
Oxidative-Stress-Associated Proteostasis Disturbances and Increased DNA Damage in the Hippocampal Granule Cells of the Ts65Dn Model of Down Syndrome
 , and
, and 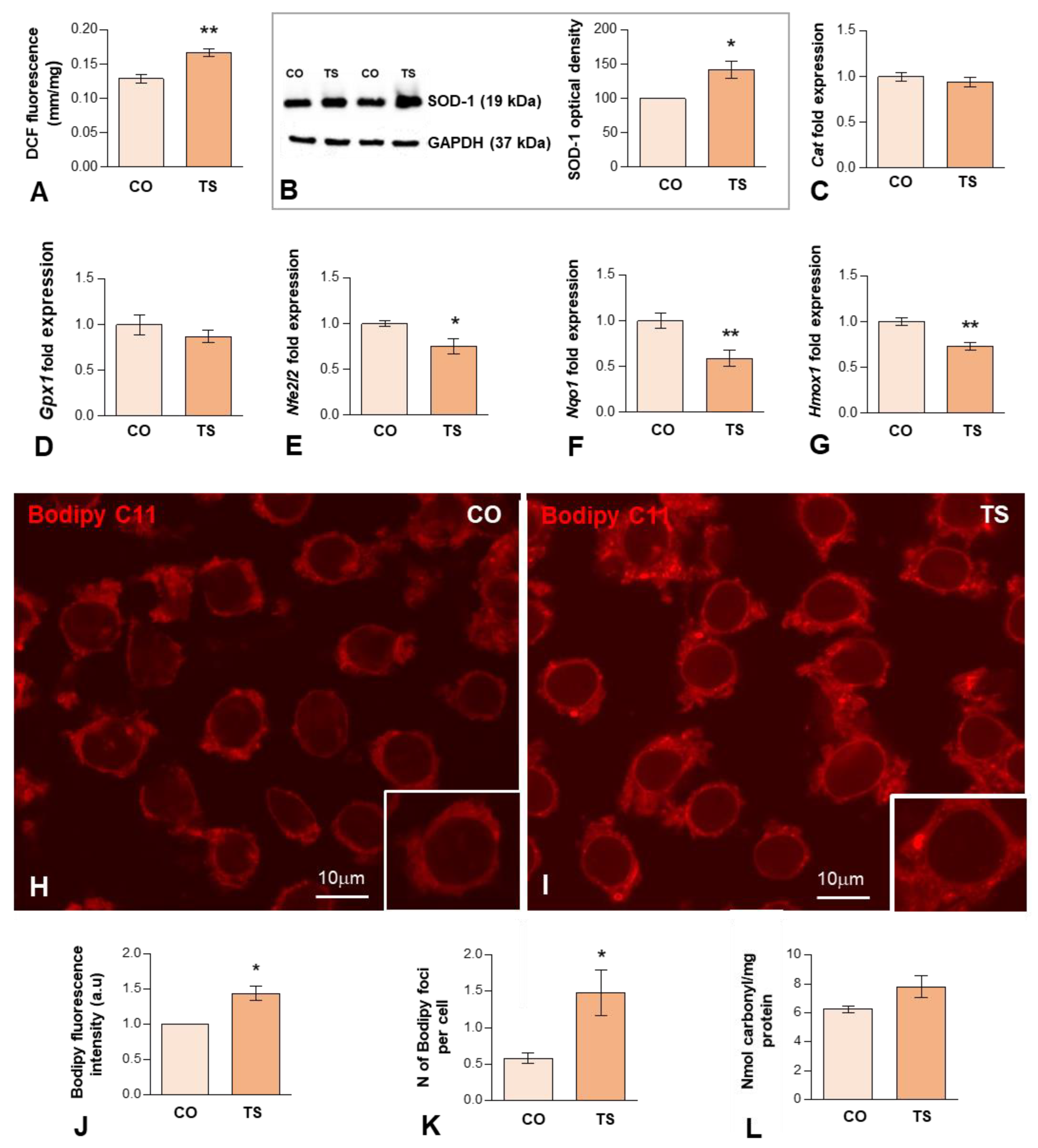{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Immunofluorescence and Confocal Microscopy
2.3. Conventional Electron Microscopy
2.4. Detection of ROS Levels
2.5. In Situ Determination of Oxidized Lipids
2.6. Determination of Protein Oxidation: Protein Carbonyl Content
2.7. Real-Time Quantitative PCR
2.8. Western Blotting
2.9. Proteasome Activity Assay
2.10. Statistical Analysis
3. Results
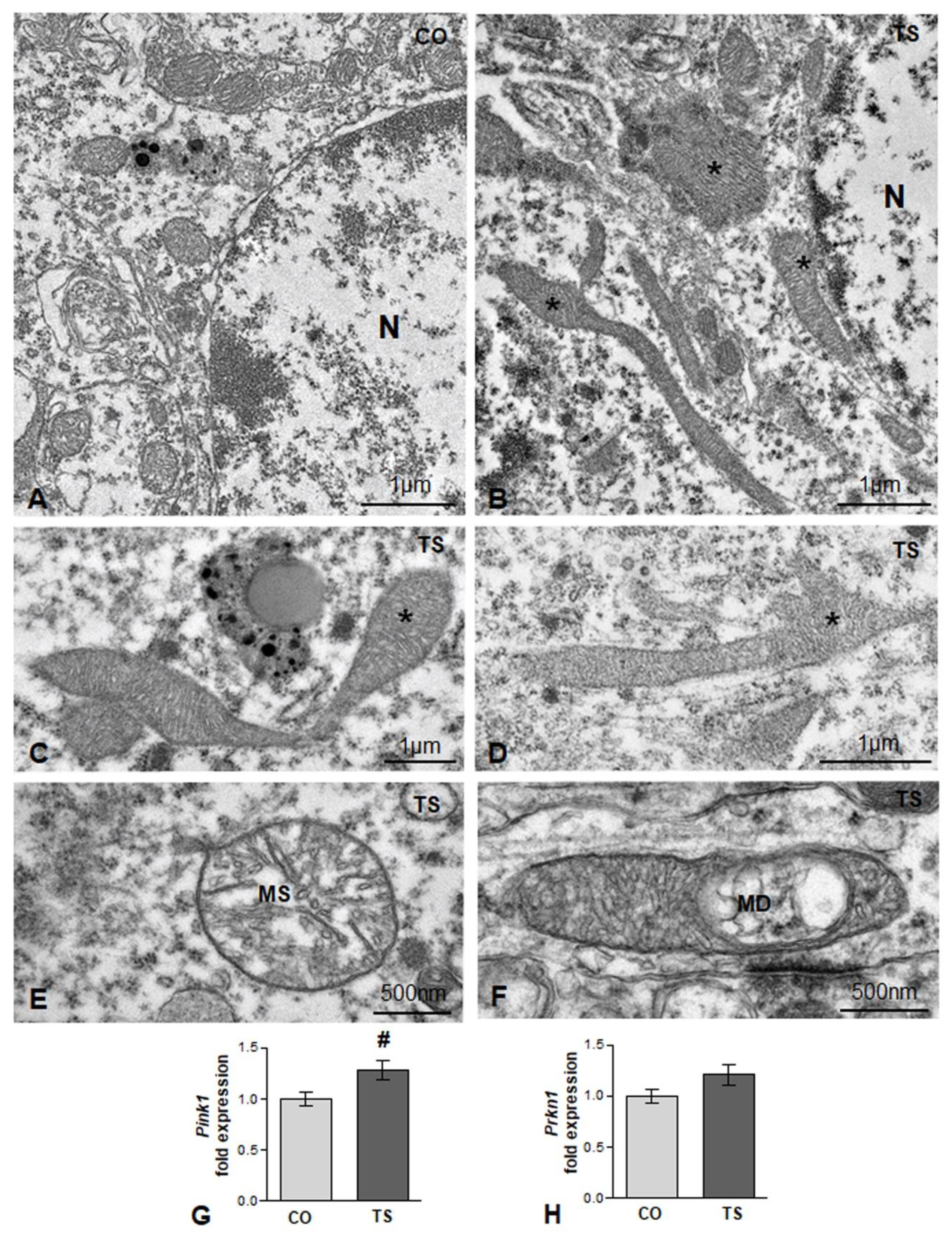
3.1. Increased OS in the Hippocampi of TS Mice Is Associated with Increased ROS Generation, Altered Antioxidant Response, Oxidative Damage to Lipids and Proteins, and Mitochondrial Anomalies in TS GCs
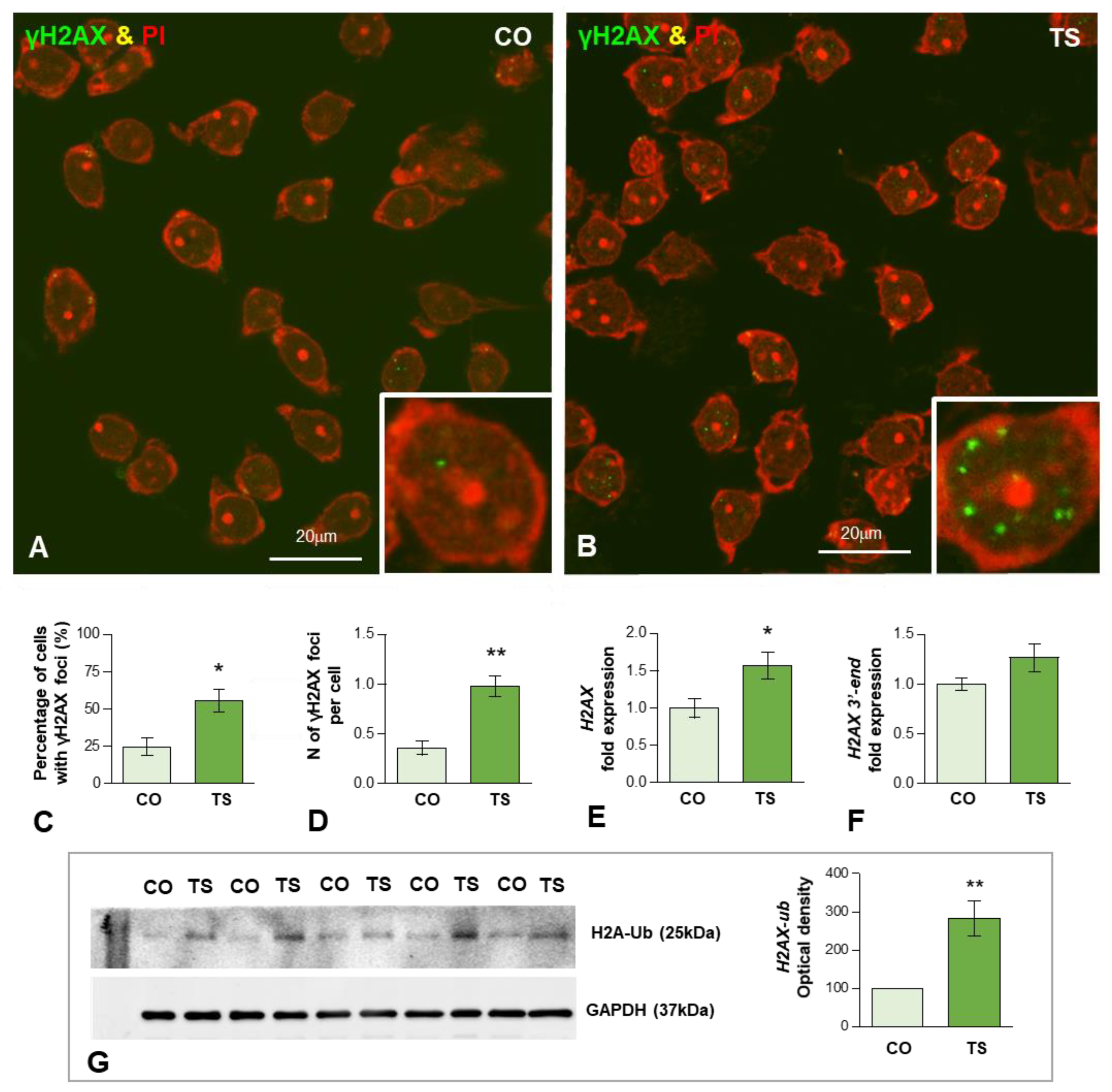
3.2. Oxidative Stress in TS GCs Induces DNA Damage
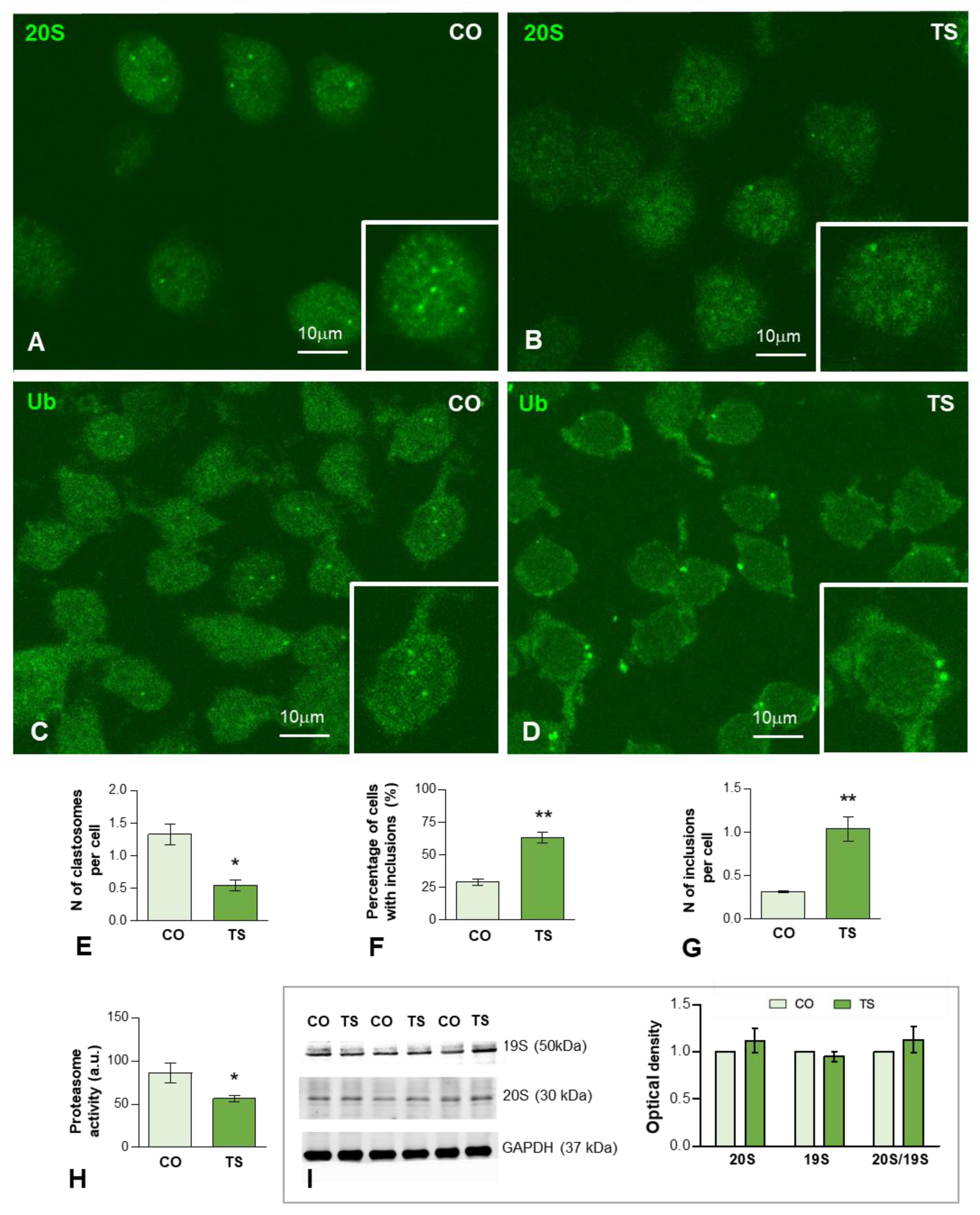
3.3. Increased OS Is Associated with Proteostasis Disturbances in TS GCs
3.3.1. Dysfunction of the Proteasome
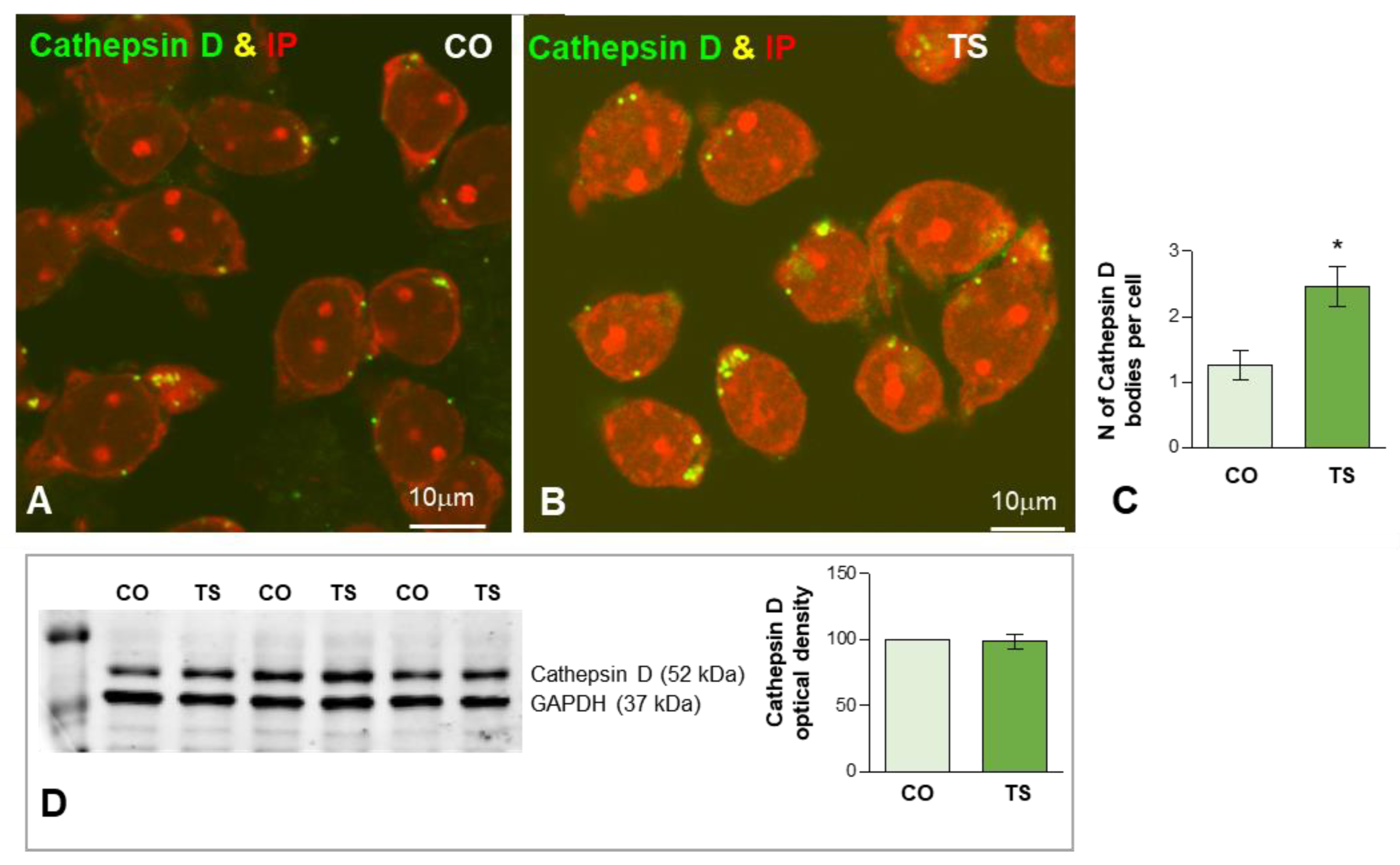
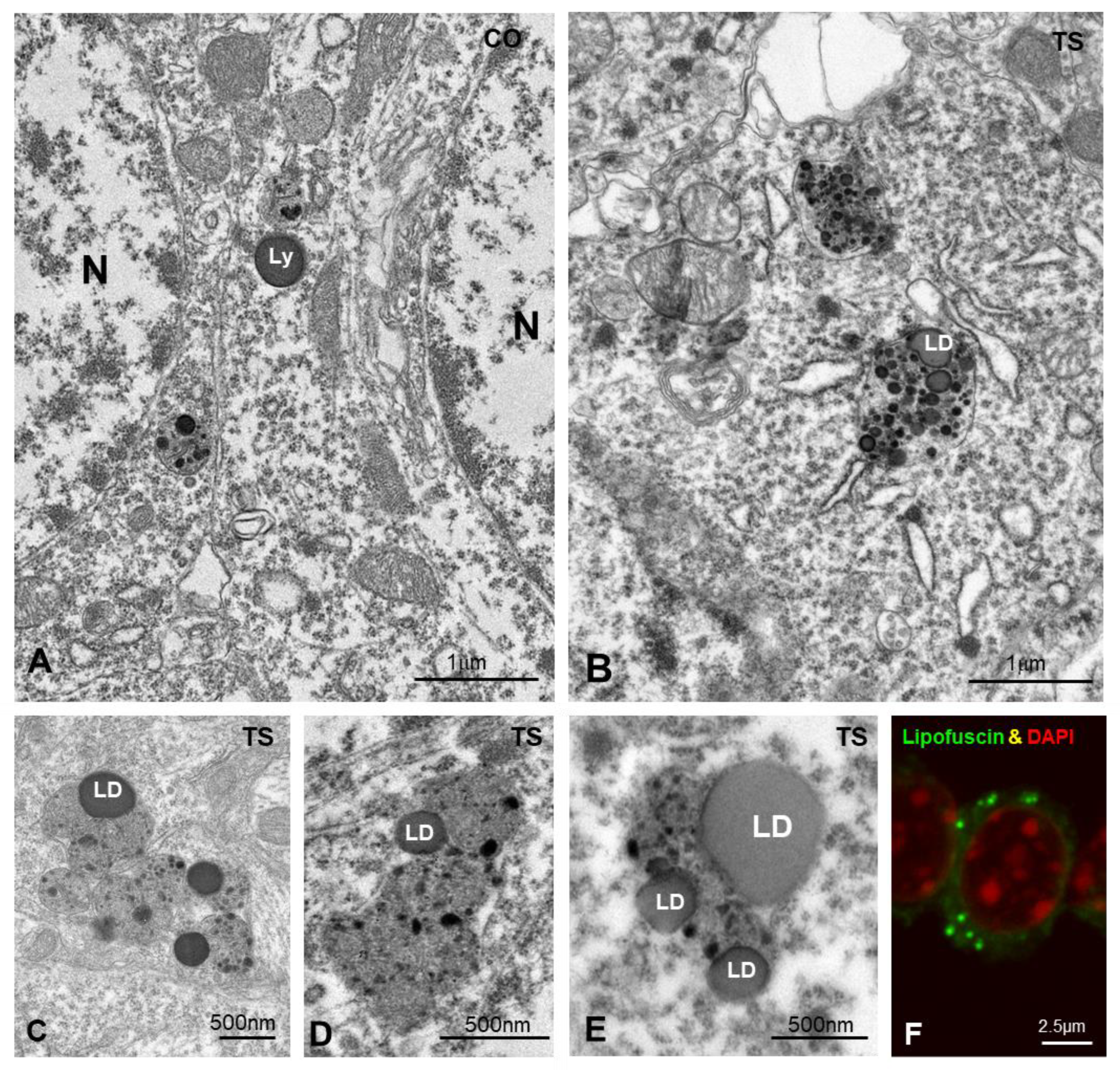
3.3.2. Dysfunction of Lysosomal System
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shin, M.; Besser, L.M.; Kucik, J.E.; Lu, C.; Siffel, C.; Correa, A. Prevalence of Down syndrome among children and adolescents in 10 regions of the United States. Pediatrics 2009, 124, 1565–1571. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, S.E.; Skotko, B.G.; Rafii, M.; Strydom, A.; Pape, S.E.; Bianchi, D.; Sherman, S.L.; Reeves, R.H. Down syndrome. Nat. Rev. Dis. Primers 2020, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Cué, C.; Bartesaghi, R. Fatty Acids: A Safe Tool for Improving Neurodevelopmental Alterations in Down Syndrome? Nutrients 2022, 14, 2880. [Google Scholar] [CrossRef] [PubMed]
- Lott, I.T.; Dierssen, M. Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neurol. 2010, 9, 623–633. [Google Scholar] [CrossRef]
- Wilcock, D.M.; Griffin, W.S. Down’s syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J. Neuroinflamm. 2013, 10, 84. [Google Scholar] [CrossRef] [Green Version]
- Perluigi, M.; Butterfield, D.A. Oxidative Stress and Down Syndrome: A Route toward Alzheimer-Like Dementia. Curr. Gerontol. Geriatr. Res. 2012, 2012, 724904. [Google Scholar] [CrossRef] [Green Version]
- Rueda, N.; Martinez-Cue, C. Antioxidants in Down syndrome: From preclinical studies to clinical trials. Antioxidants 2020, 9, 692. [Google Scholar] [CrossRef]
- Wiseman, F.K.; Al-Janabi, T.; Hardy, J.; Karmiloff-Smith, A.; Nizetic, D.; Tybulewicz, V.L.; Fisher, E.M.; Strydom, A. A genetic cause of Alzheimer disease: Mechanistic insights from Down syndrome. Nat. Rev. Neurosci. 2015, 16, 564–574. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Font, M.F.; Sebastià, J.; Sanfeliu, C.; Cristòfol, R.; Marfany, G.; Gonzàlez-Duarte, R. Peroxiredoxin 2 (PRDX2), an antioxidant enzyme, is under-expressed in Down syndrome fetal brains. Cell. Mol. Life Sci. 2003, 60, 1513–1523. [Google Scholar] [CrossRef]
- Gimeno, A.; García-Giménez, J.L.; Audí, L.; Toran, N.; Andaluz, P.; Dasí, F.; Viña, J.; Pallardó, F.V. Decreased cell proliferation and higher oxidative stress in fibroblasts from Down syndrome fetuses. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 116–125. [Google Scholar] [CrossRef]
- Hamed, R.R.; Maharem, T.M.; Abdel-Meguid, N.; Sabry, G.M.; Abdalla, A.M.; Guneidy, R.A. Purification and biochemical characterization of glutathione S-transferase from Down syndrome and normal children erythrocytes: A comparative study. Res. Dev. Disabil. 2011, 32, 1470–1482. [Google Scholar] [CrossRef] [PubMed]
