
Unraveling and Targeting Myocardial Regeneration Deficit in Diabetes

 ,
, 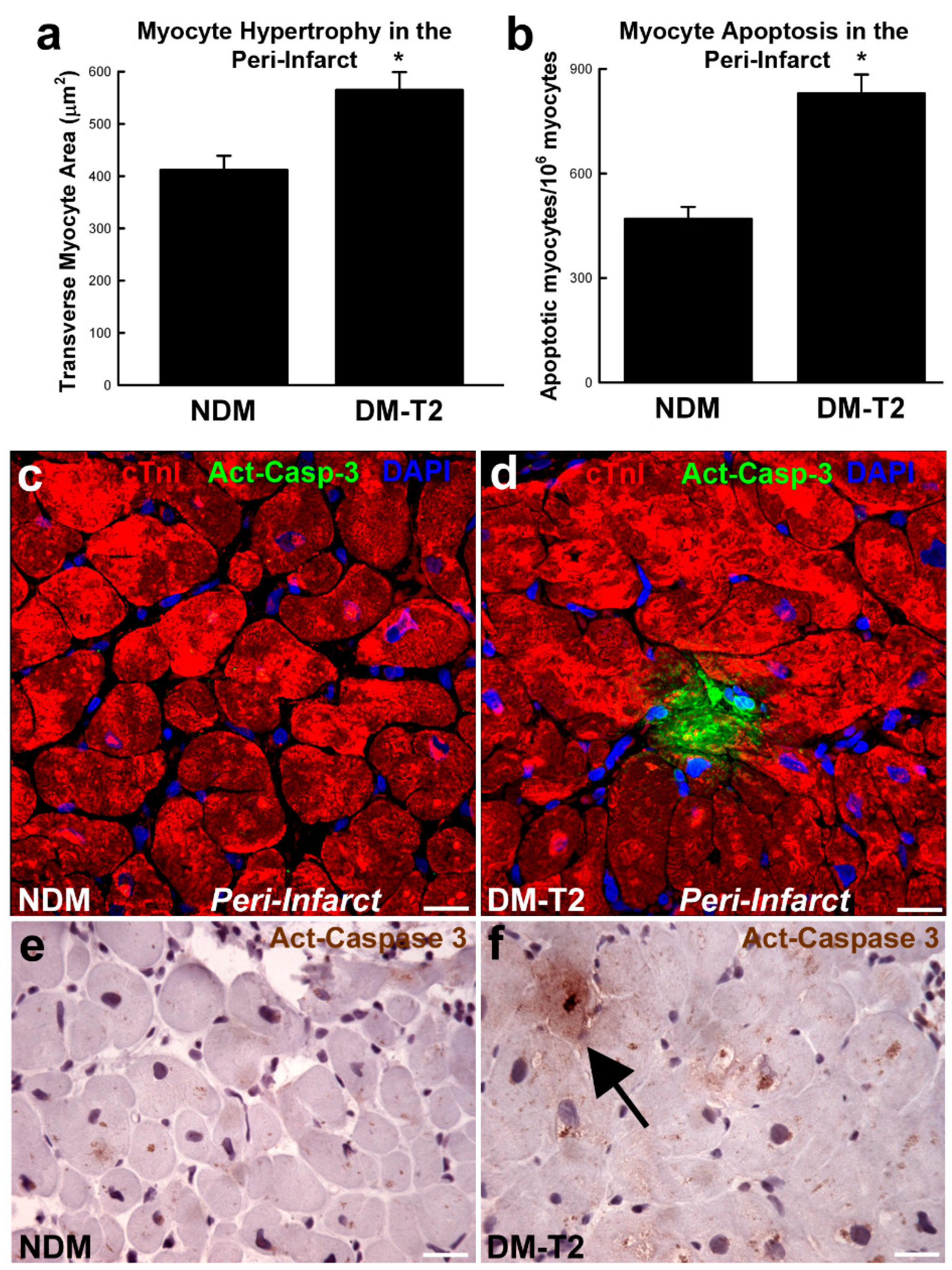{kind=link}
Abstract
:1. Introduction
2. Myocardial Pathophysiology in Diabetic Cardiomyopathy
2.1. Inflammation
2.2. Neurohormonal Activation
2.3. Nitric Oxide (NO) and Reactive Oxygen Species (ROS)
2.4. Endothelial Dysfunction
2.5. Glucose Transport and Calcium Homeostasis
2.6. Myocardial Fibrosis
2.7. Myocardial Cell Loss
2.8. Cell Senescence
3. Diabetes and Adult Stem Cell Function
4. Diabetes and Cardiac Stem Cell Biology
5. Cell Therapy for Diabetic Cardiomyopathy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- American Diabetes Association. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 2011, 34 (Suppl S1), S62–S69. [Google Scholar] [CrossRef] [Green Version]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef]
- Rahman, S.; Rahman, T.; Ismail, A.A.; Rashid, A.R. Diabetes-associated macrovasculopathy: Pathophysiology and pathogenesis. Diabetes Obes. Metab. 2007, 9, 767–780. [Google Scholar] [CrossRef]
- Shindler, D.M.; Kostis, J.B.; Yusuf, S.; Quinones, M.A.; Pitt, B.; Stewart, D.; Pinkett, T.; Ghali, J.K.; Wilson, A.C.; The SOLVD Investigators. Diabetes mellitus, a predictor of morbidity and mortality in the studies of left ventricular dysfunction (SOLVD) trials and registry. Am. J. Cardiol. 1996, 77, 1017–1020. [Google Scholar] [CrossRef]
- Ryden, L.; Armstrong, P.W.; Cleland, J.G.; Horowitz, J.D.; Massie, B.M.; Packer, M.; Poole-Wilson, P.A. Efficacy and safety of high-dose lisinopril in chronic heart failure patients at high cardiovascular risk, including those with diabetes mellitus. Results from the ATLAS trial. Eur. Heart J. 2000, 21, 1967–1978. [Google Scholar] [CrossRef] [PubMed]
- Thrainsdottir, I.S.; Aspelund, T.; Thorgeirsson, G.; Gudnason, V.; Hardarson, T.; Malmberg, K.; Sigurdsson, G.; Rydén, L. The Association Between Glucose Abnormalities and Heart Failure in the Population-Based Reykjavík Study. Diabetes Care 2005, 28, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2013, 62, e147–e239. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.M.; Perry, I.J. Does Inflammation Determine Metabolic Health Status in Obese and Nonobese Adults? J. Clin. Endocrinol. Metab. 2013, 98, E1610–E1619. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; DeMarco, V.G.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2016, 12, 144–153. [Google Scholar] [CrossRef]
- Deten, A.; Volz, H.C.; Briest, W.; Zimmer, H.-G. Cardiac cytokine expression is upregulated in the acute phase after myocardial infarction. Experimental studies in rats. Cardiovasc. Res. 2002, 55, 329–340. [Google Scholar] [CrossRef] [Green Version]
- Irwin, M.W.; Mak, S.; Mann, D.L.; Qu, R.; Penninger, J.M.; Yan, A.; Dawood, F.; Wen, W.-H.; Shou, Z.; Liu, P. Tissue Expression and Immunolocalization of Tumor Necrosis Factor-α in Postinfarction Dysfunctional Myocardium. Circulation 1999, 99, 1492–1498. [Google Scholar] [CrossRef] [Green Version]
- Gwechenberger, M.; Mendoza, L.H.; Youker, K.A.; Frangogiannis, N.G.; Smith, C.W.; Michael, L.H.; Entman, M.L. Cardiac Myocytes Produce Interleukin-6 in Culture and in Viable Border Zone of Reperfused Infarctions. Circulation 1999, 99, 546–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herder, C.; Kolb, H.; Koenig, W.; Haastert, B.; Muller-Scholze, S.; Rathmann, W.; Holle, R.; Thorand, B.; Wichmann, H.-E. Association of Systemic Concentrations of Macrophage Migration Inhibitory Factor With Impaired Glucose Tolerance and Type 2 Diabetes: Results from the Cooperative Health Research in the Region of Augsburg, Survey 4 (KORA S4). Diabetes Care 2006, 29, 368–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.Y.; Chen, H.M.; Liang, J.L.; Lin, Q.X.; Tan, H.H.; Fu, Y.H.; Liu, X.Y.; Shan, Z.X.; Li, X.H.; Yang, H.Z.; et al. Hyperglycemic Myocardial Damage Is Mediated by Proinflammatory Cytokine: Macrophage Migration Inhibitory Factor. PLoS ONE 2011, 6, e16239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.-Z.; Zhang, X.; Wang, L.; Zhang, F.; Qiu, Q.; Liu, M.-L.; Zhang, G.-R.; Wu, X.