
Reactive Oxygen Species (ROS) and Antioxidants as Immunomodulators in Exercise: Implications for Heme Oxygenase and Bilirubin

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Effect of Exercise on Weight Management and Inflammation
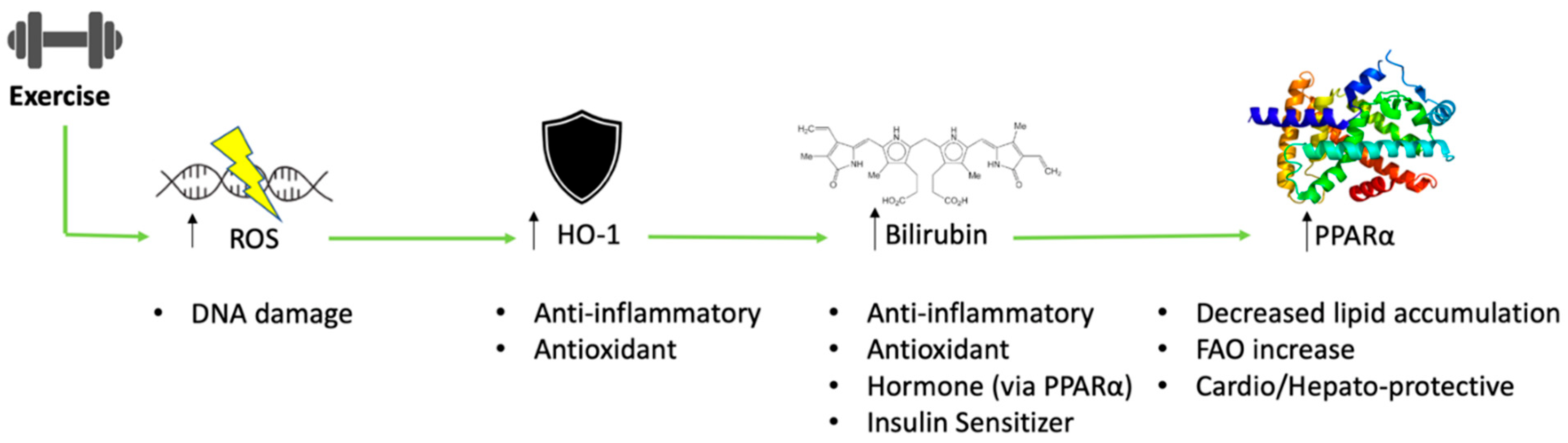
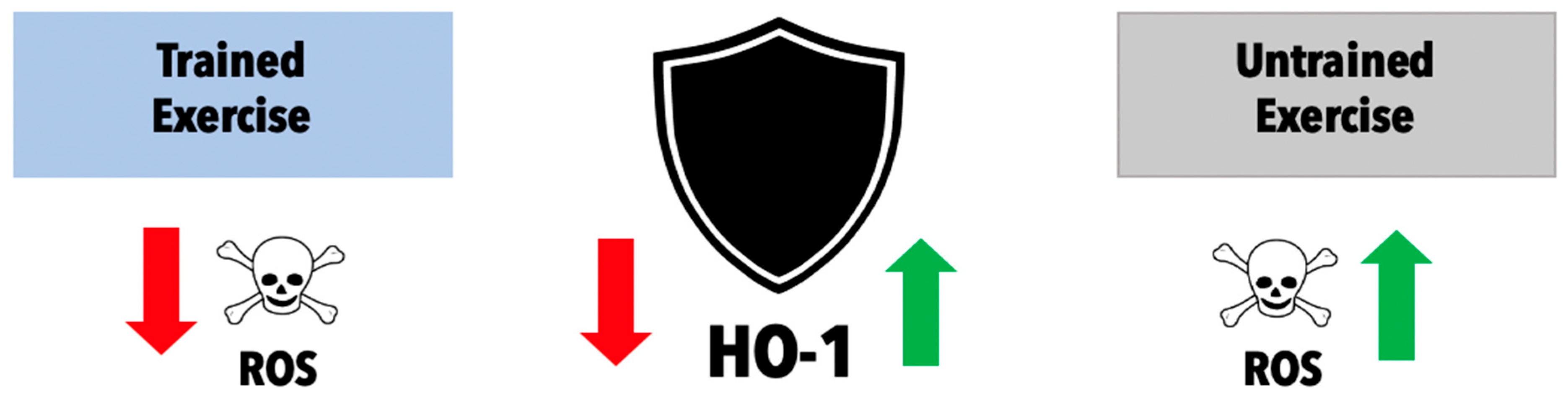
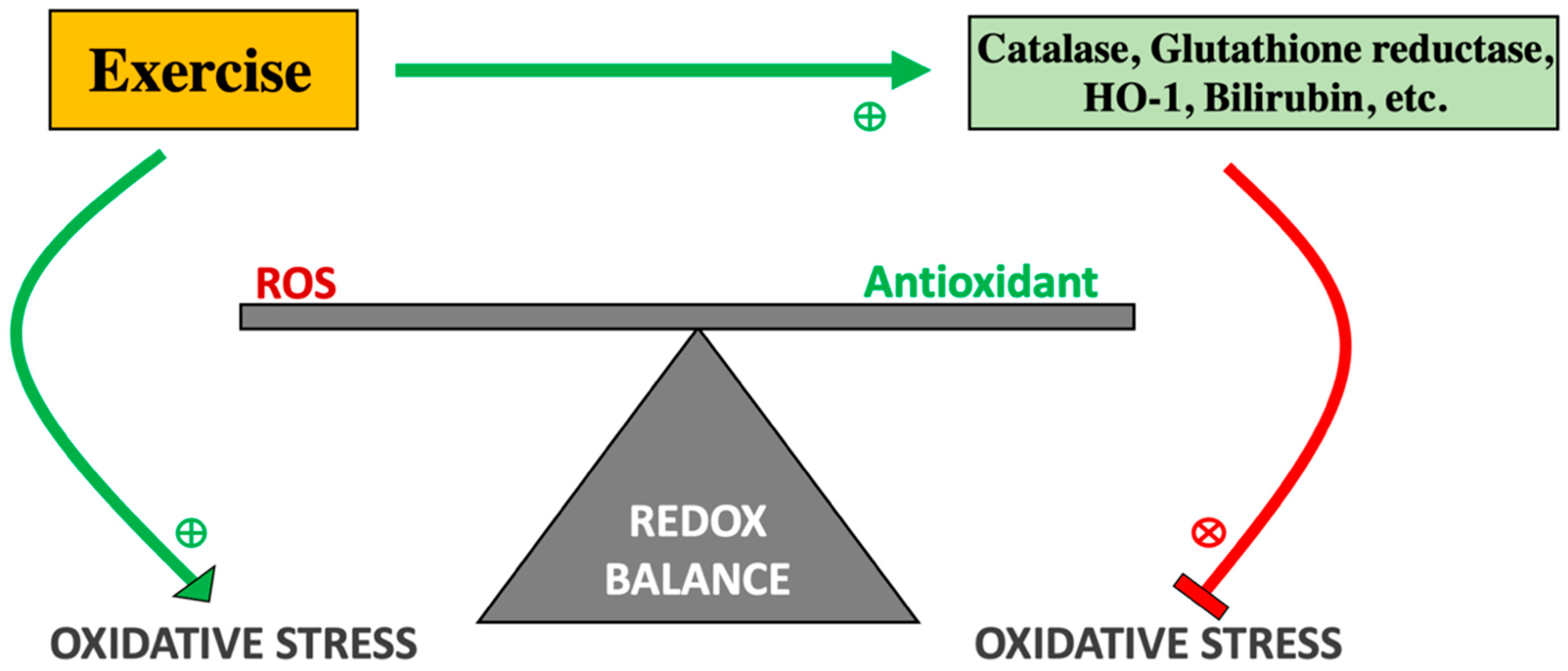
Exercise-Induced Formation of Reactive Oxygen Species and Select Antioxidant Defense Mechanisms
3. HO-1, BVRA, and Bilirubin as Inflammatory Mediators
3.1. Exercise-Induced HO-1 as a Mediator of Immune System Responses
3.2. The Emerging Role of Biliverdin Reductase in Immune Response
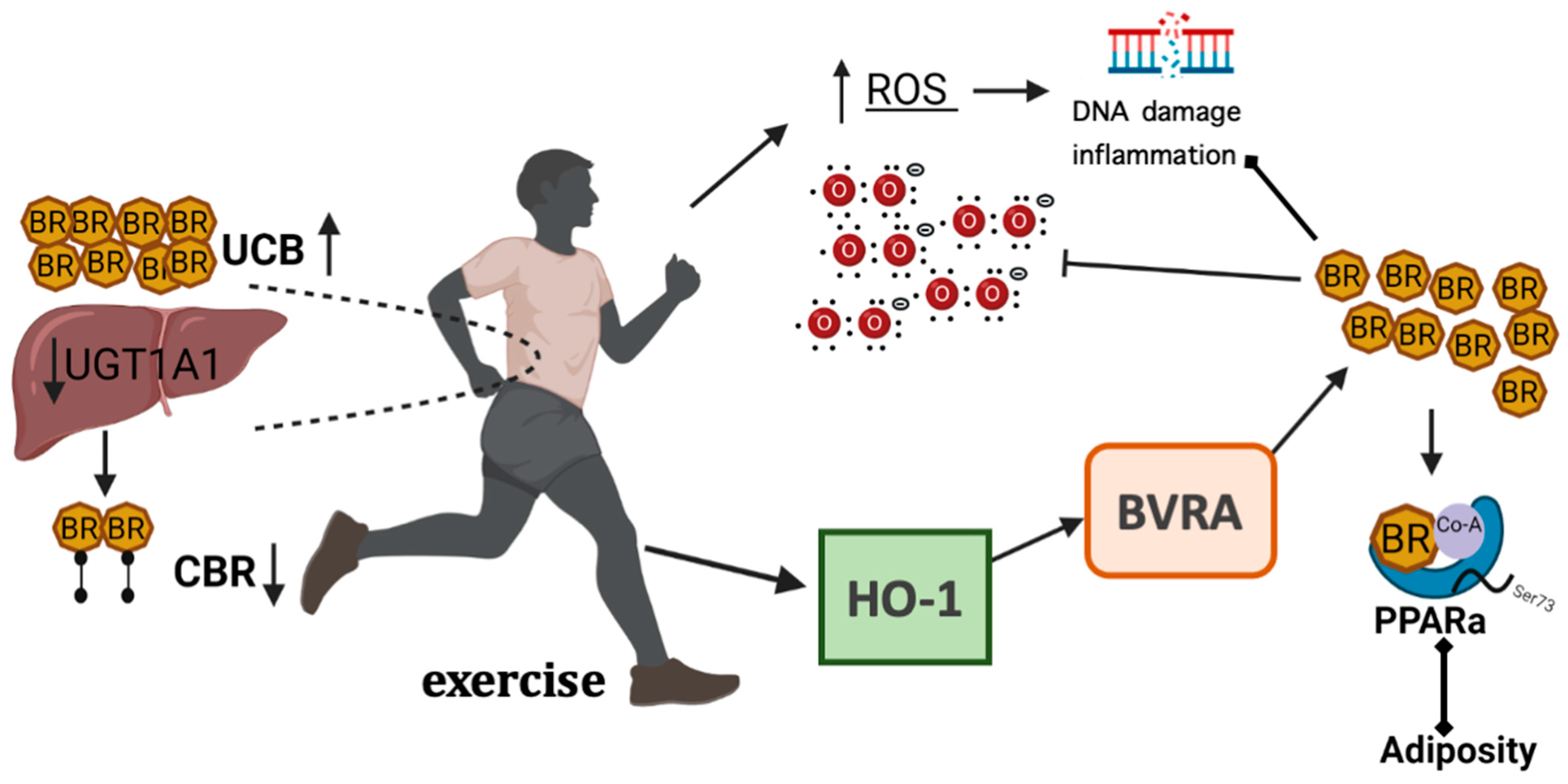
3.3. The Effect of Exercise on Bilirubin and Its Actions
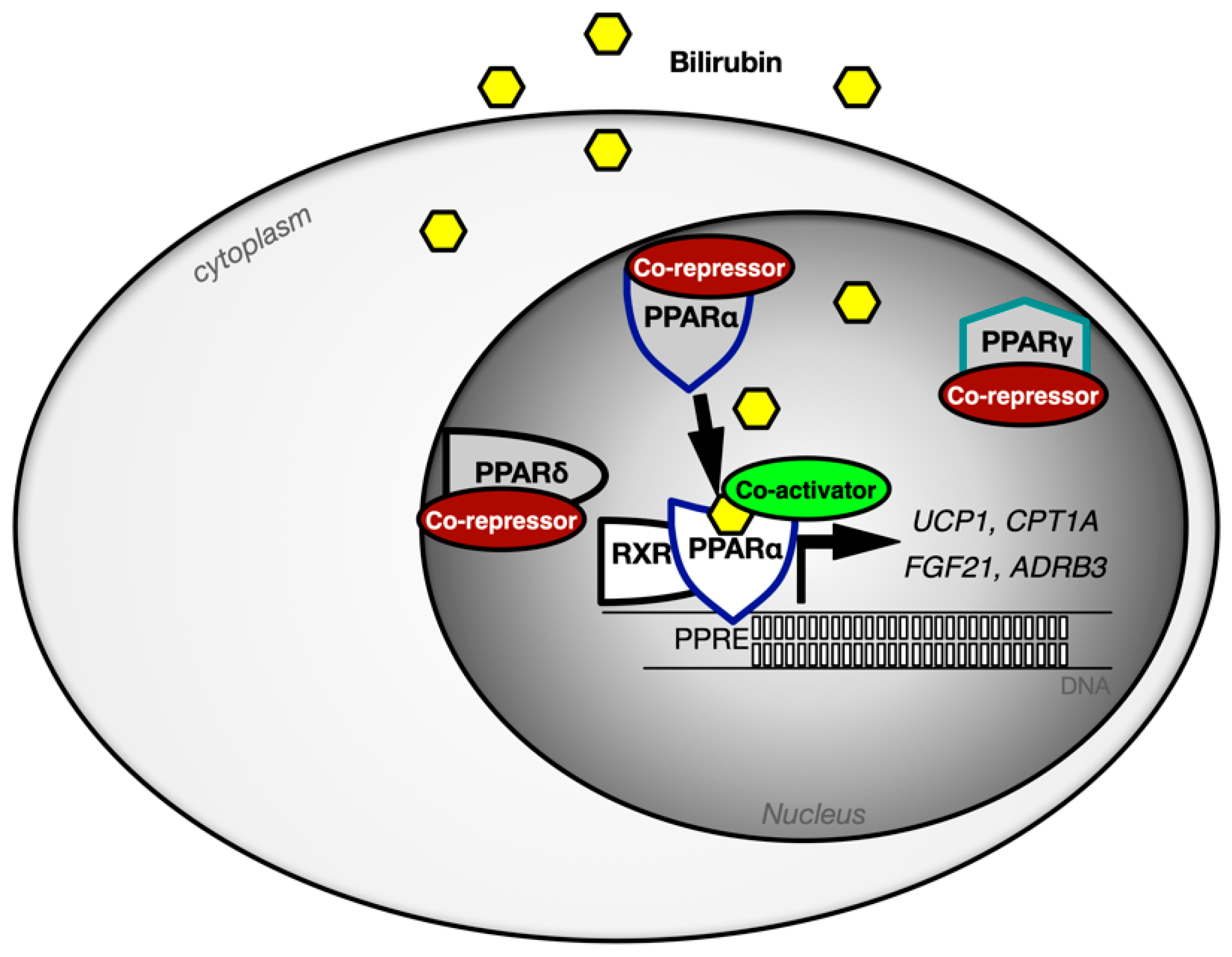
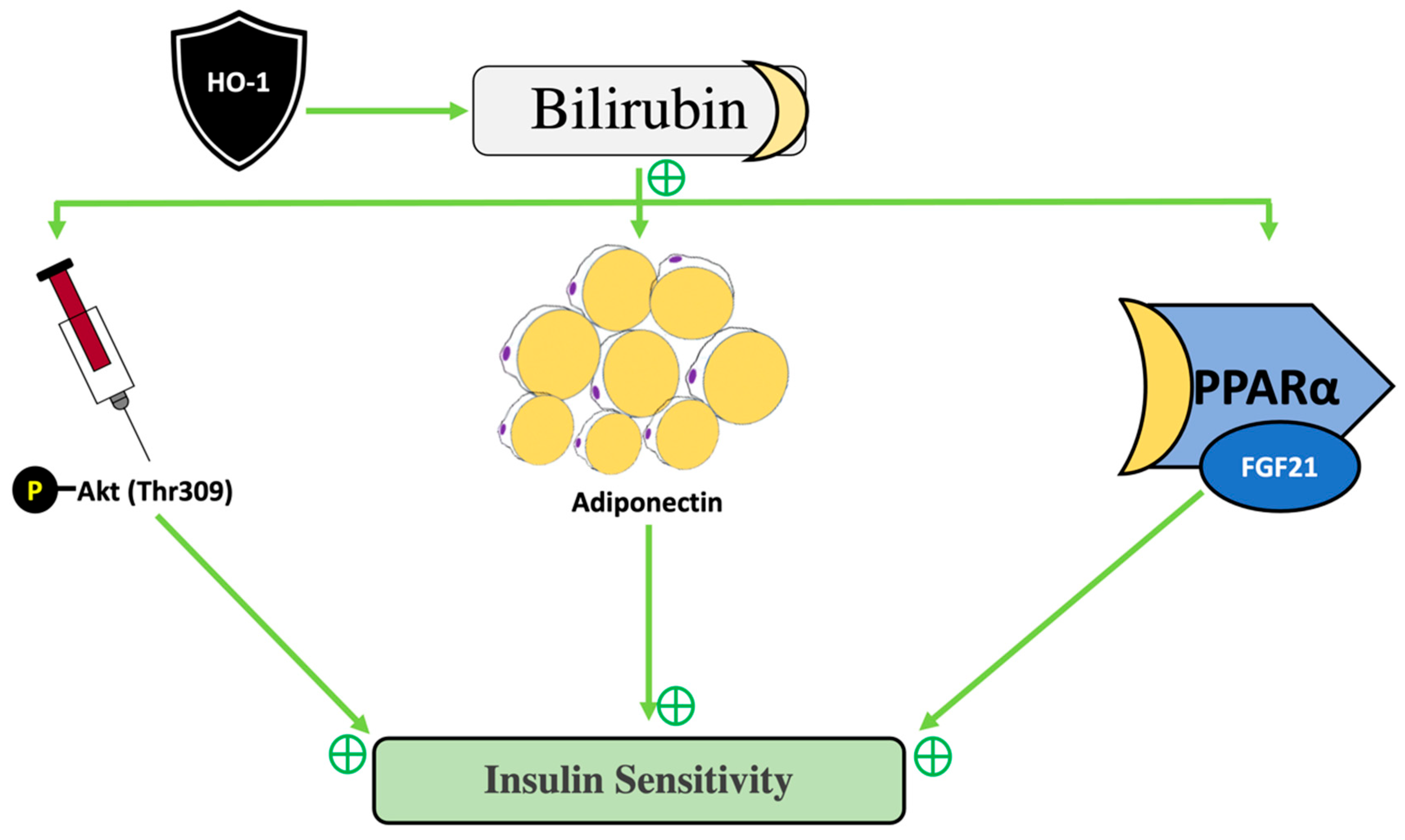
3.4. The Hormonal Function of Bilirubin in Exercise and the Impact of PPAR Signaling
4. The Signaling Mechanisms of Heme Oxygenase and Bilirubin in Metabolism
4.1. Generation and Catabolism of Bilirubin
4.2. Biliverdin Reductase and Metabolism
4.3. Bilirubin and Metabolic Dysfunction
5. Strategies to Improve Metabolic Outcomes through Nutraceuticals
5.1. The Influence of Diet on Antioxidants
5.2. The Benefits of Moderately Raising Plasma Bilirubin
5.3. Vitamin D Repletion
5.4. Nitrate from Foods and Dietary Supplements