- Lanzillotta, C.; Zuliani, I.; Tramutola, A.; Barone, E.; Blarzino, C.; Folgiero, V.; Caforio, M.; Valentini, D.; Villani, A.; Locatelli, F.; et al. Chronic PERK induction promotes Alzheimer-like neuropathology in Down syndrome: Insights for therapeutic intervention. Prog. Neurobiol. 2021, 196, 101892. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.; Mollo, N.; Nitti, M.; Paladino, S.; Calì, G.; Genesio, R.; Bonfiglio, F.; Cicatiello, R.; Barbato, M.; Sarnataro, V.; et al. Mitochondrial dysfunction in down syndrome: Molecular mechanisms and therapeutic targets. Mol. Med. 2018, 24, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordi, M.; Darji, S.; Sato, Y.; Mellen, M.; Berg, M.J.; Kumar, A.; Jiang, Y.; Nixon, R.A. mTOR hyperactivation in Down Syndrome underlies deficits in autophagy induction, autophagosome formation, and mitophagy. Cell Death Dis. 2019, 10, 563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollo, N.; Cicatiello, R.; Aurilia, M.; Scognamiglio, R.; Genesio, R.; Charalambous, M.; Paladino, S.; Conti, A.; Nitsch, L.; Izzo, A. Targeting Mitochondrial Network Architecture in Down Syndrome and Aging. Int. J. Mol. Sci. 2020, 21, 3134. [Google Scholar] [CrossRef]
- Zamponi, E.; Zamponi, N.; Coskun, P.; Quassollo, G.; Lorenzo, A.; Cannas, S.A.; Pigino, G.; Chialvo, D.R.; Gardiner, K.; Busciglio, J.; et al. Nrf2 stabilization prevents critical oxidative damage in Down syndrome cells. Aging Cell 2018, 17, e12812. [Google Scholar] [CrossRef] [Green Version]
- Bayona-Bafaluy, M.P.; Garrido-Pérez, N.; Meade, P.; Iglesias, E.; Jiménez-Salvador, I.; Montoya, J.; Martínez-Cué, C.; Ruiz-Pesini, E. Down syndrome is an oxidative phosphorylation disorder. Redox Biol. 2021, 41, 101871. [Google Scholar] [CrossRef]
- Izzo, A.; Nitti, M.; Mollo, N.; Paladino, S.; Procaccini, C.; Faicchia, D.; Cali, G.; Genesio, R.; Bonfiglio, F.; Cicatiello, R.; et al. Metformin restores the mitochondrial network and reverses mitochondrial dysfunction in Down syndrome cells. Hum. Mol. Genet. 2017, 26, 1056–1069. [Google Scholar] [CrossRef] [Green Version]
- Odetti, P.; Angelini, G.; Dapino, D.; Zaccheo, D.; Garibaldi, S.; Dagna-Bricarelli, F.; Piombo, G.; Perry, G.; Smith, M.; Traverso, N.; et al. Early glycoxidation damage in brains from Down’s syndrome. Biochem. Biophys. Res. Commun. 1998, 243, 849–851. [Google Scholar] [CrossRef]
- Busciglio, J.; Yankner, B.A. Apoptosis and increased generation of reactive oxygen species in Down’s syndrome neurons In Vitro. Nature 1995, 378, 776–779. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Pappolla, M.A.; Friedland, R.P.; Hirai, K.; Chiba, S.; Smith, M.A. Neuronal oxidative stress precedes amyloid-β deposition in down syndrome. J. Neuropathol. Exp. Neurol. 2000, 59, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Perluigi, M.; Reed, T.; Muharib, T.; Hughes, C.P.; Robinson, R.A.; Sultana, R. Redox proteomics in selected neurodegenerative disorders: From its infancy to future applications. Antioxid. Redox Signal. 2012, 17, 1610–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perluigi, M.; Di Domenico, F.; Buttterfield, D.A. Unraveling the complexity of neurodegeneration in brains of subjects with Down syndrome: Insights from proteomics. Proteom. Clin. Appl. 2014, 8, 73–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Domenico, F.; Coccia, R.; Cocciolo, A.; Murphy, M.P.; Cenini, G.; Head, E.; Butterfield, D.A.; Giorgi, A.; Schinina, M.E.; Mancuso, C.; et al. Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer’s disease neuropathology: Redox proteomics analysis of human brain. Biochim. Biophys. Acta 2013, 1832, 1249–1259. [Google Scholar] [CrossRef]
- Zana, M.; Janka, Z.; a Kálmán, J. Oxidative Stress: A Bridge between Down’s Syndrome and Alzheimer’s Disease. Neurobiol. Aging 2007, 28, 648–676. [Google Scholar] [CrossRef] [PubMed]
- Morawiec, Z.; Janik, K.; Kowalski, M.; Stetkiewicz, T.; Szaflik, J.; Morawiec-Bajda, A.; Sobczuk, A.; Blasiak, J. DNA damage and repair in children with Down’s syndrome. Mutat. Res. 2008, 637, 118–123. [Google Scholar] [CrossRef]
