the response of Saccharomyces cerevisiae to oxidative stress by hydroperoxides. EMBO J. 1994, 13, 655–664. [Google Scholar] [CrossRef]
- Kuge, S.; Jones, N.; Nomoto, A. Regulation of yAP-1 nuclear localization in response to oxidative stress. EMBO J. 1997, 16, 1710–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaunay, A.; Isnard, A.D.; Toledano, M.B. H2O2 sensing through oxidation of the Yap1 transcription factor. EMBO J. 2000, 19, 5157–5166. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, S.; Tachibana, T.; Naganuma, A.; Mano, N.; Kuge, S. Multistep disulfide bond formation in Yap1 is required for sensing and transduction of H2O2 stress signal. Mol. Cell 2007, 27, 675–688. [Google Scholar] [CrossRef]
- Delaunay, A.; Pflieger, D.; Barrault, M.B.; Vinh, J.; Toledano, M.B. A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell 2002, 111, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Maeda, Y.; Oku, M.; Sakai, Y. A defect of the vacuolar putative lipase Atg15 accelerates degradation of lipid droplets through lipolysis. Autophagy 2015, 11, 1247–1258. [Google Scholar] [CrossRef] [Green Version]
- Ramya, V.; Rajasekharan, R. ATG15 encodes a phospholipase and is transcriptionally regulated by YAP1 in Saccharomyces cerevisiae. FEBS Lett. 2016, 590, 3155–3167. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, M.E.; Zaffagnini, M.; Marchand, C.H.; Crespo, J.L.; Lemaire, S.D. The yeast autophagy protease Atg4 is regulated by thioredoxin. Autophagy 2014, 10, 1953–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherz-Shouval, R.; Elazar, Z. Monitoring starvation-induced reactive oxygen species formation. Methods Enzymol. 2009, 452, 119–130. [Google Scholar] [CrossRef]
- Mungai, P.T.; Waypa, G.B.; Jairaman, A.; Prakriya, M.; Dokic, D.; Ball, M.K.; Schumacker, P.T. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol. Cell. Biol. 2011, 31, 3531–3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djavaheri-Mergny, M.; Amelotti, M.; Mathieu, J.; Besançon, F.; Bauvy, C.; Souquère, S.; Pierron, G.; Codogno, P. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J. Biol. Chem. 2006, 281, 30373–30382. [Google Scholar] [CrossRef] [Green Version]
- Djavaheri-Mergny, M.; Javelaud, D.; Wietzerbin, J.; Besançon, F. NF-kappaB activation prevents apoptotic oxidative stress via an increase of both thioredoxin and MnSOD levels in TNFalpha-treated Ewing sarcoma cells. FEBS Lett. 2004, 578, 111–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [Green Version]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [Green Version]
- Alexander, A.; Kim, J.; Walker, C.L. ATM engages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy 2010, 6, 672–673. [Google Scholar] [CrossRef] [Green Version]
- Cirotti, C.; Rizza, S.; Giglio, P.; Poerio, N.; Allega, M.F.; Claps, G.; Pecorari, C.; Lee, J.H.; Benassi, B.; Barilà, D.; et al. Redox activation of ATM enhances GSNOR translation to sustain mitophagy and tolerance to oxidative stress. EMBO Rep. 2021, 22, e50500. [Google Scholar] [CrossRef] [PubMed]
- Dames, S.A.; Mulet, J.M.; Rathgeb-Szabo, K.; Hall, M.N.; Grzesiek, S. The solution structure of the FATC domain of the protein kinase target of rapamycin suggests a role for redox-dependent structural and cellular stability. J. Biol. Chem. 2005, 280, 20558–20564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensaad, K.; Cheung, E.C.; Vousden, K.H. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009, 28, 3015–3026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClung, J.M.; Judge, A.R.; Powers, S.K.; Yan, Z. p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am. J. Physiol. Cell Physiol. 2010, 298, C542–C549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajares, M.; Cuadrado, A.; Engedal, N.; Jirsova, Z.; Cahova, M. The Role of Free Radicals in Autophagy Regulation: Implications for Ageing. Oxid. Med. Cell. Longev. 