5.5. Vitamin E Supplementation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Budui, S.L.; Rossi, A.P.; Zamboni, M. The pathogenetic bases of sarcopenia. Clin. Cases Miner. Bone Metab. 2015, 12, 22–26. [Google Scholar] [CrossRef]
- Dewidar, B.; Kahl, S.; Pafili, K.; Roden, M. Metabolic liver disease in diabetes—From mechanisms to clinical trials. Metabolism 2020, 111, 154299. [Google Scholar] [CrossRef] [PubMed]
- Takada, S.; Sabe, H.; Kinugawa, S. Abnormalities of Skeletal Muscle, Adipocyte Tissue, and Lipid Metabolism in Heart Failure: Practical Therapeutic Targets. Front. Cardiovasc. Med. 2020, 12, 79. [Google Scholar] [CrossRef] [PubMed]
- Allison, D.B.; Fontaine, K.R.; Manson, J.E.; Stevens, J.; VanItallie, T.B. Annual deaths attributable to obesity in the United States. JAMA 1999, 282, 1530–1538. [Google Scholar] [CrossRef]
- Williams, E.P.; Mesidor, M.; Winters, K.; Dubbert, P.M.; Wyatt, S.B. Overweight and Obesity: Prevalence, Consequences, and Causes of a Growing Public Health Problem. Curr. Obes. Rep. 2015, 4, 363–370. [Google Scholar] [CrossRef]
- Ogden, C.L.; Carroll, M.D.; Kit, B.K.; Flegal, K.M. Prevalence of obesity among adults: United States, 2011–2012. NCHS Data Brief 2013, 131, 1–8. [Google Scholar]
- National Center for Health Statistics. Health, United States, 2016: With Chart Book on Long-Term Trends in Health; National Center for Health Statistics: Hyattsville, MD, USA, 2017.