- Necchi, D.; Pinto, A.; Tillhon, M.; Dutto, I.; Serafini, M.M.; Lanni, C.; Govoni, S.; Racchi, M.; Prosperi, E. Defective DNA repair and increased chromatin binding of DNA repair factors in Down syndrome fibroblasts. Mutat. Res. 2015, 780, 15–23. [Google Scholar] [CrossRef]
- Tiano, L.; Padella, L.; Santoro, L.; Carnevali, P.; Principi, F.; Brugè, F.; Gabrielli, O.; Littarru, G.P. Prolonged coenzyme Q10 treatment in Down syndrome patients: Effect on DNA oxidation. Neurobiol. Aging 2012, 33, 626.e1–626.e8. [Google Scholar] [CrossRef]
- Lanzillota, C.; Di Dominico, F. Stress responses in Down syndrome neurodegeneration: State of the arte and therapeutic molecules. Biomolecules 2021, 11, 266. [Google Scholar] [CrossRef]
- Aivazidis, S.; Coughlan, C.M.; Rauniyar, A.K.; Jiang, H.; Liggett, L.A.; Maclean, K.N.; Roede, J.R. The burden of trisomy 21disrupts the proteostasis network in Down syndrome. PLoS ONE 2017, 12, e0176307. [Google Scholar] [CrossRef]
- Tramutola, A.; Di Domenico, F.; Barone, E.; Arena, A.; Giorgi, A.; di Francesco, L.; Schininà, M.E.; Coccia, R.; Head, E.; Butterfield, D.A.; et al. Polyubiquitinylation Profile in Down Syndrome Brain Before and After the Development of Alzheimer Neuropathology. Antioxid. Redox Signal. 2017, 26, 280–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Sato, Y.; Im, E.; Berg, M.; Bordi, M.; Darji, S.; Kumar, A.; Mohan, P.S.; Bandyopadhyay, U.; Diaz, A.; et al. Lysosomal dysfunction in Down syndrome is APP-dependent and mediated by APP-ßCTF (C99). J. Neurosci. 2019, 39, 5255–5268. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; di Domenico, F.; Fiorini, A.; Cocciolo, A.; Giorgi, A.; Foppoli, C.; Butterfield, D.A.; Giorlandino, M.; Giorlandino, C.; Schininà, M.E.; et al. Oxidative stress occurs early in Down syndrome pregnancy: A redox proteomics analysis of amniotic fluid. Proteom. Clin. Appl. 2011, 5, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Tramutola, A.; Pagnotta, S.; Barone, E.; Butterfield, D.A. The BACH1/Nrf2 Axis in Brain in Down Syndrome and Transition to Alzheimer Disease-Like Neuropathology and Dementia. Antioxidants 2020, 9, 779. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Cué, C.; Rueda, N. Cellular Senescence in Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 16. [Google Scholar] [CrossRef]
- Rueda, N.; Flórez, J.; Martínez-Cué, C. Mouse models of Down syndrome as a tool to unravel the causes of mental disabilities. Neural Plast. 2012, 2012, 584071. [Google Scholar] [CrossRef] [Green Version]
- Corrales, A.; Vidal, R.; García, S.; Vidal, V.; Martínez, P.; García, E.; Flórez, J.; Sanchez-Barceló, E.J.; Martínez-Cué, C.; Rueda, N. Chronic melatonin treatment rescues electrophysiological and neuromorphological deficits in a mouse model of Down syndrome. J. Pineal Res. 2014, 56, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Corrales, A.; Parisotto, E.B.; Vidal, V.; García-Cerro, S.; Lantigua, S.; Diego, M.; Wilhem-Filho, D.; Sanchez-Barceló, E.J.; Martínez-Cué, C.; Rueda, N. Pre- and post-natal melatonin administration partially regulates brain oxidative stress but does not improve cognitive or histological alterations in the Ts65Dn mouse model of Down syndrome. Behav. Brain Res. 2018, 334, 142–154. [Google Scholar] [CrossRef] [Green Version]
- Rueda, N.; Vidal, V.; García-Cerro, S.; Narcís, J.O.; Llorens-Martín, M.; Corrales, A.; Lantigua, S.; Iglesias, M.; Merino, J.; Merino, R.; et al. Anti-IL17 treatment ameliorates Down syndrome phenotypes in mice. Brain Behav. Immun. 2018, 73, 235–251. [Google Scholar] [CrossRef]
- Uguagliati, B.; Al-Absi, A.R.; Stagni, F.; Emili, M.; Giacomini, A.; Guidi, S.; Nyengaard, J.R.; Bartesaghi, R. Early appearance of developmental alterations in the dendritic tree of the hippocampal granule cells in the Ts65Dn model of Down syndrome. Hippocampus 2021, 31, 435–447. [Google Scholar] [CrossRef]