-L. An Increased Circulating Angiotensin II Concentration is Associated with Hypoadiponectinemia and Postprandial Hyperglycemia in Men with Nonalcoholic Fatty Liver Disease. Intern. Med. 2013, 52, 855–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnett, A.H.; Bain, S.C.; Bouter, P.; Karlberg, B.; Madsbad, S.; Jervell, J.; Mustonen, J. Angiotensin-Receptor Blockade versus Converting–Enzyme Inhibition in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2004, 351, 1952–1961. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.Y.; Chen, Y.W.; Chenier, I.; Tran Sle, M.; Zhang, S.L. Angiotensin II Type II Receptor Deficiency Accelerates the Development of Nephropathy in Type I Diabetes via Oxidative Stress and ACE2. Exp. Diabetes Res. 2011, 2011, 521076. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.C.; Burghi, V.; Miquet, J.G.; Cervino, I.A.; Quiroga, D.T.; Mazziotta, L.; Dominici, F.P. Chronic blockade of the AT2 receptor with PD123319 impairs insulin signaling in C57BL/6 mice. Peptides 2017, 88, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Baudrand, R.; Gupta, N.; Garza, A.E.; Vaidya, A.; Leopold, J.A.; Hopkins, P.N.; Jeunemaitre, X.; Ferri, C.; Romero, J.R.; Williams, J.; et al. Caveolin 1 Modulates Aldosterone-Mediated Pathways of Glucose and Lipid Homeostasis. J. Am. Heart Assoc. 2016, 5, e003845. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.; Baugé, E.; Lalloyer, F.; Lefebvre, P.; Staels, B. Ketone Body Therapy Protects From Lipotoxicity and Acute Liver Failure Upon Pparα Deficiency. Mol. Endocrinol. 2015, 29, 1134–1143. [Google Scholar] [CrossRef] [Green Version]
- Bisognano, J.D.; Weinberger, H.D.; Bohlmeyer, T.J.; Pende, A.; Raynolds, M.V.; Sastravaha, A.; Roden, R.; Asano, K.; Blaxall, B.C.; Wu, S.C.; et al. Myocardial-Directed Overexpression of the Human β1-Adrenergic Receptor in Transgenic Mice. J. Mol. Cell. Cardiol. 2000, 32, 817–830. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Petrie, J.R.; Ueda, S.; Webb, D.J.; Elliott, H.L.; Connell, J.M. Endothelial Nitric Oxide Production and Insulin Sensitivity. A physiological link with implications for pathogenesis of cardiovascular disease. Circulation 1996, 93, 1331–1333. [Google Scholar] [CrossRef]
- Federici, M.; Menghini, R.; Mauriello, A.; Hribal, M.L.; Ferrelli, F.; Lauro, D.; Sbraccia, P.; Spagnoli, L.G.; Sesti, G.; Lauro, R. Insulin-Dependent Activation of Endothelial Nitric Oxide Synthase Is Impaired by O-Linked Glycosylation Modification of Signaling Proteins in Human Coronary Endothelial Cells. Circulation 2002, 106, 466–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, S.B.; Cusco, J.A.; Roddy, M.-A.; Johnstone, M.T.; Creager, M.A. Impaired nitric oxide-mediated vasodilation in patients with non-insulin-dependent diabetes mellitus. J. Am. Coll. Cardiol. 1996, 27, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Fiorentino, T.V.; Prioletta, A.; Zuo, P.; Folli, F. Hyperglycemia-induced Oxidative Stress and its Role in Diabetes Mellitus Related Cardiovascular Diseases. Curr. Pharm. Des. 2013, 19, 5695–5703. [Google Scholar] [CrossRef]
- Lee, A.Y.; Chung, S.S. Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1999, 13, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, P.; Winhofer, Y.; Krssak, M.; Smajis, S.; Harreiter, J.; Kosi-Trebotic, L.; Fürnsinn, C.; Anderwald, C.-H.; Baumgartner-Parzer, S.; Trattnig, S.; et al. Suppression of plasma free fatty acids reduces myocardial lipid content and systolic function in type 2 diabetes. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandesara, P.B.; O’Neal, W.T.; Kelli, H.M.; Samman-Tahhan, A.; Hammadah, M.; Quyyumi, A.A.; Sperling, L.S. The Prognostic Significance of Diabetes and Microvascular Complications in Patients With Heart Failure With Preserved Ejection Fraction. Diabetes Care 2018, 41, 150–155. [Google Scholar] [CrossRef] [Green Version]
- Torella, D.; Iaconetti, C.; Tarallo, R.; Marino, F.; Giurato, G.; Veneziano, C.; Aquila, I.; Scalise, M.; Mancuso, T.; Cianflone, E.; et al. miRNA Regulation of the Hyperproliferative Phenotype of Vascular Smooth Muscle Cells in Diabetes. Diabetes 2018, 67, 2554–2568. [Google Scholar] [CrossRef] [Green Version]