- Thomas, D.M.; Bouchard, C.; Church, T.; Slentz, C.; Kraus, W.E.; Redman, L.M.; Martin, C.K.; Silva, A.M.; Vossen, M.; Westerterp, K.; et al. Why do individuals not lose more weight from an exercise intervention at a defined dose? An energy balance analysis. Obes. Rev. 2012, 13, 835–847. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.M.; Kyle, T.K.; Stanford, F.C. The gap between expectations and reality of exercise-induced weight loss is associated with discouragement. Prev. Med. 2015, 81, 357–360. [Google Scholar] [CrossRef] [PubMed]
- King, N.A.; Caudwell, P.; Hopkins, M.; Byrne, N.M.; Colley, R.; Hills, A.P.; Stubbs, J.R.; Blundell, J.E. Metabolic and behavioral compensatory responses to exercise interventions: Barriers to weight loss. Obesity 2007, 15, 1373–1383. [Google Scholar] [CrossRef]
- Kirwan, J.P.; Sacks, J.; Nieuwoudt, S. The essential role of exercise in the management of type 2 diabetes. Clevel. Clin. J. Med. 2017, 84, S15–S21. [Google Scholar] [CrossRef]
- Winzer, E.B.; Woitek, F.; Linke, A. Physical Activity in the Prevention and Treatment of Coronary Artery Disease. J. Am. Heart Assoc. 2018, 7, e007725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitsett, M.; VanWagner, L.B. Physical activity as a treatment of non-alcoholic fatty liver disease: A systematic review. World J. Hepatol. 2015, 7, 2041–2052. [Google Scholar] [CrossRef] [PubMed]
- Vina, J.; Sanchis-Gomar, F.; Martinez-Bello, V.; Gomez-Cabrera, M.C. Exercise acts as a drug; the pharmacological benefits of exercise. Br. J. Pharmacol. 2012, 167, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Singh, R. The importance of exercise as a therapeutic agent. Malays. J. Med. Sci. 2002, 9, 7–16. [Google Scholar]
- Bird, S.R.; Hawley, J.A. Update on the effects of physical activity on insulin sensitivity in humans. BMJ. Open Sport Exerc. Med. 2017, 2, e000143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, L.; Hosick, P.A.; John, K.; Stec, D.E.; Hinds, T.D., Jr. Biliverdin reductase isozymes in metabolism. Trends Endocrinol. Metab. 2015, 26, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Colberg, S.R.; Sigal, R.J.; Fernhall, B.; Regensteiner, J.G.; Blissmer, B.J.; Rubin, R.R.; Chasan-Taber, L.; Albright, A.L.; Braun, B.; American College of Sports, M.; et al. Exercise and type 2 diabetes: The American College of Sports Medicine and the American Diabetes Association: Joint position statement. Diabetes Care 2010, 33, e147–e167. [Google Scholar] [CrossRef] [Green Version]
- Bruning, R.S.; Sturek, M. Benefits of exercise training on coronary blood flow in coronary artery disease patients. Prog. Cardiovasc. Dis. 2015, 57, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Gilbertson, N.M.; Eichner, N.Z.M.; Heiston, E.M.; Gaitan, J.M.; Francois, M.E.; Mehaffey, J.H.; Hassinger, T.E.; Hallowell, P.T.; Weltman, A.; Malin, S.K. A low-calorie diet with or without interval exercise training improves adiposopathy in obese women. Appl. Physiol. Nutr. Metab. 2019, 44, 1057–1064. [Google Scholar] [CrossRef]
- Sargeant, J.A.; Gray, L.J.; Bodicoat, D.H.; Willis, S.A.; Stensel, D.J.; Nimmo, M.A.; Aithal, G.P.; King, J.A. The effect of exercise training on intrahepatic triglyceride and hepatic insulin sensitivity: A systematic review and meta-analysis. Obes. Rev. 2018, 19, 1446–1459. [Google Scholar] [CrossRef]
- Francois, M.E.; Gilbertson, N.M.; Eichner, N.Z.M.; Heiston, E.M.; Fabris, C.; Breton, M.; Mehaffey, J.H.; Hassinger, T.; Hallowell, P.T.; Malin, S.K. Combining Short-Term Interval Training with Caloric Restriction Improves ss-Cell Function in Obese Adults. Nutrients 2018, 10, 717. [Google Scholar] [CrossRef] [Green Version]
- Mora-Rodriguez, R.; Ortega, J.F.; Ramirez-Jimenez, M.; Moreno-Cabanas, A.; Morales-Palomo, F. Insulin sensitivity improvement with exercise training is mediated by body weight loss in subjects with metabolic syndrome. Diabetes Metab. 2019, 46, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Haskell, W.L.; Lee, I.M.; Pate, R.R.; Powell, K.E.; Blair, S.N.; Franklin, B.A.; Macera, C.A.; Heath, G.W.; Thompson, P.D.; Bauman, A. Physical activity and public health: Updated recommendation for adults from the American College of Sports Medicine and the American Heart Association. Med. Sci. Sports Exerc. 2007, 39, 1423–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnelly, J.E.; Blair, S.N.; Jakicic, J.M.; Manore, M.M.; Rankin, J.W.; Smith, B.K.; American College of Sports Medicine. American College of Sports Medicine Position Stand. Appropriate physical activity intervention strategies for weight loss and prevention of weight regain for adults. Med. Sci. Sports Exerc. 2009, 41, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Prevention Program Research, G. Long-term effects of lifestyle intervention or metformin on diabetes development and microvascular complications over 15-year follow-up: The Diabetes Prevention Program Outcomes Study. Lancet Diabetes Endocrinol. 2015, 3, 866–875. [Google Scholar] [CrossRef] [Green Version]
- De Vries, T.I.; Dorresteijn, J.A.N.; van der Graaf, Y.; Visseren, F.L.J.; Westerink, J. Heterogeneity of Treatment Effects From an Intensive Lifestyle Weight Loss Intervention on Cardiovascular Events in Patients with Type 2 Diabetes: Data From the Look AHEAD Trial. Diabetes Care 2019, 42, 1988–1994. [Google Scholar] [CrossRef] [PubMed]
- Flack, K.D.; Hays, H.M.; Moreland, J.; Long, D.E. Exercise for Weight Loss: Further Evaluating Energy Compensation with Exercise. Med. Sci. Sports Exerc. 2020, 52, 2466–2475. [Google Scholar] [CrossRef]
- Flack, K.D.; Ufholz, K.; Johnson, L.; Fitzgerald, J.S.; Roemmich, J.N. Energy compensation in response to aerobic exercise training in overweight adults. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R619–R626. [Google Scholar] [CrossRef]
- Rosenkilde, M.; Auerbach, P.; Reichkendler, M.H.; Ploug, T.; Stallknecht, B.M.; Sjodin, A. Body fat loss and compensatory mechanisms in response to different doses of aerobic exercise—A randomized controlled trial in overweight sedentary males. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R571–R579. [Google Scholar] [CrossRef] [Green Version]
- Church, T.S.; Martin, C.K.; Thompson, A.M.; Earnest, C.P.; Mikus, C.R.; Blair, S.N. Changes in weight, waist circumference and compensatory responses with different doses of exercise among sedentary, overweight postmenopausal women. PLoS ONE 2009, 4, e4515. [Google Scholar] [CrossRef]
- King, N.A.; Hopkins, M.; Caudwell, P.; Stubbs, R.J.; Blundell, J.E. Individual variability following 12 weeks of supervised exercise: Identification and characterization of compensation for exercise-induced weight loss. Int. J. Obes. 2008, 32, 177–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, N.A.; Tremblay, A.; Blundell, J.E. Effects of exercise on appetite control: Implications for energy balance. Med. Sci. Sports Exerc. 1997, 29, 1076–1089. [Google Scholar] [CrossRef] [PubMed]
- Tryfidou, D.V.; McClean, C.; Nikolaidis, M.G.; Davison, G.W. NA Damage Following Acute Aerobic Exercise: A Systematic Review and Meta-analysis. Sports Med. 2020, 50, 103–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbacher, P.; Eckl, P. Impact of oxidative stress on exercising skeletal muscle. Biomolecules 2015, 5, 356–377. [Google Scholar] [CrossRef]
- Fittipaldi, S.; Mercatelli, I.; Caporossi, D. Role of exercise-induced reactive oxygen species in the modulation of heat shock protein response. Free Radic. Res. 2014, 48, 52–70. [Google Scholar] [CrossRef]
- Kawamura, T.; Muraoka, I. Exercise-Induced Oxidative Stress and the Effects of Antioxidant Intake from a Physiological Viewpoint. Antioxidants 2018, 7, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Cabrera, M.C.; Domenech, E.; Vina, J. Moderate exercise is an antioxidant: Upregulation of antioxidant genes by training. Free Radic. Biol. Med. 2008, 44, 126–131. [Google Scholar] [CrossRef] [PubMed]