- Puente-Bedia, A.; Berciano, M.T.; Tapia, O.; Martinez-Cué, C.; Lafarga, M.; Rueda, N. Nuclear reorganization in hippocampal granule cell neurons from a mouse model of Down syndrome: Changes in chromatin configuration, nucleoli and Cajal bodies. Int. J. Mol. Sci. 2021, 22, 1259. [Google Scholar] [CrossRef]
- Villarroya, O.; Ballestín, R.; López-Hidalgo, R.; Mulet, M.; Blasco-Ibáñez, J.M.; Crespo, C.; Nacher, J.; Gilabert-Juan, J.; Varea, E. Morphological alterations in the hippocampus of the Ts65Dn mouse model for Down Syndrome correlate with structural plasticity markers. Histol. Histopathol. 2018, 33, 101–115. [Google Scholar] [PubMed]
- Stagni, F.; Salvalai, M.E.; Giacomini, A.; Emili, M.; Uguagliati, B.; Xia, E.; Grilli, M.; Bartesaghi, R.; Guidi, S. Neonatal treatment with cyclosporine A restores neurogenesis and spinogenesis in the Ts65Dn model of Down syndrome. Neurobiol. Dis. 2019, 129, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Lockrow, J.; Prakasam, A.; Huang, P.; Bimonte-Nelson, H.; Sambamurti, K.; Granholm, A.C. Cholinergic degeneration and memory loss delayed by vitamin E in a Down syndrome mouse model. Exp. Neurol. 2009, 216, 278–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shichiri, M.; Yoshida, Y.; Ishida, N.; Hagihara, Y.; Iwahashi, H.; Tamai, H.; Niki, E. α-Tocopherol suppresses lipid peroxidation and behavioral and cognitive impairments in the Ts65Dn mouse model of Down syndrome. Free Radic. Biol. Med. 2011, 50, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Parisotto, E.B.; Vidal, V.; García-Cerro, S.; Lantigua, S.; Wilhelm Filho, D.; Sanchez-Barceló, E.J.; Martínez-Cué, C.; Rueda, N. Chronic Melatonin Administration Reduced Oxidative Damage and Cellular Senescence in the Hippocampus of a Mouse Model of Down Syndrome. Neurochem. Res. 2016, 41, 2904–2913. [Google Scholar] [CrossRef] [Green Version]
- Valenti, D.; de Bari, L.; de Rasmo, D.; Signorile, A.; Henrion-Caude, A.; Contestabile, A.; Vacca, R.A. The polyphenols resveratrol and epigallocatechin-3-gallate restore the severe impairment of mitochondria in hippocampal progenitor cells from a Down syndrome mouse model. Biochim. Biophys. Acta 2016, 1862, 1093–1104. [Google Scholar] [CrossRef]
- Alldred, M.J.; Lee, S.H.; Stutzmann, G.E.; Ginsberg, S.D. Oxidative Phosphorylation Is Dysregulated Within the Basocortical Circuit in a 6-month old Mouse Model of Down Syndrome and Alzheimer’s Disease. Front Aging Neurosci. 2021, 13, 707950. [Google Scholar] [CrossRef]
- Tramutola, A.; Pupo, G.; Di Domenico, F.; Barone, E.; Arena, A.; Lanzillotta, C.; Brokeaart, D.; Blarzino, C.; Head, E.; Butterfield, D.A.; et al. Activation of p53 in Down syndrome and in the Ts65Dn mouse brain is associated with a proapoptotic phenotype. J. Alzheimers Dis. 2016, 52, 359–371. [Google Scholar] [CrossRef] [Green Version]
- Corrales, A.; Martínez, P.; García, S.; Vidal, V.; García, E.; Flórez, J.; Sánchez-Barceló, E.J.; Martínez-Cué, C.; Rueda, N. Long-term oral administration of melatonin improves spatial learning and memory and protects against cholinergic degeneration in middle-aged Ts65Dn mice, a model of Down syndrome. J. Pineal Res. 2013, 54, 346–358. [Google Scholar] [CrossRef]
- García-Cerro, S.; Rueda, N.; Vidal, V.; Lantigua, S.; Martínez-Cué, C. Normalizing the gene dosage of Dyrk1A in a mouse model of Down syndrome rescues several Alzheimer’s disease phenotypes. Neurobiol. Dis. 2017, 106, 76–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palkovits, M. Isolated removal of hypothalamic and other brain nuclei of the rat. Brain Res. 1973, 59, 449–450. [Google Scholar] [CrossRef] [PubMed]
- Pena, E.; Berciano, M.T.; Fernandez, R.; Ojeda, J.L.; Lafarga, M. Neuronal body size correlates with the number of nucleoli and Cajal bodies, and with the organization of the splicing machinery in rat trigeminal ganglion neurons. J. Comp. Neurol. 2001, 430, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; Stagni, F.; Emili, M.; Guidi, S.; Bartesaghi, R.; Vacca, R.A. Impaired Brain Mitochondrial Bioenergetics in the Ts65Dn Mouse Model of Down Syndrome Is Restored by Neonatal Treatment with the Polyphenol 7,8-Dihydroxyflavone. Antioxidants 2021, 11, 62. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrouh, N.H.; Farr, A.L.; Randall, R.J. Protein Measurement with the Folin Phenol Reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.N.; Hanni, K.B.; Markesbery, W.R. Impaired proteasome function in Alzheimer’s disease. J. Neurochem. 2000, 75, 436–439. [Google Scholar] [CrossRef]