- Reinstadler, S.J.; Stiermaier, T.; Eitel, C.; Metzler, B.; De Waha, S.; Fuernau, G.; Desch, S.; Thiele, H.; Eitel, I. Relationship between diabetes and ischaemic injury among patients with revascularized ST -elevation myocardial infarction. Diabetes Obes. Metab. 2017, 19, 1706–1713. [Google Scholar] [CrossRef]
- Kristensen, S.L.; Rørth, R.; Jhund, P.S.; Shen, L.; Lee, M.M.Y.; Petrie, M.C.; Køber, L.; McMurray, J.J.; on behalf of the BEST Investigators. Microvascular complications in diabetes patients with heart failure and reduced ejection fraction-insights from the Beta-blocker Evaluation of Survival Trial. Eur. J. Heart Fail. 2018, 20, 1549–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, M.A.; Clerk, L.H.; Lindner, J.R.; Klibanov, A.L.; Clark, M.G.; Rattigan, S.; Barrett, E.J. Microvascular Recruitment Is an Early Insulin Effect That Regulates Skeletal Muscle Glucose Uptake In Vivo. Diabetes 2004, 53, 1418–1423. [Google Scholar] [CrossRef] [Green Version]
- Widyantoro, B.; Emoto, N.; Nakayama, K.; Anggrahini, D.W.; Adiarto, S.; Iwasa, N.; Yagi, K.; Miyagawa, K.; Rikitake, Y.; Suzuki, T.; et al. Endothelial Cell–Derived Endothelin-1 Promotes Cardiac Fibrosis in Diabetic Hearts Through Stimulation of Endothelial-to-Mesenchymal Transition. Circulation 2010, 121, 2407–2418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thijssen, D.H.; Torella, D.; Hopman, M.T.; Ellison, G.M. The role of endothelial progenitor and cardiac stem cells in the cardiovascular adaptations to age and exercise. Front. Biosci. 2009, 14, 4685–4702. [Google Scholar] [CrossRef] [Green Version]
- Marino, F.; Scalise, M.; Cianflone, E.; Salerno, L.; Cappetta, D.; Salerno, N.; De Angelis, A.; Torella, D.; Urbanek, K. Physical Exercise and Cardiac Repair: The Potential Role of Nitric Oxide in Boosting Stem Cell Regenerative Biology. Antioxidants 2021, 10, 1002. [Google Scholar] [CrossRef]
- Randriamboavonjy, V.; Pistrosch, F.; Bolck, B.; Schwinger, R.H.; Dixit, M.; Badenhoop, K.; Cohen, R.A.; Busse, R.; Fleming, I. Platelet Sarcoplasmic Endoplasmic Reticulum Ca2+ -ATPase and μ-Calpain Activity Are Altered in Type 2 Diabetes Mellitus and Restored by Rosiglitazone. Circulation 2008, 117, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Gherasim, L.; Taşcă, C.; Havriliuc, C.; Vasilescu, C. A morphological quantitative study of small vessels in diabetic cardiomyopathy. Morphol. Embryol. 1985, 31, 191–195. [Google Scholar]
- Anderson, E.J.; Kypson, A.P.; Rodriguez, E.; Anderson, C.A.; Lehr, E.J.; Neufer, P.D. Substrate-Specific Derangements in Mitochondrial Metabolism and Redox Balance in the Atrium of the Type 2 Diabetic Human Heart. J. Am. Coll. Cardiol. 2009, 54, 1891–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, E.J.; Rodriguez, E.; Anderson, C.A.; Thayne, K.; Chitwood, W.R.; Kypson, A.P. Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H118–H124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frustaci, A.; Kajstura, J.; Chimenti, C.; Jakoniuk, I.; Leri, A.; Maseri, A.; Nadal-Ginard, B.; Anversa, P. Myocardial Cell Death in Human Diabetes. Circ. Res. 2000, 87, 1123–1132. [Google Scholar] [CrossRef]
- Kuethe, F.; Sigusch, H.H.; Bornstein, S.R.; Hilbig, K.; Kamvissi, V.; Figulla, H.R. Apoptosis in Patients with Dilated Cardiomyopathy and Diabetes: A Feature of Diabetic Cardiomyopathy? Horm. Metab. Res. 2007, 39, 672–676. [Google Scholar] [CrossRef]
- Torella, D.; Ellison, G.M.; Torella, M.; Vicinanza, C.; Aquila, I.; Iaconetti, C.; Scalise, M.; Marino, F.; Henning, B.J.; Lewis, F.C.; et al. Carbonic Anhydrase Activation Is Associated With Worsened Pathological Remodeling in Human Ischemic Diabetic Cardiomyopathy. J. Am. Heart Assoc. 2014, 3, e000434. [Google Scholar] [CrossRef] [Green Version]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Tuttle, C.S.L.; Waaijer, M.E.C.; Slee-Valentijn, M.S.; Stijnen, T.; Westendorp, R.; Maier, A.B. Cellular senescence and chronological age in various human tissues: A systematic review and meta-analysis. Aging Cell 2020, 19, e13083. [Google Scholar] [CrossRef] [Green Version]
- Ksiazek, K.; Passos, J.F.; Olijslagers, S.; von Zglinicki, T. Mitochondrial dysfunction is a possible cause of accelerated senescence of mesothelial cells exposed to high glucose. Biochem. Biophys. Res. Commun. 2008, 366, 793–799. [Google Scholar] [CrossRef]