- John, K.; Marino, J.S.; Sanchez, E.R.; Hinds, T.D., Jr. The glucocorticoid receptor: Cause of or cure for obesity? Am. J. Physiol. Endocrinol. Metab. 2016, 310, E249–E257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, F.L.; Song, W.; Jang, Y.C.; Liu, Y.; Sabia, M.; Richardson, A.; Van Remmen, H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1159–R1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creeden, J.F.; Gordon, D.M.; Stec, D.E.; Hinds, T.D., Jr. Bilirubin as a metabolic hormone: The physiological relevance of low levels. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E191–E207. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Stec, D.E. Bilirubin, a Cardiometabolic Signaling Molecule. Hypertension 2018, 72, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Ndisang, J.F. Role of Heme Oxygenase in Inflammation, Insulin-Signalling, Diabetes and Obesity. Mediat. Inflamm. 2010, 2010, 359732. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Huang, H.; Yun, X.; Kim, D.-s.; Yue, Y.; Wu, H.; Sutter, A.; Chavin, K.D.; Otterbein, L.E.; Adams, D.B.; et al. Bilirubin increases insulin sensitivity in leptin-receptor deficient and diet-induced obese mice through suppression of ER stress and chronic inflammation. Endocrinology 2014, 155, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Takei, R.; Inoue, T.; Sonoda, N.; Kohjima, M.; Okamoto, M.; Sakamoto, R.; Inoguchi, T.; Ogawa, Y. Bilirubin reduces visceral obesity and insulin resistance by suppression of inflammatory cytokines. PLoS ONE 2019, 14, e0223302. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Creeden, J.F.; Gordon, D.M.; Stec, D.F.; Donald, M.C.; Stec, D.E. Bilirubin Nanoparticles Reduce Diet-Induced Hepatic Steatosis, Improve Fat Utilization, and Increase Plasma beta-Hydroxybutyrate. Front. Pharm. 2020, 11, 594574. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Hosick, P.A.; Hankins, M.W.; Nestor-Kalinoski, A.; Stec, D.E. Mice with hyperbilirubinemia due to Gilbert’s Syndrome polymorphism are resistant to hepatic steatosis by decreased serine 73 phosphorylation of PPARalpha. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E244–E252. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Nakamura, K.; Kageyama, S.; Lawal, A.O.; Gong, K.W.; Bhetraratana, M.; Fujii, T.; Sulaiman, D.; Hirao, H.; Bolisetty, S.; et al. Myeloid HO-1 modulates macrophage polarization and protects against ischemia-reperfusion injury. JCI Insight 2018, 3, e120596. [Google Scholar] [CrossRef]
- Kato, H.; Amersi, F.; Buelow, R.; Melinek, J.; Coito, A.J.; Ke, B.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Heme Oxygenase-1 Overexpression Protects Rat Livers from Ischemia/Reperfusion Injury with Extended Cold Preservation. Am. J. Transplant. 2001, 1, 121–128. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Burns, K.A.; Hosick, P.A.; McBeth, L.; Nestor-Kalinoski, A.; Drummond, H.A.; AlAmodi, A.A.; Hankins, M.W.; Vanden Heuvel, J.P.; Stec, D.E. Biliverdin reductase A attenuates hepatic steatosis by inhibition of glycogen synthase kinase (GSK) 3beta phosphorylation of serine 73 of peroxisome proliferator-activated receptor (PPAR) alpha. J. Biol. Chem. 2016, 291, 25179–25191. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Tumanov, S.; Fazakerley, D.J.; Cantley, J.; James, D.E.; Dunn, L.L.; Shaik, T.; Suarna, C.; Stocker, R. Bilirubin deficiency renders mice susceptible to hepatic steatosis in the absence of insulin resistance. Redox Biol. 2021, 47, 102152. [Google Scholar] [CrossRef]
- Gobert, A.P.; Verriere, T.; Asim, M.; Barry, D.P.; Piazuelo, M.B.; de Sablet, T.; Delgado, A.G.; Bravo, L.E.; Correa, P.; Peek, R.M., Jr.; et al. Heme oxygenase-1 dysregulates macrophage polarization and the immune response to Helicobacter pylori. J. Immunol. 2014, 193, 3013–3022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Kim, D.H.; Tsenovoy, P.L.; Peterson, S.J.; Rezzani, R.; Rodella, L.F.; Aronow, W.S.; Ikehara, S.; Abraham, N.G. Treatment of obese diabetic mice with a heme oxygenase inducer reduces visceral and subcutaneous adiposity, increases adiponectin levels, and improves insulin sensitivity and glucose tolerance. Diabetes 2008, 57, 1526–1535. [Google Scholar] [CrossRef] [Green Version]
- Ndisang, J.F.; Lane, N.; Syed, N.; Jadhav, A. Up-regulating the heme oxygenase system with hemin improves insulin sensitivity and glucose metabolism in adult spontaneously hypertensive rats. Endocrinology 2010, 151, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Yu, C.-H.; Jen, C.-Y.; Cheng, C.-F.; Chou, Y.; Chang, C.-C.; Juan, S.-H. Adiponectin-mediated heme oxygenase-1 induction protects against iron-induced liver injury via a PPARα dependent mechanism. Am. J. Pathol. 2010, 177, 1697–1709. [Google Scholar] [CrossRef]
- Yang, M.; Kimura, M.; Ng, C.; He, J.; Keshvari, S.; Rose, F.J.; Barclay, J.L.; Whitehead, J.P. Induction of heme-oxygenase-1 (HO-1) does not enhance adiponectin production in human adipocytes: Evidence against a direct HO-1—Adiponectin axis. Mol. Cell. Endocrinol. 2015, 413, 209–216. [Google Scholar] [CrossRef]
- Araujo, J.A.; Zhang, M.; Yin, F. Heme oxygenase-1, oxidation, inflammation, and atherosclerosis. Front. Pharm. 2012, 3, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jais, A.; Einwallner, E.; Sharif, O.; Gossens, K.; Lu, T.T.-H.; Soyal, S.M.; Medgyesi, D.; Neureiter, D.; Paier-Pourani, J.; Dalgaard, K.; et al. Heme oxygenase-1 drives metaflammation and insulin resistance in mouse and man. Cell 2014, 158, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Ghio, A.J.; Case, M.W.; Soukup, J.M. Heme oxygenase activity increases after exercise in healthy volunteers. Free Radic. Res. 2018, 52, 267–272. [Google Scholar] [CrossRef]
- Niess, A.M.; Passek, F.; Lorenz, I.; Schneider, E.M.; Dickhuth, H.-H.; Northoff, H.; Fehrenbach, E. Expression of the antioxidant stress protein heme oxygenase-1 (HO-1) in human leukocytes: Acute and adaptational responses to endurance exercise. Free Radic. Biol. Med. 1999, 26, 184–192. [Google Scholar] [CrossRef]
- Kitamuro, T.; Takahashi, K.; Ogawa, K.; Udono-Fujimori, R.; Takeda, K.; Furuyama, K.; Nakayama, M.; Sun, J.; Fujita, H.; Hida, W.; et al. Bach1 functions as a hypoxia-inducible repressor for the heme oxygenase-1 gene in human cells. J. Biol. Chem. 2003, 278, 9125–9133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichard, J.F.; Sartor, M.A.; Puga, A. BACH1 is a specific repressor of HMOX1 that is inactivated by arsenite. J. Biol. Chem. 2008, 283, 22363–22370. [Google Scholar] [CrossRef] [Green Version]
- Stec, D.E.; Hinds, T.D., Jr. Natural Product Heme Oxygenase Inducers as Treatment for Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 9493. [Google Scholar] [CrossRef]
- Kurata, S.; Matsumoto, M.; Nakajima, H. Transcriptional control of the heme oxygenase gene in mouse M1 cells during their TPA-induced differentiation into macrophages. J. Cell. Biochem. 1996, 62, 314–324. [Google Scholar] [CrossRef]
- Falone, S.; Mirabilio, A.; Pennelli, A.; Cacchio, M.; Di Baldassarre, A.; Gallina, S.; Passerini, A.; Amicarelli, F. Differential impact of acute bout of exercise on redox- and oxidative damage-related profiles between untrained subjects and amateur runners. Physiol. Res. 2010, 59, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, B.; Baty, C.J.; Gallo, D.; Csizmadia, E.; Scott, J.R.; Akhavan, A.; Chin, B.Y.; Kaczmarek, E.; Alam, J.; Bach, F.H.; et al. Cell surface biliverdin reductase mediates biliverdin-induced anti-inflammatory effects via phosphatidylinositol 3-kinase and Akt. J. Biol. Chem. 2009, 284, 21369–21378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stechschulte, L.A.; Wuescher, L.; Marino, J.S.; Hill, J.W.; Eng, C.; Hinds, T.D., Jr. Glucocorticoid receptor beta stimulates Akt1 growth pathway by attenuation of PTEN. J. Biol. Chem. 2014, 289, 17885–17894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.Z.; Pei, G.C.; Wang, P.G.; Yang, J.; Zhu, F.M.; Guo, Y.J.; Wang, M.; Yao, Y.; Zeng, R.; Liao, W.H.; et al. Biliverdin Reductase A (BVRA) Mediates Macrophage Expression of Interleukin-10 in Injured Kidney. Int. J. Mol. Sci. 2015, 16, 22621–22635. [Google Scholar] [CrossRef]
- Bisht, K.; Canesin, G.; Cheytan, T.; Li, M.; Nemeth, Z.; Csizmadia, E.; Woodruff, T.M.; Stec, D.E.; Bulmer, A.C.; Otterbein, L.E.; et al. Deletion of Biliverdin Reductase A in Myeloid Cells Promotes Chemokine Expression and Chemotaxis in Part via a Complement C5a-C5aR1 Pathway. J. Immunol. 2019, 202, 2982–2990. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Creeden, J.F.; Gordon, D.M.; Spegele, A.C.; Britton, S.L.; Koch, L.G.; Stec, D.E. Rats Genetically Selected for High Aerobic Exercise Capacity Have Elevated Plasma Bilirubin by Upregulation of Hepatic Biliverdin Reductase-A (BVRA) and Suppression of UGT1A1. Antioxidants 2020, 9, 889. [Google Scholar] [CrossRef]
- Swift, D.L.; Johannsen, N.M.; Earnest, C.P.; Blair, S.N.; Church, T.S. Effect of different doses of aerobic exercise training on total bilirubin levels. Med. Sci. Sports Exerc. 2012, 44, 569–574. [Google Scholar] [CrossRef] [Green Version]
- Witek, K.; Scislowska, J.; Turowski, D.; Lerczak, K.; Lewandowska-Pachecka, S.; Pokrywka, A. Total bilirubin in athletes, determination of reference range. Biol. Sport 2017, 34, 45–48. [Google Scholar] [CrossRef]
- Priest, J.B.; Oei, T.O.; Moorehead, W.R. Exercise-induced changes in common laboratory tests. Am. J. Clin. Pathol. 1982, 77, 285–289. [Google Scholar] [CrossRef] [Green Version]
- Loprinzi, P.D.; Abbott, K. Physical activity and total serum bilirubin levels among insulin sensitive and insulin resistant U.S. adults. J. Diabetes Metab. Disord. 2014, 13, 47. [Google Scholar] [CrossRef] [Green Version]