- Casafont, I.; Berciano, M.T.; Lafarga, M. Bortezomib induces the formation of nuclear poly(A) RNA granules enriched in Sam68 and PABPN1 in sensory ganglia neurons. Neurotox. Res. 2010, 17, 167–178. [Google Scholar] [CrossRef]
- Chi, L.; Ke, Y.; Luo, C.; Gozal, D.; Liu, R. Depletion of reduced glutathione enhances motor neuron degeneration in vitro and in vivo. Neuroscience 2007, 144, 991–1003. [Google Scholar] [CrossRef] [Green Version]
- Fritz, K.S.; Petersen, D.R. An overview of the chemistry and biology of reactive aldehydes. Free Radic. Biol. Med. 2013, 59, 85–91. [Google Scholar] [CrossRef]
- Ruiz-Soto, M.; Riancho, J.; Tapia, O.; Lafarga, M.; Berciano, M.T. Satellite glial cells of the dorsal root ganglion: A new “guest/physiopathological target” in ALS. Front. Aging Neurosci. 2020, 12, 595751. [Google Scholar] [CrossRef] [PubMed]
- Schuchmann, S.; Heinemann, U. Increased mitochondrial superoxide generation in neurons from trisomy 16 mice: A model of Down’s Syndrome. Free Radic. Biol. Med. 2000, 28, 235–250. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 2004, 6, 168–170. [Google Scholar] [CrossRef]
- Coppedè, F.; Migliore, L. DNA damage in neurodegenerative diseases. Mutat. Res. 2015, 776, 84–97. [Google Scholar] [CrossRef]
- Fernandez-Capetillo, O.; Lee, A.; Nussenzweig, M.; Nussenzweig, A. H2AX: The histone guardian of genome. DNA Repair 2004, 3, 959–967. [Google Scholar] [CrossRef] [Green Version]
- Lobrich, M.; Shibata, A.; Beucher, A.; Fisher, A.; Ensminger, M.; Goodarzi, A.A.; Barton, O.; Jeggo, P.A. Gamma H2AX foci analysis for monitoring DNA double-strand break repair: Strengths, limitations and optimization. Cell Cycle 2010, 9, 662–669. [Google Scholar] [CrossRef] [Green Version]
- Palanca, A.R.; Casafont, I.; Berciano, M.T.; Lafarga, M. Reactive nucleolar and Cajal body responses to proteasome inhibition in sensory ganglion neurons. Biochim. Biophys. Acta 2014, 1842, 848–859. [Google Scholar] [CrossRef]
- Pessina, F.; Giavazzi, F.; Yin, Y.; Gioia, U.; Vitelli, V.; Galbiati, A.; Barozzi, S.; Garre, M.; Oldani, A.; Flaus, A.; et al. Functional transcription promoters at DNA double-strand breaks mediate RNA-driven phase separation of damage-response factors. Nat. Cell Biol. 2019, 21, 1286–1299. [Google Scholar] [CrossRef]
- Mehta, S.; Zhang, J. Liquid-liquid phase separation drives cellular function and dysfunction in cancer. Nat. Rev Cancer 2022, 22, 239–252. [Google Scholar] [CrossRef]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. GammaH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Marzluff, W.F.; Wagner, E.J.; Duronio, R.J. Metabolism and regulation of canonical histone mRNAs: Life without a poly(A) tail. Nat. Rev. Genet. 2008, 9, 843–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tisdale, S.; Lotti, F.; Saieva, L.; Van Meerbeke, J.P.; Crawford, T.O.; Sumner, C.J.; Mentis, G.Z.; Pellizzoni, L. SMN is essential for the biogenesis of U7 small nuclear ribonucleoprotein and 3′-end formation of histone mRNAs. Cell Rep. 2013, 5, 1187–1195. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.R.; Peng, G.; Hung, W.C.; Lin, S.Y. Monoubiquitination of H2AX protein regulates DNA damage response signaling. J. Biol. Chem. 2011, 286, 28599–28607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Huen, M.S.; Lu, L.Y.; Ye, L.; Dou, Y.; Ljungman, M.; Chen, J.; Yu, X. Histone ubiquitination associates with BRCA1-dependent DNA damage response. Mol. Cell Biol. 2009, 29, 849–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekker-Jensen, S.; Rendtlew, D.J.; Fugger, K.; Gromova, I.; Nerstedt, A.; Bartek, J.; Lukas, J.; Mailand, N. HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat. Cell Biol. 2010, 12, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafarga, M.; Berciano, M.T.; Pena, E.; Mayo, I.; Castano, J.G.; Bohmann, D.; Rodrigues, J.P.; Tavanez, J.P.; Carmo-Fonseca, M. Clastosome: A subtype of nuclear body enriched in 19S and 20S proteasomes, ubiquitin, and protein substrates of proteasome. Mol. Biol. Cell 2002, 13, 2771–2782. [Google Scholar] [CrossRef]