- Peppa, M.; Uribarri, J.; Vlassara, H. Glucose, Advanced Glycation End Products, and Diabetes Complications: What Is New and What Works. Clin. Diabetes 2003, 21, 186–187. [Google Scholar] [CrossRef] [Green Version]
- Davalos, A.R.; Kawahara, M.; Malhotra, G.K.; Schaum, N.; Huang, J.; Ved, U.; Beausejour, C.M.; Coppe, J.-P.; Rodier, F.; Campisi, J. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol. 2013, 201, 613–629. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utsal, L.; Tillmann, V.; Zilmer, M.; Mäestu, J.; Purge, P.; Jürimäe, J.; Saar, M.; Lätt, E.; Maasalu, K.; Jürimäe, T. Elevated Serum IL-6, IL-8, MCP-1, CRP, and IFN-? Levels in 10- to 11-Year-Old Boys with Increased BMI. Horm. Res. Paediatr. 2012, 78, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.D.; Manson, J.E.; Rifai, N.; Buring, J.E.; Ridker, P.M. C-Reactive Protein, Interleukin 6, and Risk of Developing Type 2 Diabetes Mellitus. JAMA 2001, 286, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.; Bergholz, J.; Zhang, H.; He, H.; Wang, Y.; Zhang, Y.; Li, Q.; Kirkland, J.L.; Xiao, Z. Insulin-like growth factor-1 regulates the SIRT1-p53 pathway in cellular senescence. Aging Cell 2014, 13, 669–678. [Google Scholar] [CrossRef]
- Minamino, T.; Miyauchi, H.; Tateno, K.; Kunieda, T.; Komuro, I. Akt-Induced Cellular Senescence: Implication for Human Disease. Cell Cycle 2004, 3, 447–449. [Google Scholar] [CrossRef] [Green Version]
- Tchkonia, T.; Corkey, B.E.; Kirkland, J.L. Current Views of the Fat Cell as an Endocrine Cell: Lipotoxicity. In Overweight and the Metabolic Syndrome; Bray, G.A., Ryan, D.H., Eds.; Endocrine Updates; Springer: Boston, MA, USA, 2006; Volume 26, pp. 105–123. [Google Scholar]
- Cable, J.; Fuchs, E.; Weissman, I.; Jasper, H.; Glass, D.; Rando, T.; Blau, H.; Debnath, S.; Oliva, A.; Park, S.; et al. Adult stem cells and regenerative medicine—A symposium report. Ann. N. Y. Acad. Sci. 2020, 1462, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Wu, Z.; Xu, X.; Liao, L.; Chen, J.; Huang, L.; Wu, W.; Luo, F.; Wu, C.; Pugliese, A.; et al. Umbilical Cord Mesenchymal Stromal Cell With Autologous Bone Marrow Cell Transplantation in Established Type 1 Diabetes: A Pilot Randomized Controlled Open-Label Clinical Study to Assess Safety and Impact on Insulin Secretion. Diabetes Care 2016, 39, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Fadini, G.P.; Avogaro, A. Diabetes impairs mobilization of stem cells for the treatment of cardiovascular disease: A meta-regression analysis. Int. J. Cardiol. 2013, 168, 892–897. [Google Scholar] [CrossRef]
- Vecellio, M.; Spallotta, F.; Nanni, S.; Colussi, C.; Cencioni, C.; Derlet, A.; Bassetti, B.; Tilenni, M.; Carena, M.C.; Farsetti, A.; et al. The Histone Acetylase Activator Pentadecylidenemalonate 1b Rescues Proliferation and Differentiation in the Human Cardiac Mesenchymal Cells of Type 2 Diabetic Patients. Diabetes 2014, 63, 2132–2147. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Han, J.W.; Lee, J.Y.; Choi, Y.J.; Sohn, Y.-D.; Song, M.; Yoon, Y.-S. Diabetic Mesenchymal Stem Cells Are Ineffective for Improving Limb Ischemia Due to Their Impaired Angiogenic Capability. Cell Transplant. 2015, 24, 1571–1584. [Google Scholar] [CrossRef] [Green Version]
- Fadini, G.P.; Albiero, M.; De Kreutzenberg, S.V.; Boscaro, E.; Cappellari, R.; Marescotti, M.; Poncina, N.; Agostini, C.; Avogaro, A. Diabetes Impairs Stem Cell and Proangiogenic Cell Mobilization in Humans. Diabetes Care 2013, 36, 943–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, P.; Zhang, X.; Wu, Y.; Li, L.; Yin, Q.; Zheng, L.; Zhang, H.; Sun, C. Streptozotocin-Induced Diabetic Rat–Derived Bone Marrow Mesenchymal Stem Cells Have Impaired Abilities in Proliferation, Paracrine, Antiapoptosis, and Myogenic Differentiation. Transplant. Proc. 2010, 42, 2745–2752. [Google Scholar] [CrossRef] [PubMed]