- Fallon, K.E. The clinical utility of screening of biochemical parameters in elite athletes: Analysis of 100 cases. Br. J. Sports Med. 2008, 42, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Hammouda, O.; Chtouror, H.; Chaouachi, A.; Chahed, H.; Ferchichi, S.; Kallel, C.; Chamari, K.; Souissi, N. Effect of short-term maximal exercise on biochemical markers of muscle damage, total antioxidant status, and homocysteine levels in football players. Asian J. Sports Med. Clin. N. Am. 2012, 3, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Kratz, A.; Lewandowski, K.B.; Siegel, A.J.; Chun, K.Y.; Flood, J.G.; van Cott, E.M. Effect of marathon running on hematologic and biochemical laboratory parameters, including cardiac markers. Am. J. Clin. Pathol. 2002, 188, 856–863. [Google Scholar] [CrossRef]
- Drukalec-Michalski, K.; Nowaczyk, P.; Glowka, N.; Ziobrowska, A.; Podgorski, T. Is a Four-Week Ketogenic Diet an Effective Nutritional Strategy in CrossFit-Trained Female and Male Athletes? Nutrients 2021, 13, 864. [Google Scholar] [CrossRef]
- Schoonjans, K.; Martin, G.; Staels, B.; Auwerx, J. Peroxisome proliferator-activated receptors, orphans with ligands and functions. Curr. Opin. Lipidol. 1997, 8, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, K.; Puri, N.; Kim, D.H.; Hinds, T.D.; Stechschulte, L.A.; Favero, G.; Rodella, L.; Shapiro, J.I.; Jude, D.; Abraham, N.G. PPARdelta binding to heme oxygenase 1 promoter prevents angiotensin II-induced adipocyte dysfunction in Goldblatt hypertensive rats. Int. J. Obes. 2014, 38, 456–465. [Google Scholar] [CrossRef] [Green Version]
- Fedorova, L.V.; Sodhi, K.; Gatto-Weis, C.; Puri, N.; Hinds, T.D., Jr.; Shapiro, J.I.; Malhotra, D. Peroxisome proliferator-activated receptor delta agonist, HPP593, prevents renal necrosis under chronic ischemia. PLoS ONE 2013, 8, e64436. [Google Scholar] [CrossRef]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Smedlund, K.B.; Sanchez, E.R.; Hinds, T.D., Jr. FKBP51 and the molecular chaperoning of metabolism. Trends Endocrinol. Metab. 2021, 32, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D.; John, K.; McBeth, L.; Trabbic, C.J.; Sanchez, E.R. Timcodar (VX-853) Is a Non-FKBP12 Binding Macrolide Derivative That Inhibits PPARγand Suppresses Adipogenesis. PPAR Res. 2016, 2016, 6218637. [Google Scholar] [CrossRef] [Green Version]
- Hinds, T.D., Jr.; Stechschulte, L.A.; Cash, H.A.; Whisler, D.; Banerjee, A.; Yong, W.; Khuder, S.S.; Kaw, M.K.; Shou, W.; Najjar, S.M.; et al. Protein phosphatase 5 mediates lipid metabolism through reciprocal control of glucocorticoid receptor and peroxisome proliferator-activated receptor-gamma (PPARgamma). J. Biol. Chem. 2011, 286, 42911–42922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, M.; McBeth, L.; Sindhwani, P.; Hinds, T.D. Deciphering the Roles of Thiazolidinediones and PPARgamma in Bladder Cancer. PPAR Res. 2017, 2017, 4810672. [Google Scholar] [CrossRef] [Green Version]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stec, D.E.; Gordon, D.M.; Hipp, J.A.; Hong, S.; Mitchell, Z.L.; Franco, N.R.; Robison, J.W.; Anderson, C.D.; Stec, D.F.; Hinds, T.D., Jr. The loss of hepatic PPARalpha promotes inflammation and serum hyperlipidemia in diet-induced obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R733–R745. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Kipp, Z.A.; Xu, M.; Yiannikouris, F.B.; Morris, A.J.; Stec, D.F.; Wahli, W.; Stec, D.E. Adipose-Specific PPARalpha Knockout Mice Have Increased Lipogenesis by PASK-SREBP1 Signaling and a Polarity Shift to Inflammatory Macrophages in White Adipose Tissue. Cells 2021, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Marino, J.S.; Stechschulte, L.A.; Stec, D.E.; Nestor-Kalinoski, A.; Coleman, S.; Hinds, T.D., Jr. Glucocorticoid receptor beta induces hepatic steatosis by augmenting inflammation and inhibition of the peroxisome proliferator-activated receptor (PPAR) alpha. J. Biol. Chem. 2016, 291, 25776–25788. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.M.; Hong, S.H.; Kipp, Z.A.; Hinds, T.D., Jr. Identification of Binding Regions of Bilirubin in the Ligand-Binding Pocket of the Peroxisome Proliferator-Activated Receptor-A (PPARalpha). Molecules 2021, 26, 2975. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.M.; Neifer, K.L.; Hamoud, A.A.; Hawk, C.F.; Nestor-Kalinoski, A.L.; Miruzzi, S.A.; Morran, M.P.; Adeosun, S.O.; Sarver, J.G.; Erhardt, P.W.; et al. Bilirubin remodels murine white adipose tissue by reshaping mitochondrial activity and the coregulator profile of peroxisome proliferator-activated receptor alpha. J. Biol. Chem. 2020, 295, 9804–9822. [Google Scholar] [CrossRef]
- Gordon, D.M.; Blomquist, T.M.; Miruzzi, S.A.; McCullumsmith, R.; Stec, D.E.; Hinds, T.D., Jr. RNA sequencing in human HepG2 hepatocytes reveals PPAR-alpha mediates transcriptome responsiveness of bilirubin. Physiol. Genom. 2019, 51, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Stec, D.E.; John, K.; Trabbic, C.J.; Luniwal, A.; Hankins, M.W.; Baum, J.; Hinds, T.D., Jr. Bilirubin Binding to PPARα Inhibits Lipid Accumulation. PLoS ONE 2016, 11, e0153427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenson, R.S. Fenofibrate: Treatment of hyperlipidemia and beyond. Expert Rev. Cardiovasc. 2008, 6, 1319–1330. [Google Scholar] [CrossRef]
- Iemitsu, M.; Miyauchi, T.; Maeda, S.; Tanabe, T.; Takanashi, M.; Irukayama-Tomobe, Y.; Sakai, S.; Ohmori, H.; Matsuda, M.; Yamaguchi, I. Aging-induced decrease in the PPAR-alpha level in hearts is improved by exercise training. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1750–H1760. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.; Li, Q.; Dong, X.; Hu, H.; Hu, R.; Ye, H.; Wu, Y.; Hu, R.; Li, Y. Exercise improved rat metabolism by raising PPAR-α. Int. J. Sports Med. 2011, 32, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Stanislaus, S.; Chinookoswong, N.; Lau, Y.Y.; Hager, T.; Patel, J.; Ge, H.; Weiszmann, J.; Lu, S.-C.; Graham, M.; et al. Acute glucose-lowering and insulin-sensitizing action of FGF21 in insulin-resistant mouse models—Association with liver and adipose tissue effects. Am. J. Physiol.-Endocrinol. Metab. 2009, 297, E1105–E1114. [Google Scholar] [CrossRef] [Green Version]
- Tezze, C.; Romanello, V.; Sandri, M. FGF21 as Modulator of Metabolism in Health and Disease. Front. Physiol. 2019, 10, 419. [Google Scholar] [CrossRef]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular Actions of PPARα in Lipid Metabolism and Inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras, A.V.; Torres, N.; Tovar, A.R. PPAR-α as a Key Nutritional and Environmental Sensor for Metabolic Adaptation. Adv. Nutr. 2013, 4, 439–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; He, Y.; Chung, P.K.; Tong, T.K.; Fu, F.H.; Chen, Y.; Jiao, G. Effects of 12 Weeks of Exercise on Hepatic TNF-α and PPARα in an Animal Model of High-Fat Diet-Induced Nonalcoholic Steatohepatitis. J. Exerc. Sci. Fit. 2009, 7, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Tanimura, Y.; Aoi, W.; Takanami, Y.; Kawai, Y.; Mizushima, K.; Naito, Y.; Yoshikawa, T. Acute exercise increases fibroblast growth factor 21 in metabolic organs and circulation. Physiol. Rep. 2016, 4, e12828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muoio, D.M.; MacLean, P.S.; Lang, D.B.; Li, S.; Houmard, J.A.; Way, J.M.; Winegar, D.A.; Corton, J.C.; Dohm, G.L.; Kraus, W.E. Fatty acid homeostasis and induction of lipid regulatory genes in skeletal muscles of peroxisome proliferator-activated receptor (PPAR) alpha knock-out mice. Evidence for compensatory regulation by PPAR delta. J. Biol. Chem. 2002, 277, 26089–26097. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Leon, S.; Tuvblad, C.; Forero, D.A. Sports genetics: The PPARA gene and athletes’ high ability in endurance sports. A systematic review and meta-analysis. Biol. Sport 2016, 33, 3–6. [Google Scholar] [CrossRef]
- Russell, A.P.; Feilchenfeldt, J.; Schreiber, S.; Praz, M.; Crettenand, A.; Gobelet, C.; Meier, C.A.; Bell, D.R.; Kralli, A.; Giacobino, J.P.; et al. Endurance training in humans leads to fiber type-specific increases in levels of peroxisome proliferator-activated receptor-gamma coactivator-1 and peroxisome proliferator-activated receptor-alpha in skeletal muscle. Diabetes 2003, 52, 2874–2881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritz, T.; Krämer, D.K.; Karlsson, H.K.; Galuska, D.; Engfeldt, P.; Zierath, J.R.; Krook, A. Low-intensity exercise increases skeletal muscle protein expression of PPARdelta and UCP3 in type 2 diabetic patients. Diabetes Metab. Res. Rev. 2006, 22, 492–498. [Google Scholar] [CrossRef]
- Kannisto, K.; Chibalin, A.; Glinghammar, B.; Zierath, J.R.; Hamsten, A.; Ehrenborg, E. Differential expression of peroxisomal proliferator activated receptors alpha and delta in skeletal muscle in response to changes in diet and exercise. Int. J. Mol. Med. 2006, 17, 45–52. [Google Scholar]
- Rachid, T.L.; Silva-Veiga, F.M.; Graus-Nunes, F.; Bringhenti, I.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. Differential actions of PPAR-α and PPAR-β/δ on beige adipocyte formation: A study in the subcutaneous white adipose tissue of obese male mice. PLoS ONE 2018, 13, e0191365. [Google Scholar] [CrossRef] [PubMed]