- Janer, A.; Martin, E.; Muriel, M.P.; Latouche, M.; Fujigasaki, H.; Ruberg, M.; Brice, A.; Trottier, Y.; Sittler, A. PML clastosomes prevent nuclear accumulation of mutant ataxin-7 and other polyglutamine proteins. J. Cell Biol. 2006, 174, 65–76. [Google Scholar] [CrossRef]
- Spector, D. SnapShot: Cellular bodies. Cell 2006, 127, 1071. [Google Scholar] [CrossRef]
- Carmo-Fonseca, M.; Berciano, M.T.; Lafarga, M. Orphan nuclear bodies. Cold Spring Harb. Perspect. Biol. 2010, 2, A000703. [Google Scholar]
- Lü, S.; Wang, J. The resistence mechanism of proteasome inhibitor bortezomib. Biomark. Res. 2013, 1, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botté, A.; Lainé, J.; Xicota, L.; Heiligenstein, X.; Fontaine, G.; Kasri, A.; Rivals, I.; Goh, P.; Faklaris, O.; Cossec, J.-C.; et al. Ultrastructural and dynamic studies of the endosomal compartment in Down syndrome. Acta Neuropathol. Commun. 2020, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Filippone, A.; Praticó, D. Endosome dysregulation in Down Syndrome: A potential contributos to Alzheimer disease pathology. Ann. Neurol. 2021, 90, 4–14. [Google Scholar] [CrossRef]
- Yang, C.; Wang, X. Lysosome biogenesis: Regulation and functions. J. Cell Biol. 2021, 220, e202102001. [Google Scholar] [CrossRef] [PubMed]
- Moreno-García, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An overview of the role of lipofuscin in age-related neurodegeneration. Front. Neurosci. 2018, 12, 464. [Google Scholar] [CrossRef] [Green Version]
- Hainmueller, T.; Bartos, M. Dentate gyrus circuits for encoding, retrieval and discrimination of episodic memories. Nat. Rev. Neurosci. 2020, 21, 153–168. [Google Scholar] [CrossRef]
- Day, S.M.; Yang, W.; Wang, X.; Stern, J.E.; Zhou, X.; Macauley, S.L.; Ma, T. Glucagon-Like Peptide-1 Cleavage Product Improves Cognitive Function in a Mouse Model of Down Syndrome. eNeuro 2019, 16, ENEURO.0031-19.2019. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, K.; Amano, K.; Takaki, E.; Ebrahim, A.S.; Shimohata, A.; Shibazaki, N.; Inoue, I.; Takaki, M.; Ueda, Y.; Sago, H.; et al. Increased lipid peroxidation in Down’s syndrome mouse models. J. Neurochem. 2009, 110, 1965–1976. [Google Scholar] [CrossRef]
- Di Domenico, F.; Barone, E.; Mancuso, C.; Perluigi, M.; Cocciolo, A.; Mecocci, P.; Butterfield, D.A.; Coccia, R. HO-1/BVR-a system analysis in plasma from probable Alzheimer’s disease and mild cognitive impairment subjects: A potential biochemical marker for the prediction of the disease. J. Alzheimers Dis. 2012, 32, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Buendia, I.; Michalska, P.; Navarro, E.; Gameiro, I.; Egea, J.; León, R. Nrf2-ARE pathway: An emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol. Ther. 2016, 157, 84–104. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Pupo, G.; Mancuso, C.; Barone, E.; Paolini, F.; Arena, A.; Blarzino, C.; Schmitt, F.A.; Head, E.; Butterfield, D.A.; et al. Bach1 overexpression in Down syndrome correlates with the alteration of the HO-1/BVR-a system: Insights for transition to Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 1107–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukkur, E.A.; Shimohata, A.; Akagi, T.; Yu, W.; Yamaguchi, M.; Murayama, M.; Chui, D.; Takeuchi, T.; Amano, K.; Subramhanya, K.H.; et al. Mitochondrial dysfunction and tau hyperphosphorylation in Ts1Cje, a mouse model for Down syndrome. Hum. Mol. Genet. 