2018, 2018, 2450748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madden, S.K.; Itzhaki, L.S. Structural and mechanistic insights into the Keap1-Nrf2 system as a route to drug discovery. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140405. [Google Scholar] [CrossRef]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Fuse, Y.; Kobayashi, M. Conservation of the Keap1-Nrf2 System: An Evolutionary Journey through Stressful Space and Time. Molecules 2017, 22, 436. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, S.C.; Li, X.; Henzl, M.T.; Beamer, L.J.; Hannink, M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006, 25, 3605–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, M.; Xiong, Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell. Biol. 2005, 25, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Muramatsu, A.; Saito, R.; Iso, T.; Shibata, T.; Kuwata, K.; Kawaguchi, S.I.; Iwawaki, T.; Adachi, S.; Suda, H.; et al. Molecular Mechanism of Cellular Oxidative Stress Sensing by Keap1. Cell Rep. 2019, 28, 746–758.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hourihan, J.M.; Kenna, J.G.; Hayes, J.D. The gasotransmitter hydrogen sulfide induces nrf2-target genes by inactivating the keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between cys-226 and cys-613. Antioxid. Redox Signal. 2013, 19, 465–481. [Google Scholar] [CrossRef]
- Fourquet, S.; Guerois, R.; Biard, D.; Toledano, M.B. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J. Biol. Chem. 2010, 285, 8463–8471. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [Green Version]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copple, I.M.; Lister, A.; Obeng, A.D.; Kitteringham, N.R.; Jenkins, R.E.; Layfield, R.; Foster, B.J.; Goldring, C.E.; Park, B.K. Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J. Biol. Chem. 2010, 285, 16782–16788. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Tang, Z.; Chen, D.; Moughon, D.; Ding, X.; Chen, S.; Zhu, M.; Zhong, Q. Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy 2010, 6, 614–621. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, Á.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef] [Green Version]
- Pajares, M.; Rojo, A.I.; Arias, E.; Díaz-Carretero, A.; Cuervo, A.M.; Cuadrado, A. Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 2018, 14, 1310–1322. [Google Scholar] [CrossRef]
- Meng, F.; Yao, D.; Shi, Y.; Kabakoff, J.; Wu, W.; Reicher, J.; Ma, Y.; Moosmann, B.; Masliah, E.; Lipton, S.A.; et al. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol. Neurodegener. 2011, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Vandiver, M.S.; Paul, B.D.; Xu, R.; Karuppagounder, S.; Rao, F.; Snowman, A.M.; Ko, H.S.; Lee, Y.I.; Dawson, V.L.; Dawson, T.M.; et al. Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 2013, 4, 1626. [Google Scholar] [CrossRef] [Green Version]
- Astuti, R.I.; Nasuno, R.; Takagi, H. Nitric oxide signaling in yeast. Appl. Microbiol. Biotechnol. 2016, 100, 9483–9497. [Google Scholar] [CrossRef] [PubMed]
- Tillmann, A.; Gow, N.A.; Brown, A.J. Nitric oxide and nitrosative stress tolerance in yeast. Biochem. Soc. Trans. 2011, 39, 219–223. [Google Scholar] [CrossRef]
- Shahani, N.; Sawa, A. Nitric oxide signaling and nitrosative stress in neurons: Role for S-nitrosylation. Antioxid. Redox Signal. 2011, 14, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Sueta, G.; Campolo, N.; Trujillo, M.; Bartesaghi, S.; Carballal, S.; Romero, N.; Alvarez, B.; Radi, R. Biochemistry of Peroxynitrite and Protein Tyrosine Nitration. Chem. Rev. 2018, 118, 1338–1408. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Protein tyrosine nitration: Biochemical mechanisms and structural basis of functional effects. Acc. Chem. Res. 2013, 46, 550–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Yuan, Q.; Chen, F.; Pang, J.; Pan, C.; Xu, F.; Chen, Y. Fundamental Mechanisms of the Cell Death Caused by Nitrosative Stress. Front. Cell Dev. Biol. 2021, 9, 742483. [Google Scholar] [CrossRef]