- Varga, T.; Czimmerer, Z.; Nagy, L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim. Biophys. Acta 2011, 1812, 1007–1022. [Google Scholar] [CrossRef]
- Thomas, A.W.; Davies, N.A.; Moir, H.; Watkeys, L.; Ruffino, J.S.; Isa, S.A.; Butcher, L.R.; Hughes, M.G.; Morris, K.; Webb, R. Exercise-associated generation of PPARγ ligands activates PPARγ signaling events and upregulates genes related to lipid metabolism. J. Appl Physiol. 2012, 112, 806–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Wang, D.; Zhao, W.; Xu, L. Deciphering the Roles of PPARγ in Adipocytes via Dynamic Change of Transcription Complex. Front. Endocrinol. 2018, 9, 473. [Google Scholar] [CrossRef] [Green Version]
- Vidal-Puig, A.J.; Considine, R.V.; Jimenez-Liñan, M.; Werman, A.; Pories, W.J.; Caro, J.F.; Flier, J.S. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J. Clin. Investig. 1997, 99, 2416–2422. [Google Scholar] [CrossRef] [PubMed]
- Verreth, W.; Keyzer, D.D.; Pelat, M.; Verhamme, P.; Ganame, J.; Bielicki, J.K.; Mertens, A.; Quarck, R.; Benhabilès, N.; Marguerie, G.; et al. Weight Loss–Associated Induction of Peroxisome Proliferator–Activated Receptor-α;and Peroxisome Proliferator–Activated Receptor-γ Correlate with Reduced Atherosclerosis and Improved Cardiovascular Function in Obese Insulin-Resistant Mice. Circulation 2004, 110, 3259–3269. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Mullican, S.E.; DiSpirito, J.R.; Peed, L.C.; Lazar, M.A. Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARγ. Proc. Natl. Acad. Sci. USA 2013, 110, 18656–18661. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.R.; Barrick, C.; Kim, K.-A.; Lindner, J.; Blondeau, B.; Fujimoto, Y.; Shiota, M.; Kesterson, R.A.; Kahn, B.B.; Magnuson, M.A. Deletion of PPARgamma in adipose tissues of mice protects against high fat diet-induced obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2005, 102, 6207–6212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Dong, H.; Zhang, Y.; Cao, M.; Song, L.; Pan, Q.; Bulmer, A.; Adams, D.B.; Dong, X.; Wang, H. Bilirubin Increases Insulin Sensitivity by Regulating Cholesterol Metabolism, Adipokines and PPARγ Levels. Sci. Rep. 2015, 5, 9886. [Google Scholar] [CrossRef] [Green Version]
- Shiels, R.G.; Vidimce, J.; Pearson, A.G.; Matthews, B.; Wagner, K.-H.; Battle, A.R.; Sakellaris, H.; Bulmer, A.C. Unprecedented Microbial Conversion of Biliverdin into Bilirubin-10-sulfonate. Sci. Rep. 2019, 9, 2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinds, T.D., Jr.; Stec, D.E. Bilirubin Safeguards Cardiorenal and Metabolic Diseases: A Protective Role in Health. Curr. Hypertens. Rep. 2019, 21, 87. [Google Scholar] [CrossRef]
- Weaver, L.; Hamoud, A.R.; Stec, D.E.; Hinds, T.D., Jr. Biliverdin reductase and bilirubin in hepatic disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G668–G676. [Google Scholar] [CrossRef]
- Sundararaghavan, V.L.; Binepal, S.; Stec, D.E.; Sindhwani, P.; Hinds, T.D., Jr. Bilirubin, a new therapeutic for kidney transplant? Transpl. Rev. 2018, 32, 234–240. [Google Scholar] [CrossRef]
- Hamoud, A.R.; Weaver, L.; Stec, D.E.; Hinds, T.D., Jr. Bilirubin in the Liver-Gut Signaling Axis. Trends Endocrinol. Metab. 2018, 29, 140–150. [Google Scholar] [CrossRef]
- Adeosun, S.O.; Moore, K.H.; Lang, D.M.; Nwaneri, A.C.; Hinds, T.D., Jr.; Stec, D.E. A Novel Fluorescence-Based Assay for the Measurement of Biliverdin Reductase Activity. React. Oxyg. Species 2018, 5, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, D.M.; Adeosun, S.O.; Ngwudike, S.I.; Anderson, C.D.; Hall, J.E.; Hinds, T.D., Jr.; Stec, D.E. CRISPR Cas9-mediated deletion of biliverdin reductase A (BVRA) in mouse liver cells induces oxidative stress and lipid accumulation. Arch. Biochem. Biophys. 2019, 672, 108072. [Google Scholar] [CrossRef]
- Adeosun, S.O.; Gordon, D.M.; Weeks, M.F.; Moore, K.H.; Hall, J.E.; Hinds, T.D., Jr.; Stec, D.E. Loss of biliverdin reductase-A promotes lipid accumulation and lipotoxicity in mouse proximal tubule cells. Am. J. Physiol. Ren. Physiol. 2018, 315, F323–F331. [Google Scholar] [CrossRef]
- Sundararaghavan, V.L.; Sindhwani, P.; Hinds, T.D., Jr. Glucuronidation and UGT isozymes in bladder: New targets for the treatment of uroepithelial carcinomas? Oncotarget 2017, 8, 3640–3648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stec, D.E.; Gordon, D.M.; Nestor-Kalinoski, A.L.; Donald, M.C.; Mitchell, Z.L.; Creeden, J.F.; Hinds, T.D., Jr. Biliverdin Reductase A (BVRA) Knockout in Adipocytes Induces Hypertrophy and Reduces Mitochondria in White Fat of Obese Mice. Biomolecules 2020, 10, 387. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, V.; Barchetta, I.; Cimini, F.A.; Bertoccini, L.; Chiappetta, C.; Capoccia, D.; Carletti, R.; Di Cristofano, C.; Silecchia, G.; Fontana, M.; et al. Reduced Biliverdin Reductase-A Expression in Visceral Adipose Tissue is Associated with Adipocyte Dysfunction and NAFLD in Human Obesity. Int. J. Mol. Sci. 2020, 21, 9091. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Di Domenico, F.; Cassano, T.; Arena, A.; Tramutola, A.; Lavecchia, M.A.; Coccia, R.; Butterfield, D.A.; Perluigi, M. Impairment of biliverdin reductase-A promotes brain insulin resistance in Alzheimer disease: A new paradigm. Free Radic. Biol. Med. 2016, 91, 127–142. [Google Scholar] [CrossRef]
- Triani, F.; Tramutola, A.; Di Domenico, F.; Sharma, N.; Butterfield, D.A.; Head, E.; Perluigi, M.; Barone, E. Biliverdin reductase-A impairment links brain insulin resistance with increased Abeta production in an animal model of aging: Implications for Alzheimer disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3181–3194. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Tramutola, A.; Triani, F.; Calcagnini, S.; Di Domenico, F.; Ripoli, C.; Gaetani, S.; Grassi, C.; Butterfield, D.A.; Cassano, T.; et al. Biliverdin Reductase-A Mediates the Beneficial Effects of Intranasal Insulin in Alzheimer Disease. Mol. Neurobiol. 2019, 56, 2922–2943. [Google Scholar] [CrossRef]
- McCarty, M.F. Serum bilirubin may serve as a marker for increased heme oxygenase activity and inducibility in tissues—A rationale for the versatile health protection associated with elevated plasma bilirubin. Med. Hypotheses 2013, 81, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Chang, P.F.; Hu, F.C.; Chang, M.H.; Ni, Y.H. Variants in the UGT1A1 gene and the risk of pediatric nonalcoholic fatty liver disease. Pediatrics 2009, 124, e1221–e1227. [Google Scholar] [CrossRef] [PubMed]
- Verlaan, S.; Maier, A.B.; Bauer, J.M.; Bautmans, I.; Brandt, K.; Donini, L.M.; Maggio, M.; McMurdo, M.E.; Mets, T.; Seal, C.; et al. Sufficient levels of 25-hydroxyvitamin D and protein intake required to increase muscle mass in sarcopenic older adults—The PROVIDE study. Clin. Nutr. 2017, 37, 551–557. [Google Scholar] [CrossRef]
- Lee, M.J.; Jung, C.H.; Kang, Y.M.; Hwang, J.Y.; Jang, J.E.; Leem, J.; Park, J.Y.; Kim, H.K.; Lee, W.J. Serum bilirubin as a predictor of incident metabolic syndrome: A 4-year retrospective longitudinal study of 6205 initially healthy Korean men. Diabetes Metab. 2014, 40, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Turfan, M.; Duran, M.; Poyraz, F.; Yayla, C.; Akboga, M.K.; Sahinarslan, A.; Tavil, Y.; Pasaoglu, H.; Boyaci, B. Inverse relationship between serum total bilirubin levels and severity of disease in patients with stable coronary artery disease. Coron. Artery Dis. 2013, 24, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Jang, B.K. Elevated serum bilirubin levels are inversely associated with nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2012, 18, 357–359. [Google Scholar] [CrossRef]
- Higuchi, S.; Kabeya, Y.; Uchida, J.; Kato, K.; Tsukada, N. Low Bilirubin Levels Indicate a High Risk of Cerebral Deep White Matter Lesions in Apparently Healthy Subjects. Sci. Rep. 2018, 8, 6473. [Google Scholar] [CrossRef] [PubMed]
- Garde, E.; Mortensen, E.L.; Krabbe, K.; Rostrup, E.; Larsson, H.B. Relation between age-related decline in intelligence and cerebral white-matter hyperintensities in healthy octogenarians: A longitudinal study. Lancet 2000, 356, 628–634. [Google Scholar] [CrossRef]
- Verdelho, A.; Madureira, S.; Moleiro, C.; Ferro, J.M.; Santos, C.O.; Erkinjuntti, T.; Pantoni, L.; Fazekas, F.; Visser, M.; Waldemar, G.; et al. White matter changes and diabetes predict cognitive decline in the elderly: The LADIS study. Neurology 2010, 75, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Debette, S.; Markus, H.S. The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: Systematic review and meta-analysis. BMJ 2010, 341, c3666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benzie, I.F. Evolution of dietary antioxidants. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2003, 136, 113–126. [Google Scholar] [CrossRef]
- Watzl, B.; Kulling, S.E.; Moseneder, J.; Barth, S.W.; Bub, A. A 4-wk intervention with high intake of carotenoid-rich vegetables and fruit reduces plasma C-reactive protein in healthy, nonsmoking men. Am. J. Clin. Nutr. 2005, 82, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Nanri, A.; Moore, M.A.; Kono, S. Impact of C-reactive protein on disease risk and its relation to dietary factors. Asian Pac. J. Cancer Prev. 2007, 8, 167–177. [Google Scholar]