2006, 15, 2752–2762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccoli, C.; Izzo, A.; Scrima, R.; Bonfiglio, F.; Manco, R.; Negri, R.; Quarato, G.; Cela, O.; Ripoli, M.; Prisco, M.; et al. Chronic pro-oxidative state and mitochondrial dysfunctions are more pronounced in fibroblasts from Down syndrome foeti with congenital heart defects. Hum. Mol. Genet. 2013, 22, 1218–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, P.M.J.; Moreira, P.I.; Ambrósio, A.F.; Alves, C.H. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol. Commun. 2020, 8, 189. [Google Scholar] [CrossRef]
- Tramutola, A.; Lanzillotta, C.; Barone, E.; Arena, A.; Zuliani, I.; Mosca, L.; Blarzino, C.; Butterfield, D.A.; Perluigi, M.; Di Domenico, F. Intranasal rapamycin ameliorates Alzheimer-like cognitive decline in a mouse model of Down syndrome. Transl. Neurodegener. 2018, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Casafont, I.; Palanca, A.; Lafarga, V.; Berciano, M.T.; Lafarga, M. Effect of ionizing radiation in sensory ganglion neurons: Organization and dynamics of nuclear compartments of DNA damage/repair and their relationship with transcription and cell cycle. Acta Neuropathol. 2011, 122, 481–493. [Google Scholar] [CrossRef]
- Crowe, S.L.; Tsukerman, S.; Gale, K.; Jorgensen, T.J.; Kondratyev, A.D. Phosphorylation of histone H2A.X as an early marker of neuronal endangerment following seizures in the adult rat brain. J. Neurosci. 2011, 31, 7648–7656. [Google Scholar] [CrossRef] [Green Version]
- Chang, I.Y.; Kim, J.H.; Cho, K.W.; Yoon, S.P. Acute responses of DNA repair proteins and StarD6 in rat hippocampus after domoic acid-induced excitotoxicity. Acta Histochem. 2013, 115, 234–239. [Google Scholar] [CrossRef]
- Sharma, V.; Collins, L.B.; Chen, T.-H.; Herr, N.; Takeda, S.; Sun, W.; Swenberg, J.A.; Nakamura, J. Oxidative stress at low levels can induce clustered DNA lesions leading to NHEJ mediated mutations. Oncotarget 2016, 7, 25377–25390. [Google Scholar] [CrossRef]
- Suberbielle, E.; E Sanchez, P.; Kravitz, A.; Wang, X.; Ho, K.; Eilertson, K.; Devidze, N.; Kreitzer, A.C.; Mucke, L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat. Neurosci. 2013, 16, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Gao, F.; Pfenning, A.R.; Pan, L.; Yamakawa, S.; Seo, J.; Rueda, R.; Phan, T.X.; Yamakawa, H.; Pao, P.-C.; et al. Activity-induced DNA breaks govern the expression of neuronal early-responsegenes. Cell 2015, 161, 1592–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katyal, S.; Lee, Y.; Nitiss, K.C.; Downing, S.M.; Li, Y.; Shimada, M.; Zhao, J.; Russell, H.R.; Petrini, J.H.J.; Nitiss, J.L.; et al. Aberrant topoisomerase-1 DNA lesions are pathogenic inneurodegenerative genome instability syndromes. Nat. Neurosci. 2014, 17, 813–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herreros, F.G.; Schuurs-Hoeijmakers, J.H.M.; McCormack, M.; Greally, M.T.; Rulten, S.; Romero-Granados, R.; Counihan, T.J.; Chaila, E.; Conroy, J.; Ennis, S.; et al. TDP2 protects transcription from abortive topoisomerase activity and is required for normal neural function. Nat. Genet. 2014, 46, 516–521. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Penney, J.; Tsai, L.H. Chromatin regulation of DNA damage repair and genome integrity in the central nervous system. J. Mol. Biol. 2014, 426, 3376–3388. [Google Scholar] [CrossRef] [Green Version]
- Chow, H.M.; Herrup, K. Genomic integrity and the ageing brain. Nat. Rev. Neurosci. 2015, 16, 672–684. [Google Scholar] [CrossRef]
- Thadathil, N.; Delotterie, D.F.; Xiao, J.; Hori, R.; McDonald, M.P.; Khan, M.M. DNA double-strand breaks accumulation in Alzheimer’s disease: Evidence from experimental models and postmorten human brains. Mol. Neurobiol. 2021, 58, 118–131. [Google Scholar] [CrossRef]
- Shadfar, S.; Brocardo, M.; Atkin, J.D. The complex mechanism by which neurons die following DNA damage in neurodegenerative diseases. Int. J. Mol. Sci. 2022, 23, 2484. [Google Scholar] [CrossRef]