- Spinetti, G.; Cordella, D.; Fortunato, O.; Sangalli, E.; Losa, S.; Gotti, A.; Carnelli, F.; Rosa, F.; Riboldi, S.; Sessa, F.; et al. Global Remodeling of the Vascular Stem Cell Niche in Bone Marrow of Diabetic Patients: Implication of the microRNA-155/FOXO3a signaling pathway. Circ. Res. 2013, 112, 510–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boniakowski, A.E.; Kimball, A.S.; Jacobs, B.N.; Kunkel, S.L.; Gallagher, K.A. Macrophage-Mediated Inflammation in Normal and Diabetic Wound Healing. J. Immunol. 2017, 199, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, K.A.; Joshi, A.; Carson, W.F.; Schaller, M.; Allen, R.; Mukerjee, S.; Kittan, N.; Feldman, E.L.; Henke, P.K.; Hogaboam, C.; et al. Epigenetic Changes in Bone Marrow Progenitor Cells Influence the Inflammatory Phenotype and Alter Wound Healing in Type 2 Diabetes. Diabetes 2015, 64, 1420–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barman, P.K.; Urao, N.; Koh, T.J. Diabetes induces myeloid bias in bone marrow progenitors associated with enhanced wound macrophage accumulation and impaired healing. J. Pathol. 2019, 249, 435–446. [Google Scholar] [CrossRef]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–966. [Google Scholar] [CrossRef]
- Yoon, Y.-S.; Uchida, S.; Masuo, O.; Cejna, M.; Park, J.-S.; Gwon, H.-C.; Kirchmair, R.; Bahlman, F.; Walter, D.; Curry, C.; et al. Progressive Attenuation of Myocardial Vascular Endothelial Growth Factor Expression Is a Seminal Event in Diabetic Cardiomyopathy: Restoration of microvascular homeostasis and recovery of cardiac function in diabetic cardiomyopathy after replenishment of local vascular endothelial growth factor. Circulation 2005, 111, 2073–2085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, K.A.; Liu, Z.-J.; Xiao, M.; Chen, H.; Goldstein, L.J.; Buerk, D.G.; Nedeau, A.; Thom, S.R.; Velazquez, O.C. Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1α. J. Clin. Investig. 2007, 117, 1249–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, J.M.; Zalos, G.; Halcox, J.P.J.; Schenke, W.H.; Waclawiw, M.A.; Quyyumi, A.A.; Finkel, T. Circulating Endothelial Progenitor Cells, Vascular Function, and Cardiovascular Risk. N. Engl. J. Med. 2003, 348, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Tepper, O.M.; Galiano, R.D.; Capla, J.M.; Kalka, C.; Gagne, P.J.; Jacobowitz, G.R.; Levine, J.P.; Gurtner, G.C. Human Endothelial Progenitor Cells From Type II Diabetics Exhibit Impaired Proliferation, Adhesion, and Incorporation Into Vascular Structures. Circulation 2002, 106, 2781–2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, Y.; Ii, M.; Qin, G.; Hamada, H.; Asai, J.; Takenaka, H.; Sekiguchi, H.; Renault, M.-A.; Jujo, K.; Katoh, N.; et al. CXCR4 Antagonist AMD3100 Accelerates Impaired Wound Healing in Diabetic Mice. J. Investig. Dermatol. 2012, 132, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Yong, X.; Li, C.; Lü, M.; Liu, D.; Chen, L.; Hu, J.; Teng, M.; Zhang, D.; Fan, Y.; et al. CXCL12/CXCR4 axis promotes mesenchymal stem cell mobilization to burn wounds and contributes to wound repair. J. Surg. Res. 2013, 183, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Badillo, A.T.; Chung, S.; Zhang, L.; Zoltick, P.; Liechty, K.W. Lentiviral Gene Transfer of SDF-1α to Wounds Improves Diabetic Wound Healing. J. Surg. Res. 2007, 143, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Henderson, P.W.; Singh, S.P.; Krijgh, D.D.; Yamamoto, M.; Rafii, D.C.; Sung, J.J.; Rafii, S.; Rabbany, S.Y.; Spector, J.A. Stromal-derived factor-1 delivered via hydrogel drug-delivery vehicle accelerates wound healing in vivo. Wound Repair Regen. 2011, 19, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Beltrami, A.P.; Barlucchi, L.; Torella, D.; Baker, M.; Limana, F.; Chimenti, S.; Kasahara, H.; Rota, M.; Musso, E.; Urbanek, K.; et al. Adult Cardiac Stem Cells Are Multipotent and Support Myocardial Regeneration. Cell 2003, 114, 763–776. [Google Scholar] [CrossRef] [Green Version]
- Ellison, G.M.; Vicinanza, C.; Smith, A.J.; Aquila, I.; Leone, A.; Waring, C.D.; Henning, B.J.; Stirparo, G.G.; Papait, R.; Scarfò, M.; et al. Adult c-kitpos Cardiac Stem Cells Are Necessary and Sufficient for Functional Cardiac Regeneration and Repair. Cell 2013, 154, 827–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Siena, S.; Gimmelli, R.; Nori, S.L.; Barbagallo, F.; Campolo, F.; Dolci, S.; Rossi, P.; Venneri, M.A.; Giannetta, E.; Gianfrilli, D.; et al. Activated c-Kit receptor in the heart promotes cardiac repair and regeneration after injury. Cell Death Dis. 2016, 7, e2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadal-Ginard, B.; Ellison, G.M.; Torella, D. Absence of Evidence Is Not Evidence of Absence: Pitfalls of cre knock-ins in the c-Kit locus. Circ. Res. 2014, 115, 415–418. [Google Scholar] [CrossRef]