- Esposito, K.; Marfella, R.; Ciotola, M.; Di Palo, C.; Giugliano, F.; Giugliano, G.; D’Armiento, M.; D’Andrea, F.; Giugliano, D. Effect of a mediterranean-style diet on endothelial dysfunction and markers of vascular inflammation in the metabolic syndrome: A randomized trial. JAMA 2004, 292, 1440–1446. [Google Scholar] [CrossRef] [Green Version]
- Salas-Salvado, J.; Garcia-Arellano, A.; Estruch, R.; Marquez-Sandoval, F.; Corella, D.; Fiol, M.; Gomez-Gracia, E.; Vinoles, E.; Aros, F.; Herrera, C.; et al. Components of the Mediterranean-type food pattern and serum inflammatory markers among patients at high risk for cardiovascular disease. Eur. J. Clin. Nutr. 2008, 62, 651–659. [Google Scholar] [CrossRef] [Green Version]
- Chrysohoou, C.; Panagiotakos, D.B.; Pitsavos, C.; Das, U.N.; Stefanadis, C. Adherence to the Mediterranean diet attenuates inflammation and coagulation process in healthy adults: The ATTICA Study. J. Am. Coll. Cardiol. 2004, 44, 152–158. [Google Scholar] [CrossRef] [Green Version]
- Tuttolomondo, A.; Simonetta, I.; Daidone, M.; Mogavero, A.; Ortello, A.; Pinto, A. Metabolic and Vascular Effect of the Mediterranean Diet. Int. J. Mol. Sci. 2019, 20, 4716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mentella, M.C.; Scaldaferri, F.; Ricci, C.; Gasbarrini, A.; Miggiano, G.A.D. Cancer and Mediterranean Diet: A Review. Nutrients 2019, 11, 2059. [Google Scholar] [CrossRef] [Green Version]
- Martín-Peláez, S.; Fito, M.; Castaner, O. Mediterranean Diet Effects on Type 2 Diabetes Prevention, Disease Progression, and Related Mechanisms. A Review. Nutrients 2020, 12, 2236. [Google Scholar] [CrossRef]
- Meddeb, W.; Rezig, L.; Abderrabba, M.; Lizard, G.; Mejri, M. Tunisian Milk Thistle: An Investigation of the Chemical Composition and the Characterization of Its Cold-Pressed Seed Oils. Int. J. Mol. Sci. 2017, 18, 2582. [Google Scholar] [CrossRef] [Green Version]
- Pittala, V.; Vanella, L.; Salerno, L.; Romeo, G.; Marrazzo, A.; Di Giacomo, C.; Sorrenti, V. Effects of Polyphenolic Derivatives on Heme Oxygenase-System in Metabolic Dysfunctions. Curr. Med. Chem. 2018, 25, 1577–1595. [Google Scholar] [CrossRef] [PubMed]
- Suk, J.; Jasprova, J.; Biedermann, D.; Petraskova, L.; Valentova, K.; Kren, V.; Muchova, L.; Vitek, L. Isolated Silymarin Flavonoids Increase Systemic and Hepatic Bilirubin Concentrations and Lower Lipoperoxidation in Mice. Oxid. Med. Cell. Longev. 2019, 2019, 6026902. [Google Scholar] [CrossRef] [PubMed]
- Bechynska, K.; Kosek, V.; Fenclova, M.; Muchova, L.; Smid, V.; Suk, J.; Chalupsky, K.; Sticova, E.; Hurkova, K.; Hajslova, J.; et al. The Effect of Mycotoxins and Silymarin on Liver Lipidome of Mice with Non-Alcoholic Fatty Liver Disease. Biomolecules 2021, 11, 1723. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Wang, H. Silymarin attenuated hepatic steatosis through regulation of lipid metabolism and oxidative stress in a mouse model of nonalcoholic fatty liver disease (NAFLD). Am. J. Transl. Res. 2016, 8, 1073–1081. [Google Scholar]
- Clichici, S.; Olteanu, D.; Nagy, A.L.; Oros, A.; Filip, A.; Mircea, P.A. Silymarin inhibits the progression of fibrosis in the early stages of liver injury in CCl(4)-treated rats. J. Med. Food 2015, 18, 290–298. [Google Scholar] [CrossRef]
- Gupta, N.; Singh, T.; Chaudhary, R.; Garg, S.K.; Sandhu, G.S.; Mittal, V.; Gupta, R.; Bodin, R.; Sule, S. Bilirubin in coronary artery disease: Cytotoxic or protective? World J. Gastrointest. Pharm. 2016, 7, 469–476. [Google Scholar] [CrossRef]
- Ziberna, L.; Martelanc, M.; Franko, M.; Passamonti, S. Bilirubin is an Endogenous Antioxidant in Human Vascular Endothelial Cells. Sci. Rep. 2016, 6, 29240. [Google Scholar] [CrossRef] [Green Version]
- Cesari, M.; Incalzi, R.A.; Zamboni, V.; Pahor, M. Vitamin D hormone: A multitude of actions potentially influencing the physical function decline in older persons. Geriatr. Gerontol. Int. 2011, 11, 133–142. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Iwamoto, J.; Kanoko, T.; Satoh, K. Low-dose vitamin D prevents muscular atrophy and reduces falls and hip fractures in women after stroke: A randomized controlled trial. Cerebrovasc. Dis. 2005, 20, 187–192. [Google Scholar] [CrossRef]
- Hamilton, B. Vitamin D and human skeletal muscle. Scand. J. Med. Sci. Sports 2010, 20, 182–190. [Google Scholar] [CrossRef] [Green Version]
- Girgis, C.M. Integrated therapies for osteoporosis and sarcopenia: From signaling pathways to clinical trials. Calcif. Tissue Int. 2015, 96, 243–255. [Google Scholar] [CrossRef]
- Bunout, D.; Barrera, G.; Leiva, L.; Gattas, V.; de la Maza, M.P.; Avendano, M.; Hirsch, S. Effects of vitamin D supplementation and exercise training on physical performance in Chilean vitamin D deficient elderly subjects. Exp. Gerontol. 2006, 41, 746–752. [Google Scholar] [CrossRef]
- Houston, D.K.; Tooze, J.A.; Neiberg, R.H.; Hausman, D.B.; Johnson, M.A.; Cauley, J.A.; Bauer, D.C.; Cawthon, P.M.; Shea, M.K.; Schwartz, G.G.; et al. 25-hydroxyvitamin D status and change in physical performance and strength in older adults: The Health, Aging, and Body Composition Study. Am. J. Epidemiol. 2012, 176, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Bischoff-Ferrari, H.A.; Dietrich, T.; Orav, E.J.; Hu, F.B.; Zhang, Y.; Karlson, E.W.; Dawson-Hughes, B. Higher 25-hydroxyvitamin D concentrations are associated with better lower-extremity function in both active and inactive persons aged > or =60 y. Am. J. Clin. Nutr. 2004, 80, 752–758. [Google Scholar] [CrossRef]
- Brunner, F.; Schmid, A.; Sheikhzadeh, A.; Nordin, M.; Yoon, J.; Frankel, V. Effects of aging on Type II muscle fibers: A systematic review of the literature. J. Aging Phys. Act. 2007, 15, 336–348. [Google Scholar] [CrossRef] [Green Version]
- Muir, S.W.; Montero-Odasso, M. Effect of vitamin D supplementation on muscle strength, gait and balance in older adults: A systematic review and meta-analysis. J. Am. Geriatr. Soc. 2011, 59, 2291–2300. [Google Scholar] [CrossRef]
- Ford, E.S.; Ajani, U.A.; McGuire, L.C.; Liu, S. Concentrations of serum vitamin D and the metabolic syndrome among U.S. adults. Diabetes Care 2005, 28, 1228–1230. [Google Scholar] [CrossRef] [Green Version]
- Liu, E.; Meigs, J.B.; Pittas, A.G.; McKeown, N.M.; Economos, C.D.; Booth, S.L.; Jacques, P.F. Plasma 25-hydroxyvitamin d is associated with markers of the insulin resistant phenotype in nondiabetic adults. J. Nutr. 2009, 139, 329–334. [Google Scholar] [CrossRef] [Green Version]
- Scragg, R.; Sowers, M.; Bell, C.; Third National, H.; Nutrition Examination, S. Serum 25-hydroxyvitamin D, diabetes, and ethnicity in the Third National Health and Nutrition Examination Survey. Diabetes Care 2004, 27, 2813–2818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, A.; Hollingsworth, K.G.; Ball, S.; Cheetham, T. Improving the vitamin D status of vitamin D deficient adults is associated with improved mitochondrial oxidative function in skeletal muscle. J. Clin. Endocrinol. Metab. 2013, 98, E509–E513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, D.; Hardigan, P.; Bray, N.; Penzell, D.; Savu, C. The incidence of vitamin D deficiency in the obese: A retrospective chart review. J. Community Hosp. Intern. Med. Perspect 2015, 5, 26069. [Google Scholar] [CrossRef] [PubMed]
- Vranic, L.; Mikolasevic, I.; Milic, S. Vitamin D Deficiency: Consequence or Cause of Obesity? Medicina 2019, 55, 541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, Z.C.; Craig, T.A.; Folmes, C.D.; Wang, X.; Lanza, I.R.; Schaible, N.S.; Salisbury, J.L.; Nair, K.S.; Terzic, A.; Sieck, G.C.; et al. 1alpha,25-Dihydroxyvitamin D3 Regulates Mitochondrial Oxygen Consumption and Dynamics in Human Skeletal Muscle Cells. J. Biol. Chem. 2016, 291, 1514–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagliafico, A.S.; Ameri, P.; Bovio, M.; Puntoni, M.; Capaccio, E.; Murialdo, G.; Martinoli, C. Relationship between fatty degeneration of thigh muscles and vitamin D status in the elderly: A preliminary MRI study. AJR Am. J. Roentgenol. 2010, 194, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Gilsanz, V.; Kremer, A.; Mo, A.O.; Wren, T.A.; Kremer, R. Vitamin D status and its relation to muscle mass and muscle fat in young women. J. Clin. Endocrinol. Metab. 2010, 95, 1595–1601. [Google Scholar] [CrossRef] [Green Version]