- Nakamura, T.; Tu, S.; Akhtar, M.W.; Sunico, C.R.; Okamoto, S.; Lipton, S.A. Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron 2013, 78, 596–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seneviratne, U.; Nott, A.; Bhat, V.B.; Ravindra, K.C.; Wishnok, J.S.; Tsai, L.H.; Tannenbaum, S.R. S-nitrosation of proteins relevant to Alzheimer’s disease during early stages of neurodegeneration. Proc. Natl. Acad. Sci. USA 2016, 113, 4152–4157. [Google Scholar] [CrossRef] [Green Version]
- Han, F.; Chen, Y.X.; Lu, Y.M.; Huang, J.Y.; Zhang, G.S.; Tao, R.R.; Ji, Y.L.; Liao, M.H.; Fukunaga, K.; Qin, Z.H. Regulation of the ischemia-induced autophagy-lysosome processes by nitrosative stress in endothelial cells. J. Pineal Res. 2011, 51, 124–135. [Google Scholar] [CrossRef]
- Tripathi, D.N.; Chowdhury, R.; Trudel, L.J.; Tee, A.R.; Slack, R.S.; Walker, C.L.; Wogan, G.N. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc. Natl. Acad. Sci. USA 2013, 110, E2950–E2957. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.L.; Ma, X.; Kong, F.J.; Guo, J.J.; Shi, H.T.; Zhu, J.B.; Zou, Y.Z.; Ge, J.B. Mammalian target of rapamycin inhibition attenuates myocardial ischaemia-reperfusion injury in hypertrophic heart. J. Cell. Mol. Med. 2018, 22, 1708–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S.; Datta, S.; Ghosh, S. Induction of autophagy under nitrosative stress: A complex regulatory interplay between SIRT1 and AMPK in MCF7 cells. Cell. Signal. 2019, 64, 109411. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.R.; Claude, A.; Fullam, E.F. A Study of Tissue Culture Cells by Electron Microscopy: Methods and Preliminary Observations. J. Exp. Med. 1945, 81, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Hebert, D.N.; Molinari, M. In and out of the ER: Protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007, 87, 1377–1408. [Google Scholar] [CrossRef]
- Merksamer, P.I.; Trusina, A.; Papa, F.R. Real-time redox measurements during endoplasmic reticulum stress reveal interlinked protein folding functions. Cell 2008, 135, 933–947. [Google Scholar] [CrossRef] [Green Version]
- Fujita, E.; Kouroku, Y.; Isoai, A.; Kumagai, H.; Misutani, A.; Matsuda, C.; Hayashi, Y.K.; Momoi, T. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: Ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum. Mol. Genet. 2007, 16, 618–629. [Google Scholar] [CrossRef]
- Schroder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef]
- Werner, E.D.; Brodsky, J.L.; McCracken, A.A. Proteasome-dependent endoplasmic reticulum-associated protein degradation: An unconventional route to a familiar fate. Proc. Natl. Acad. Sci. USA 1996, 93, 13797–13801. [Google Scholar] [CrossRef] [Green Version]
- Yorimitsu, T.; Nair, U.; Yang, Z.; Klionsky, D.J. Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 2006, 281, 30299–30304. [Google Scholar] [CrossRef] [Green Version]
- Bernales, S.; McDonald, K.L.; Walter, P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006, 4, e423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welihinda, A.A.; Kaufman, R.J. The unfolded protein response pathway in Saccharomyces cerevisiae: Oligomerization and trans-phosphorylation of Ire1p (Ern1p) are required for kinase activation. J. Biol. Chem. 1996, 271, 18181–18187. [Google Scholar] [PubMed] [Green Version]
- Okamura, K.; Kimata, Y.; Higashio, H.; Tsuru, A.; Kohno, K. Dissociation of Kar2p/BiP from an ER sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem. Biophys. Res. Commun. 2000, 279, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.S.; Walter, P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 1996, 87, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Sidrauski, C.; Walter, P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 1997, 90, 1031–1039. [Google Scholar] [PubMed] [Green Version]