- van Berlo, J.H.; Kanisicak, O.; Maillet, M.; Vagnozzi, R.J.; Karch, J.; Lin, S.C.; Middleton, R.C.; Marbán, E.; Molkentin, J.D. c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature 2014, 509, 337–341. [Google Scholar] [CrossRef]
- Aquila, I.; Marino, F.; Cianflone, E.; Marotta, P.; Torella, M.; Mollace, V.; Indolfi, C.; Nadal-Ginard, B.; Torella, D. The use and abuse of Cre/Lox recombination to identify adult cardiomyocyte renewal rate and origin. Pharmacol. Res. 2018, 127, 116–128. [Google Scholar] [CrossRef]
- Aquila, I.; Cianflone, E.; Scalise, M.; Marino, F.; Mancuso, T.; Filardo, A.; Smith, A.J.; Cappetta, D.; De Angelis, A.; Urbanek, K.; et al. c-kit Haploinsufficiency impairs adult cardiac stem cell growth, myogenicity and myocardial regeneration. Cell Death Dis. 2019, 10, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicinanza, C.; Aquila, I.; Cianflone, E.; Scalise, M.; Marino, F.; Mancuso, T.; Fumagalli, F.; Giovannone, E.D.; Cristiano, F.; Iaccino, E.; et al. Kitcre knock-in mice fail to fate-map cardiac stem cells. Nature 2018, 555, E1–E5. [Google Scholar] [CrossRef] [PubMed]
- Vicinanza, C.; Aquila, I.; Scalise, M.; Cristiano, F.; Marino, F.; Cianflone, E.; Mancuso, T.; Marotta, P.; Sacco, W.; Lewis, F.; et al. Adult cardiac stem cells are multipotent and robustly myogenic: C-kit expression is necessary but not sufficient for their identification. Cell Death Differ. 2017, 24, 2101–2116. [Google Scholar] [CrossRef] [Green Version]
- Scalise, M.; Marino, F.; Cianflone, E.; Mancuso, T.; Marotta, P.; Aquila, I.; Torella, M.; Nadal-Ginard, B.; Torella, D. Heterogeneity of Adult Cardiac Stem Cells. Adv. Exp. Med. Biol. 2019, 1169, 141–178. [Google Scholar] [CrossRef]
- Scalise, M.; Torella, M.; Marino, F.; Ravo, M.; Giurato, G.; Vicinanza, C.; Cianflone, E.; Mancuso, T.; Aquila, I.; Salerno, L.; et al. Atrial myxomas arise from multipotent cardiac stem cells. Eur. Heart J. 2020, 41, 4332–4345. [Google Scholar] [CrossRef] [Green Version]
- Cianflone, E.; Aquila, I.; Scalise, M.; Marotta, P.; Torella, M.; Nadal-Ginard, B.; Torella, D. Molecular basis of functional myogenic specification of Bona Fide multipotent adult cardiac stem cells. Cell Cycle 2018, 17, 927–946. [Google Scholar] [CrossRef] [Green Version]
- Scalise, M.; Marino, F.; Salerno, L.; Mancuso, T.; Cappetta, D.; Barone, A.; Parrotta, E.I.; Torella, A.; Palumbo, D.; Veltri, P.; et al. In vitro CSC-derived cardiomyocytes exhibit the typical microRNA-mRNA blueprint of endogenous cardiomyocytes. Commun. Biol. 2021, 4, 1146. [Google Scholar] [CrossRef] [PubMed]
- Rota, M.; LeCapitaine, N.; Hosoda, T.; Boni, A.; De Angelis, A.; Padin-Iruegas, M.E.; Esposito, G.; Vitale, S.; Urbanek, K.; Casarsa, C.; et al. Diabetes Promotes Cardiac Stem Cell Aging and Heart Failure, Which Are Prevented by Deletion of the p66 shc Gene. Circ. Res. 2006, 99, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Salabei, J.K.; Lorkiewicz, P.K.; Mehra, P.; Gibb, A.A.; Haberzettl, P.; Hong, K.U.; Wei, X.; Zhang, X.; Li, Q.; Wysoczynski, M.; et al. Type 2 Diabetes Dysregulates Glucose Metabolism in Cardiac Progenitor Cells. J. Biol. Chem. 2016, 291, 13634–13648. [Google Scholar] [CrossRef] [Green Version]
- Molgat, A.S.; Tilokee, E.L.; Rafatian, G.; Vulesevic, B.; Ruel, M.; Milne, R.; Suuronen, E.J.; Davis, D.R. Hyperglycemia Inhibits Cardiac Stem Cell–Mediated Cardiac Repair and Angiogenic Capacity. Circulation 2014, 130, S70–S76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migliaccio, E.; Giorgio, M.; Mele, S.; Pelicci, G.; Reboldi, P.; Pandolfi, P.P.; Lanfrancone, L.; Pelicci, P.G. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 1999, 402, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Pacini, S.; Pellegrini, M.; Migliaccio, E.; Patrussi, L.; Ulivieri, C.; Ventura, A.; Carraro, F.; Naldini, A.; Lanfrancone, L.; Pelicci, P.; et al. p66SHC Promotes Apoptosis and Antagonizes Mitogenic Signaling in T Cells. Mol. Cell. Biol. 2004, 24, 1747–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natalicchio, A.; Tortosa, F.; Perrini, S.; Laviola, L.; Giorgino, F. p66Shc, a multifaceted protein linking Erk signalling, glucose metabolism, and oxidative stress. Arch. Physiol. Biochem. 2011, 117, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Meng, K.; Pu, Y.; Wang, C.; Chen, Y.; Wang, L. Hyperglycemia Altered the Fate of Cardiac Stem Cells to Adipogenesis through Inhibiting the β-Catenin/TCF-4 Pathway. Cell. Physiol. Biochem. 2018, 49, 2254–2263. [Google Scholar] [CrossRef] [PubMed]
- Katare, R.; Oikawa, A.; Cesselli, D.; Beltrami, A.P.; Avolio, E.; Muthukrishnan, D.; Munasinghe, P.E.; Angelini, G.; Emanueli, C.; Madeddu, P. Boosting the pentose phosphate pathway restores cardiac progenitor cell availability in diabetes. Cardiovasc. Res. 2013, 97, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Vecellio, M.; Meraviglia, V.; Nanni, S.; Barbuti, A.; Scavone, A.; DiFrancesco, D.; Farsetti, A.; Pompilio, G.; Colombo, G.; Capogrossi, M.C.; et al. In Vitro Epigenetic Reprogramming of Human Cardiac Mesenchymal Stromal Cells into Functionally Competent Cardiovascular Precursors. PLoS ONE 2012, 7, e51694. [Google Scholar] [CrossRef] [Green Version]
- She, T.; Wang, X.; Gan, Y.; Kuang, D.; Yue, J.; Ni, J.; Zhao, X.; Wang, G. Hyperglycemia suppresses cardiac stem cell homing to peri-infarcted myocardium via regulation of ERK1/2 and p38 MAPK activities. Int. J. Mol. Med. 2012, 30, 1313–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Yang, C.-M.; Xi, Y.; Wu, G.; Shelat, H.; Gao, S.; Cheng, J.; Geng, Y.-J. MicroRNA-1/133 targeted dysfunction of potassium channels KCNE1 and KCNQ1 in human cardiac progenitor cells with simulated hyperglycemia. Int. J. Cardiol. 2013, 167, 1076–1078. [Google Scholar] [CrossRef]
- Messina, E.; Giacomello, A. Diabetic Cardiomyopathy: A “cardiac stem cell disease” involving p66Shc, an attractive novel molecular target for heart failure therapy. Circ. Res. 2006, 99, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Fomison-Nurse, I.; Saw, E.E.L.; Gandhi, S.; Munasinghe, P.E.; Van Hout, I.; Williams, M.J.A.; Galvin, I.; Bunton, R.; Davis, P.; Cameron, V.; et al. Diabetes induces the activation of pro-ageing miR-34a in the heart, but has differential effects on cardiomyocytes and cardiac progenitor cells. Cell Death Differ. 2018, 25, 1336–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel Aziz, M.T.; El-Asmar, M.F.; Haidara, M.; Atta, H.M.; Roshdy, N.K.; Rashed, L.A.; Sabry, D.; Youssef, M.A.; Abdel Aziz, A.T.; Moustafa, M. Effect of bone marrow-derived mesenchymal stem cells on cardiovascular complications in diabetic rats. Med. Sci. Monit. 2008, 14, BR249–BR255. [Google Scholar] [PubMed]
- Zhang, N.; Li, J.; Luo, R.; Jiang, J.; Wang, J.-A. Bone Marrow Mesenchymal Stem Cells Induce Angiogenesis and Attenuate the Remodeling of Diabetic Cardiomyopathy. Exp. Clin. Endocrinol. Diabetes 2008, 116, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Zhang, N.; Wang, J.A. Improved anti-apoptotic and anti-remodeling potency of bone marrow mesenchymal stem cells by anoxic pre-conditioning in diabetic cardiomyopathy. J. Endocrinol. Investig. 2008, 31, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Govaert, J.A.; Swijnenburg, R.-J.; Schrepfer, S.; Xie, X.; van der Bogt, K.E.; Hoyt, G.; Stein, W.; Ransohoff, K.J.; Robbins, R.C.; Wu, J.C. Poor Functional Recovery After Transplantation of Diabetic Bone Marrow Stem Cells in Ischemic Myocardium. J. Heart Lung Transplant. 2009, 28, 1158–1165.e1. [Google Scholar] [CrossRef] [Green Version]
- De Angelis, A.; Piegari, E.; Cappetta, D.; Russo, R.; Esposito, G.; Ciuffreda, L.P.; Ferraiolo, F.A.V.; Frati, C.; Fagnoni, F.; Berrino, L.; et al. SIRT1 activation rescues doxorubicin-induced loss of functional competence of human cardiac progenitor cells. Int. J. Cardiol. 2015, 189, 30–44. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, A.; Piegari, E.; Cappetta, D.; Marino, L.; Filippelli, A.; Berrino, L.; Ferreira-Martins, J.; Zheng, H.; Hosoda, T.; Rota, M.; et al. Anthracycline Cardiomyopathy Is Mediated by Depletion of the Cardiac Stem Cell Pool and Is Rescued by Restoration of Progenitor Cell Function. Circulation 2010, 121, 276–292. [Google Scholar] [CrossRef] [Green Version]