- Redzic, M.; Powell, D.K.; Thomas, D.T. Vitamin D status is related to intramyocellular lipid in older adults. Endocrine 2014, 47, 854–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, D.; Joham, A.; Teede, H.; Gibson-Helm, M.; Harrison, C.; Cassar, S.; Hutchison, S.; Ebeling, P.R.; Stepto, N.; de Courten, B. Associations of Vitamin D with Inter- and Intra-Muscular Adipose Tissue and Insulin Resistance in Women with and without Polycystic Ovary Syndrome. Nutrients 2016, 8, 774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papapostoli, P.; Lammert, F.; Stokes, C.S. Effect of Short-Term Vitamin D Correction on Hepatic Steatosis as Quantified by Controlled Attenuation Parameter (CAP). J. Gastrointest. Liver Dis. 2016, 25, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Schnell, D.M.; Walton, R.G.; Vekaria, H.J.; Sullivan, P.G.; Bollinger, L.M.; Peterson, C.A.; Thomas, D.T. Vitamin D produces a perilipin 2-dependent increase in mitochondrial function in C2C12 myotubes. J. Nutr. Biochem. 2019, 65, 83–92. [Google Scholar] [CrossRef]
- Jefferson, G.E.; Schnell, D.M.; Thomas, D.T.; Bollinger, L.M. Calcitriol concomitantly enhances insulin sensitivity and alters myocellular lipid partitioning in high fat-treated skeletal muscle cells. J. Physiol. Biochem. 2017, 73, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.T.; Schnell, D.M.; Redzic, M.; Zhao, M.; Abraha, H.; Jones, D.; Brim, H.; Yu, G. Local In Vivo Measures of Muscle Lipid and Oxygen Consumption Change in Response to Combined Vitamin D Repletion and Aerobic Training in Older Adults. Nutrients 2019, 11, 930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makanae, Y.; Ogasawara, R.; Sato, K.; Takamura, Y.; Matsutani, K.; Kido, K.; Shiozawa, N.; Nakazato, K.; Fujita, S. Acute bout of resistance exercise increases vitamin D receptor protein expression in rat skeletal muscle. Exp. Physiol. 2015, 100, 1168–1176. [Google Scholar] [CrossRef] [PubMed]
- Giulietti, A.; van Etten, E.; Overbergh, L.; Stoffels, K.; Bouillon, R.; Mathieu, C. Monocytes from type 2 diabetic patients have a pro-inflammatory profile. 1,25-Dihydroxyvitamin D(3) works as anti-inflammatory. Diabetes Res. Clin. Pract. 2007, 77, 47–57. [Google Scholar] [CrossRef]
- Di Rosa, M.; Malaguarnera, M.; Nicoletti, F.; Malaguarnera, L. Vitamin D3: A helpful immuno-modulator. Immunology 2011, 134, 123–139. [Google Scholar] [CrossRef]
- Jablonski, K.; Chonchol, M.; Pierce, G.; Walker, A.E.; Seals, D.R. 25-Hydroxyvitamin D deficiency is associated with inflammation-linked vascular endothelial dysfunction in middle-aged and older adults. Hypertension 2010, 57, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Yin, K.; Agrawal, D.K. Vitamin D and inflammatory diseases. J. Inflamm. Res. 2014, 7, 69–87. [Google Scholar] [CrossRef] [Green Version]
- Wolins, N.E.; Quaynor, B.K.; Skinner, J.R.; Tzekov, A.; Croce, M.A.; Gropler, M.C.; Varma, V.; Yao-Borengasser, A.; Rasouli, N.; Kern, P.A.; et al. OXPAT/PAT-1 is a PPAR-induced lipid droplet protein that promotes fatty acid utilization. Diabetes 2006, 55, 3418–3428. [Google Scholar] [CrossRef] [Green Version]
- Bosma, M.; Hesselink, M.K.C.; Sparks, L.M.; Timmers, S.; Ferraz, M.J.; Mattijssen, F.; van Beurden, D.; Schaart, G.; de Baets, M.H.; Verheyen, F.K.; et al. Perilipin 2 Improves Insulin Sensitivity in Skeletal Muscle Despite Elevated Intramuscular Lipid Levels. Diabetes 2012, 61, 2679–2690. [Google Scholar] [CrossRef] [Green Version]
- Shepherd, S.O.; Cocks, M.; Tipton, K.D.; Ranasinghe, A.M.; Barker, T.A.; Burniston, J.G.; Wagenmakers, A.J.; Shaw, C.S. Preferential utilization of perilipin 2-associated intramuscular triglycerides during 1 h of moderate-intensity endurance-type exercise. Exp. Physiol. 2012, 97, 970–980. [Google Scholar] [CrossRef]
- Funai, K.; Semenkovich, C.F. Skeletal muscle lipid flux: Running water carries no poison. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E245–E251. [Google Scholar] [CrossRef] [Green Version]
- Abboud, M.; Puglisi, D.A.; Davies, B.N.; Rybchyn, M.; Whitehead, N.P.; Brock, K.E.; Cole, L.; Gordon-Thomson, C.; Fraser, D.R.; Mason, R.S. Evidence for a specific uptake and retention mechanism for 25-hydroxyvitamin D (25OHD) in skeletal muscle cells. Endocrinology 2013, 154, 3022–3030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, D.T.; Erdman, K.A.; Burke, L.M. American College of Sports Medicine joint position statement: Nutrition and athletic performance. Med. Sci. Sports Exerc. 2016, 48, 543–568. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Peng, R.; Yang, Z.; Peng, Y.; Lu, N. Supplementation of dietary nitrate attenuated oxidative stress and endothelial dysfunction in diabetic vasculature through inhibition of NADPH oxidase. Nitric Oxide 2020, 96, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.M. Influence of dietary nitrate on the physiological determinants of exercise performance: A critical review. Appl. Physiol. Nutr. Metab. 2014, 39, 1019–1028. [Google Scholar] [CrossRef]
- Velmurugan, S.; Ming Gan, J.; Rathod, K.; Khambata, R.; Ghosh, M.; Hartley, A. Dietary nitrate improves vascular function in patients with hypercholesterolemia: A randomized, double-blind, placebo-controlled study. Am. J. Clin. Nutr. 2016, 103, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Basaqr, R.; Skleres, M.; Jayswal, R.; Thomas, D.T. The Effect of Dietary Nitrate and Vitamin C on Endothelial Function, Oxidative Stress, and Blood Lipids in Untreated Hypercholesterolemic Subjects: A Randomized Double-blind Crossover Study. Clin. Nutr. 2021, 40, 1851–1860. [Google Scholar] [CrossRef] [PubMed]
- Ashor, A.W.; Siervo, M.; van der Velde, F.; Willis, N.D.; Mathers, J.C. Systematic review and meta-analysis of randomised controlled trials testing the effects of vitamin C supplementation on blood lipids. Clin. Nutr. 2016, 35, 626–637. [Google Scholar] [CrossRef]
- Chambial, S.; Dwivedi, S.; Shukla, K.K.; John, P.J.; Sharma, P. Vitamin C in disease prevention and cure: An overview. Indian J. Clin. Biochem. 2013, 28, 314–328. [Google Scholar] [CrossRef] [Green Version]
- Charlton-Menys, V.; Durrington, P. Human cholesterol metabolism and therapeutic molecules. Exp. Physiol. 2008, 93, 27–42. [Google Scholar] [CrossRef]
- McRae, M. Vitamin C supplementation lowers serum low-density lipoprotein cholesterol and triglycerides: A meta-analysis of 13 randomized controlled trials. J. Chiropr. Med. 2008, 7, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Galmés, S.; Serra, F.; Palou, A. Vitamin E Metabolic Effects and Genetic Variants: A Challenge for Precision Nutrition in Obesity and Associated Disturbances. Nutrients 2018, 10, 1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, Y.K.; Omaye, S.T. Vitamin E-modulated gene expression associated with ROS generation. J. Funct. Foods 2009, 1, 241–252. [Google Scholar] [CrossRef]
- Traber, M.G. Vitamin E regulatory mechanisms. Annu. Rev. Nutr. 2007, 27, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Ulatowski, L.; Dreussi, C.; Noy, N.; Barnholtz-Sloan, J.; Klein, E.; Manor, D. Expression of the α-tocopherol transfer protein gene is regulated by oxidative stress and common single-nucleotide polymorphisms. Free Radic. Biol. Med. 2012, 53, 2318–2326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inan, C.; Kiliç, I.; Kilinç, K.; Kalayci, O.; Kotiloğlu, E. The effect of high dose antenatal vitamin E on hypoxia-induced changes in newborn rats. Pediatr. Res. 1995, 38, 685–689. [Google Scholar] [CrossRef] [Green Version]
- Campbell, S.E.; Stone, W.L.; Whaley, S.G.; Qui, M.; Krishnan, K. Gamma (gamma) tocopherol upregulates peroxisome proliferator activated receptor (PPAR) gamma (gamma) expression in SW 480 human colon cancer cell lines. BMC Cancer 2003, 3, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrier, J.-F.o.; Gouranton, E.; El Yazidi, C.; Malezet, C.; Balaguer, P.; Borel, P.; Amiot, M.-J.p. Adiponectin Expression Is Induced by Vitamin E via a Peroxisome Proliferator-Activated Receptor γ-Dependent Mechanism. Endocrinology 2009, 150, 5318–5325. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.-J.; Lee, K.-S.; Lee, S.; Park, J.-H.; Choi, H.-E.; Go, S.H.; Kwak, H.-J.; Parka, H.-Y. 15d-PGJ2 stimulates HO-1 expression through p38 MAP kinase and Nrf-2 pathway in rat vascular smooth muscle cells. Toxicol. Appl. Pharm. 2007, 223, 20–27. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kang, E.S.; Boutari, C.; Rhee, E.J.; Mantzoros, C.S. Current and emerging pharmacological options for the treatment of nonalcoholic steatohepatitis. Metabolism 2020, 111, 154203. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, D.T.; DelCimmuto, N.R.; Flack, K.D.; Stec, D.E.; Hinds, T.D., Jr. Reactive Oxygen Species (ROS) and Antioxidants as Immunomodulators in Exercise: Implications for Heme Oxygenase and Bilirubin. Antioxidants 2022, 11, 179. https://doi.org/10.3390/antiox11020179
Thomas DT, DelCimmuto NR, Flack KD, Stec DE, Hinds TD Jr. Reactive Oxygen Species (ROS) and Antioxidants as Immunomodulators in Exercise: Implications for Heme Oxygenase and Bilirubin. Antioxidants. 2022; 11(2):179. https://doi.org/10.3390/antiox11020179
Chicago/Turabian StyleThomas, David Travis, Nicholas R. DelCimmuto, Kyle D. Flack, David E. Stec, and Terry D. Hinds, Jr. 2022. "Reactive Oxygen Species (ROS) and Antioxidants as Immunomodulators in Exercise: Implications for Heme Oxygenase and Bilirubin" Antioxidants 11, no. 2: 179. https://doi.org/10.3390/antiox11020179