- Sidrauski, C.; Cox, J.S.; Walter, P. tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell 1996, 87, 405–413. [Google Scholar]
- Mori, K. Signalling pathways in the unfolded protein response: Development from yeast to mammals. J. Biochem. 2009, 146, 743–750. [Google Scholar] [CrossRef]
- Kawahara, T.; Yanagi, H.; Yura, T.; Mori, K. Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response. Mol. Biol. Cell 1997, 8, 1845–1862. [Google Scholar] [CrossRef] [Green Version]
- Patil, C.; Walter, P. Intracellular signaling from the endoplasmic reticulum to the nucleus: The unfolded protein response in yeast and mammals. Curr. Opin. Cell Biol. 2001, 13, 349–355. [Google Scholar] [CrossRef]
- Chakraborty, R.; Baek, J.H.; Bae, E.Y.; Kim, W.-Y.; Lee, S.Y.; Kim, M.G. Comparison and contrast of plant, yeast, and mammalian ER stress and UPR. Appl. Biol. Chem. 2016, 59, 337–347. [Google Scholar] [CrossRef]
- Reddy, R.K.; Mao, C.; Baumeister, P.; Austin, R.C.; Kaufman, R.J.; Lee, A.S. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: Role of ATP binding site in suppression of caspase-7 activation. J. Biol. Chem. 2003, 278, 20915–20924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, B.M.; Walter, P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 2011, 333, 1891–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 controls diverse cell type-and condition-specific transcriptional regulatory networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Margariti, A.; Li, H.; Chen, T.; Martin, D.; Vizcay-Barrena, G.; Alam, S.; Karamariti, E.; Xiao, Q.; Zampetaki, A.; Zhang, Z. XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J. Biol. Chem. 2013, 288, 859–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Lee, J.; Reno, C.M.; Sun, C.; Park, S.W.; Chung, J.; Lee, J.; Fisher, S.J.; White, M.F.; Biddinger, S.B. Regulation of glucose homeostasis through a XBP-1–FoxO1 interaction. Nat. Med. 2011, 17, 356–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, R.L.; Figueroa, A.; Court, F.A.; Thielen, P.; Molina, C.; Wirth, C.; Caballero, B.; Kiffin, R.; Segura-Aguilar, J.; Cuervo, A.M. Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Hum. Mol. Genet. 2012, 21, 2245–2262. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, X.; Cai, M.-Y.; Ma, K.; Yang, J.; Zhou, J.; Fu, W.; Wei, F.-Z.; Wang, L.; Xie, D. XBP-1u suppresses autophagy by promoting the degradation of FoxO1 in cancer cells. Cell Res. 2013, 23, 491–507. [Google Scholar] [CrossRef] [Green Version]
- Li, D.D.; Wang, L.L.; Deng, R.; Tang, J.; Shen, Y.; Guo, J.F.; Wang, Y.; Xia, L.P.; Feng, G.K.; Liu, Q.Q.; et al. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene 2009, 28, 886–898. [Google Scholar] [CrossRef] [Green Version]
- Meares, G.P.; Hughes, K.J.; Naatz, A.; Papa, F.R.; Urano, F.; Hansen, P.A.; Benveniste, E.N.; Corbett, J.A. IRE1-dependent activation of AMPK in response to nitric oxide. Mol. Cell. Biol. 2011, 31, 4286–4297. [Google Scholar] [CrossRef] [Green Version]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2α phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [Green Version]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rzymski, T.; Milani, M.; Pike, L.; Buffa, F.; Mellor, H.; Winchester, L.; Pires, I.; Hammond, E.; Ragoussis, I.; Harris, A. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 2010, 29, 4424–4435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avivar-Valderas, A.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Bardeesy, N.; Debnath, J.; Aguirre-Ghiso, J. Regulation of autophagy during ECM detachment is linked to a selective inhibition of mTORC1 by PERK. Oncogene 2013, 32, 4932–4940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brüning, A.; Rahmeh, M.; Friese, K. Nelfinavir and bortezomib inhibit mTOR activity via ATF4-mediated sestrin-2 regulation. Mol. Oncol. 2013, 7, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Yamamoto, K.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct. Funct. 2008, 33, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.-J.; Xiong, Z.; Lu, X.; Ye, M.; Han, X.; Jiang, R. ATF6 upregulates XBP1S and inhibits ER stress-mediated apoptosis in osteoarthritis cartilage. Cell. Signal. 2014, 26, 332–342. [Google Scholar] [CrossRef]