- Piegari, E.; De Angelis, A.; Cappetta, D.; Russo, R.; Esposito, G.; Costantino, S.; Graiani, G.; Frati, C.; Prezioso, L.; Berrino, L.; et al. Doxorubicin induces senescence and impairs function of human cardiac progenitor cells. Basic Res. Cardiol. 2013, 108, 334. [Google Scholar] [CrossRef]
- Cianflone, E.; Torella, M.; Biamonte, F.; De Angelis, A.; Urbanek, K.; Costanzo, F.S.; Rota, M.; Ellison-Hughes, G.M.; Torella, D. Targeting Cardiac Stem Cell Senescence to Treat Cardiac Aging and Disease. Cells 2020, 9, 1558. [Google Scholar] [CrossRef]
- Delucchi, F.; Berni, R.; Frati, C.; Cavalli, S.; Graiani, G.; Sala, R.; Chaponnier, C.; Gabbiani, G.; Calani, L.; Del Rio, D.; et al. Resveratrol Treatment Reduces Cardiac Progenitor Cell Dysfunction and Prevents Morpho-Functional Ventricular Remodeling in Type-1 Diabetic Rats. PLoS ONE 2012, 7, e39836. [Google Scholar] [CrossRef] [Green Version]
- Thirunavukkarasu, M.; Penumathsa, S.V.; Koneru, S.; Juhasz, B.; Zhan, L.; Otani, H.; Bagchi, D.; Das, D.K.; Maulik, N. Resveratrol alleviates cardiac dysfunction in streptozotocin-induced diabetes: Role of nitric oxide, thioredoxin, and heme oxygenase. Free Radic. Biol. Med. 2007, 43, 720–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulaiman, M.; Matta, M.J.; Sunderesan, N.R.; Gupta, M.P.; Periasamy, M.; Gupta, M. Resveratrol, an activator of SIRT1, upregulates sarcoplasmic calcium ATPase and improves cardiac function in diabetic cardiomyopathy. Am. J. Physiol. Circ. Physiol. 2010, 298, H833–H843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cianflone, E.; Cappetta, D.; Mancuso, T.; Sabatino, J.; Marino, F.; Scalise, M.; Albanese, M.; Salatino, A.; Parrotta, E.I.; Cuda, G.; et al. Statins Stimulate New Myocyte Formation After Myocardial Infarction by Activating Growth and Differentiation of the Endogenous Cardiac Stem Cells. Int. J. Mol. Sci. 2020, 21, 7927. [Google Scholar] [CrossRef]
- Lee, S.; Schmitt, C.A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef]
- Marino, F.; Scalise, M.; Cianflone, E.; Mancuso, T.; Aquila, I.; Agosti, V.; Torella, M.; Paolino, D.; Mollace, V.; Nadal-Ginard, B.; et al. Role of c-Kit in Myocardial Regeneration and Aging. Front. Endocrinol. 2019, 10, 371. [Google Scholar] [CrossRef]
- Khatiwala, R.V.; Zhang, S.; Li, X.; Devejian, N.; Bennett, E.; Cai, C. Inhibition of p16INK4A to Rejuvenate Aging Human Cardiac Progenitor Cells via the Upregulation of Anti-oxidant and NFκB Signal Pathways. Stem Cell Rev. Rep. 2018, 14, 612–625. [Google Scholar] [CrossRef]
- Lewis-McDougall, F.C.; Ruchaya, P.J.; Domenjo-Vila, E.; Shin Teoh, T.; Prata, L.; Cottle, B.J.; Clark, J.E.; Punjabi, P.P.; Awad, W.; Torella, D.; et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell 2019, 18, e12931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Currie, G.; Delles, C. Precision Medicine and Personalized Medicine in Cardiovascular Disease. Adv. Exp. Med. Biol. 2018, 1065, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Leopold, J.A.; Loscalzo, J. Emerging Role of Precision Medicine in Cardiovascular Disease. Circ. Res. 2018, 122, 1302–1315. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molinaro, C.; Salerno, L.; Marino, F.; Scalise, M.; Salerno, N.; Pagano, L.; De Angelis, A.; Cianflone, E.; Torella, D.; Urbanek, K. Unraveling and Targeting Myocardial Regeneration Deficit in Diabetes. Antioxidants 2022, 11, 208. https://doi.org/10.3390/antiox11020208
Molinaro C, Salerno L, Marino F, Scalise M, Salerno N, Pagano L, De Angelis A, Cianflone E, Torella D, Urbanek K. Unraveling and Targeting Myocardial Regeneration Deficit in Diabetes. Antioxidants. 2022; 11(2):208. https://doi.org/10.3390/antiox11020208
Chicago/Turabian StyleMolinaro, Claudia, Luca Salerno, Fabiola Marino, Mariangela Scalise, Nadia Salerno, Loredana Pagano, Antonella De Angelis, Eleonora Cianflone, Daniele Torella, and Konrad Urbanek. 2022. "Unraveling and Targeting Myocardial Regeneration Deficit in Diabetes" Antioxidants 11, no. 2: 208. https://doi.org/10.3390/antiox11020208