- Hirsch, I.; Weiwad, M.; Prell, E.; Ferrari, D.M. ERp29 deficiency affects sensitivity to apoptosis via impairment of the ATF6–CHOP pathway of stress response. Apoptosis 2014, 19, 801–815. [Google Scholar] [CrossRef]
- Yung, H.W.; Charnock-Jones, D.S.; Burton, G.J. Regulation of AKT phosphorylation at Ser473 and Thr308 by endoplasmic reticulum stress modulates substrate specificity in a severity dependent manner. PLoS ONE 2011, 6, e17894. [Google Scholar] [CrossRef]
- Kalvakolanu, D.V.; Gade, P. IFNG and autophagy: A critical role for the ER-stress mediator ATF6 in controlling bacterial infections. Autophagy 2012, 8, 1673–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gade, P.; Manjegowda, S.B.; Nallar, S.C.; Maachani, U.B.; Cross, A.S.; Kalvakolanu, D.V. Regulation of the death-associated protein kinase 1 expression and autophagy via ATF6 requires apoptosis signal-regulating kinase 1. Mol. Cell. Biol. 2014, 34, 4033–4048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Høyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-β, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Borodkina, A.V.; Shatrova, A.N.; Deryabin, P.I.; Griukova, A.A.; Abushik, P.A.; Antonov, S.M.; Nikolsky, N.N.; Burova, E.B. Calcium alterations signal either to senescence or to autophagy induction in stem cells upon oxidative stress. Aging 2016, 8, 3400–3418. [Google Scholar] [CrossRef] [Green Version]
- Simon, B.; Huart, A.-S.; Temmerman, K.; Vahokoski, J.; Mertens, H.D.; Komadina, D.; Hoffmann, J.-E.; Yumerefendi, H.; Svergun, D.I.; Kursula, P. Death-associated protein kinase activity is regulated by coupled calcium/calmodulin binding to two distinct sites. Structure 2016, 24, 851–861. [Google Scholar] [CrossRef] [Green Version]
- Gomes, L.R.; Menck, C.F.M.; Leandro, G.S. Autophagy Roles in the Modulation of DNA Repair Pathways. Int. J. Mol. Sci. 2017, 18, 2351. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Verma, S.; Seranova, E.; Sarkar, S.; Kumar, D. Selective Autophagy and Xenophagy in Infection and Disease. Front. Cell Dev. Biol. 2018, 6, 147. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Yao, Z.; Klionsky, D.J. How to control self-digestion: Transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015, 25, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Holczer, M.; Hajdu, B.; Lorincz, T.; Szarka, A.; Banhegyi, G.; Kapuy, O. Fine-tuning of AMPK-ULK1-mTORC1 regulatory triangle is crucial for autophagy oscillation. Sci. Rep. 2020, 10, 17803. [Google Scholar] [CrossRef]
- Ling, N.X.Y.; Kaczmarek, A.; Hoque, A.; Davie, E.; Ngoei, K.R.W.; Morrison, K.R.; Smiles, W.J.; Forte, G.M.; Wang, T.; Lie, S.; et al. mTORC1 directly inhibits AMPK to promote cell proliferation under nutrient stress. Nat. Metab. 2020, 2, 41–49. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Ghemrawi, R.; Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stress Type | Organism | Biomarkers | Detection | Reference |
|---|---|---|---|---|
| Nutrient stress | Yeast | TORC1 inactivation | Sch9 dephosphorylation | [27] |
| Mammalian EIF4EBP1 dephosphorylation in vitro | [28] | |||
| Mammals | MTORC1 inactivation | RPS6KB1 dephosphorylation | [29] | |
| EIF4EBP1 dephosphorylation | [29] | |||
| Energy stress | Yeast and mammals | Lower ATP: ADP/AMP ratio | Liquid chromatography to detect ATP, ADP and AMP level | [30] |
| ATP:ADP fluorescence reporter | [31,32] | |||
| Bioluminescent detection | [33] | |||
| Yeast | Snf1 activation | “SAMS” peptide phosphorylation | [34] | |
| Mammals | AMPK activation | AMPK phosphorylation | [35] | |
| Phosphorylation of downstream targets such as ACAC (acetyl-CoA carboxylase) | [35] | |||
| Oxidative stress | Yeast and Mammals | High level of ROS | Dichlorodihydrofluorescein fluorescence | [36] |
| PG1 or PC1 fluorescence | [37] | |||
| Calcein-acetoxymethylester (calcein-AM) fluorescence | [38] | |||
| CellROX dye | [39] | |||
| Increased GSSG:GSH ratio | High-performance liquid chromatography | [40] | ||
| Capillary electrophoresis | [40] | |||
| Bioluminescence | [41] | |||
| Genetically-encoded fluorescent sensors | [42,43] | |||
| Lipid peroxidation | Fluorescence shift of C11-BODIPY (581/591) | [44] | ||
| TBA-MDA assay | [45,46] | |||
| ER stress | Yeast | Misfolded protein accumulation | Kar2 sedimentation | [47] |
| UPR pathway activation | Transcription reporter containing a UPR element promoter driving fluorescent proteins | [47] | ||
| Ire1 clustering | [47] | |||
| Mammals | Protein aggregates | Thioflavin T (ThT) fluorescence | [48] | |
| UPR pathway activation | Spliced XBP1 mRNA detection using ER stress-activated indicator” (ERAI) construct | [49] | ||
| Upregulated expression of UPR target genes, including DDIT3 and HSPA5/GRP78 | [50] | |||
| ATF6 translocation | [51] |
| Type of Regulation | Regulatory Factors | Conditions | Effects (↑, Positive; ↓, Negative) | Target Genes or Proteins | Reference | |
|---|---|---|---|---|---|---|
| Transcriptional regulation | Pho23 | Nutrient-rich | ↓ | ATG1, 7, 8, 9, 12, 14, 29 | [69] | |
| Spt10 | Nutrient-rich | ↓ | ATG1, 7, 9,14, 32 | [62] | ||
| Fyv5 | Nutrient-rich and starvation | ↓ | ATG1, 7, 8, 9,14, 29, 32 | |||
| Sfl1 | Nutrient-rich | ↓ | ATG1, 7, 8, 9, 14, 29, 32 | |||
| Sko1 | Nutrient-rich and starvation | ↓ | ATG1, 7, 8, 32 | |||
| Zap1 | Nutrient-rich | ↓ | ATG1, 7, 8, 9, 14, 29, 32 | |||
| Swi5 | Nutrient-rich | ↑ | ATG7, 8, 9, 14, 29 | |||
| Rsc1 | Starvation | ↑ | ATG8 | [70] | ||
| Spt4/5 | Nutrient-rich | ↓ | ATG8, 41 | [71] | ||
| Starvation | ↑ | ATG41 | ||||
| Post-transcriptional regulation | Xrn1 | Nutrient-rich | ↓ | ATG1, 4, 5, 7, 8, 12, 14, 16, 29, 31 | [72] | |
| Dhh1 | Nutrient-rich | ↓ | ATG3, 7, 8, 19, 20, 22, 24 | [68] | ||
| Starvation | ↑ | ATG1,13 | [73] | |||
| Pat1 | Starvation | ↑ | ATG1, 2,7, 9 | [74] | ||
| Psp2 | Starvation | ↑ | ATG1, 13 | [75] | ||
| Ded1 | Starvation | ↑ | ATG1 | [76] | ||
| Post- translational regulation | Phosphorylation | Hrr25 | Cvt pathway and pexophagy induction | ↑ | Atg19,36 | [77] |
| Ubiquitination | Met30 | Nutrient-rich | ↓ | Atg9 | [78] | |
| Acetylation | Esa1 | Starvation | ↑ | Atg3 | [79] | |
| Deacetylation | Rpd3 | Starvation | ↓ | Atg3 | [79] | |
| Epigenetic regulation | Acetylation | Sas2 | Nutrient-rich | ↓ | Histone H4 Lys16 | [80] |
| Methylation | Unclear | Nutrient-rich | ↓ | Histone H3 Lys4 | [80] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, Y.; Huang, Y.; Wen, X.; Yin, Z.; Zhang, Z.; Klionsky, D.J. How Cells Deal with the Fluctuating Environment: Autophagy Regulation under Stress in Yeast and Mammalian Systems. Antioxidants 2022, 11, 304. https://doi.org/10.3390/antiox11020304
Lei Y, Huang Y, Wen X, Yin Z, Zhang Z, Klionsky DJ. How Cells Deal with the Fluctuating Environment: Autophagy Regulation under Stress in Yeast and Mammalian Systems. Antioxidants. 2022; 11(2):304. https://doi.org/10.3390/antiox11020304
Chicago/Turabian StyleLei, Yuchen, Yuxiang Huang, Xin Wen, Zhangyuan Yin, Zhihai Zhang, and Daniel J. Klionsky. 2022. "How Cells Deal with the Fluctuating Environment: Autophagy Regulation under Stress in Yeast and Mammalian Systems" Antioxidants 11, no. 2: 304. https://doi.org/10.3390/antiox11020304