- Murray, A.; Letourneau, A.; Canzonetta, C.; Stathaki, E.; Gimelli, S.; Sloan-Bena, F.; Abrehart, R.; Goh, P.; Lim, S.; Baldo, C.; et al. Isogenic induced pluripotent stem cell lines from an adult with mosaic Down syndrome model accelerated neuronal ageing and neurodegeneration. Stem Cells 2015, 33, 2077–2084. [Google Scholar] [CrossRef]
- Wang, Y.; Chang, J.; Shao, L.; Feng, W.; Luo, Y.; Chow, M.; Du, W.; Meng, A.; Zhou, D. Hematopoietic stem cells from Ts65Dn mice are deficient in the repair of DNA double-strand breaks. Radiat. Res. 2016, 185, 630–637. [Google Scholar] [CrossRef] [Green Version]
- Pawlikowski, B.; Betta, N.D.; Elston, T.; Williams, D.A.; Olwin, B.B. Muscle stem cell dysfunction impairs muscle regeneration in a mouse model of Down syndrome. Sci. Rep. 2018, 8, 4309. [Google Scholar] [CrossRef] [PubMed]
- Olive, P.L.; Banáth, J.P. Phosphorylation of histone H2AX as a measure of radiosensitivity. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Mata-Garrido, J.; Casafont, I.; Tapia, O.; Berciano, M.T.; Lafarga, M. Neuronal accumulation of unrepaired DNA in a novel specific chromatin domain: Structural, molecular and transcriptional characterization. Acta Neuropathol. Commun. 2016, 4, 41. [Google Scholar] [CrossRef] [Green Version]
- Shanbhag, N.J.; Evans, M.D.; Mao, W.; Nana, A.L.; Seeley, W.W.; Adame, A.; Rissman, R.A.; Masliah, E.; Mucke, L. Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathol. Commun. 2019, 7, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzi, H.A.; Reeves, R.H. Hipocampal hipocellularity in the Ts65Dn mouse originates early in development. Brain Res. 2006, 1104, 153–159. [Google Scholar] [CrossRef]
- Contestabile, A.; Fila, T.; Ceccarelli, C.; Bonasoni, P.; Bonapace, L.; Santini, D.; Bartesaghi, R.; Ciani, E. Cell cycle alteration and decreased cell proliferation in the hippocampal dentate gyrus and in the neocortical germinal matrix of fetuses with down syndrome and in Ts65Dn mice. Hippocampus 2007, 17, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Rueda, N.; Flórez, J.; Martínez-Cué, C. The Ts65Dn mouse model of Down syndrome shows reduced expression of the Bcl-X(L) antiapoptotic protein in the hippocampus not accompanied by changes in molecular or cellular markers of cell death. Int. J. Dev. Neurosci. 2011, 29, 711–716. [Google Scholar] [CrossRef]
- Rueda, N.; Flórez, J.; Martínez-Cué, C. Apoptosis in Down’s syndrome: Lessons from studies of human and mouse models. Apoptosis 2013, 18, 121–134. [Google Scholar] [CrossRef]
- Price, B.D.; D’Andrea, A.D. Chromatin remodeling at DNA double-strand breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef] [Green Version]
- Hauer, M.H.; Gasser, S.M. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 2017, 31, 2204–2221. [Google Scholar] [CrossRef] [Green Version]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Kingston, R.E. Mechanisms of polycomb gene silencing: Knowns and unknowns. Nat. Rev. Mol. Cell Biol. 2009, 10, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H.J. DNA damage, aging, and cancer. N. Eng. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Granese, B.; Scala, I.; Spatuzza, C.; Valentino, A.; Coletta, M.; Vacca, R.A.; De Luca, P.; Andria, G. Validation of microarray data in human lymphoblasts shows a role of ubiquitin-proteaome system and NF-kB in the pathogenesis of Down syndrome. BMC Med. Genom. 2013, 6, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, S.; Tsuchiya, H.; Kaiho, A.; Guo, Q.; Ikeuchi, K.; Endo, A.; Arai, N.; Ohtake, F.; Murata, S.; Inada, T.; et al. Stress- and ubiquitylation-dependent phase separation of the proteasome. Nature 2020, 578, 296–300. [Google Scholar] [CrossRef]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chetelat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Prim. 2021, 7, 33. [Google Scholar] [CrossRef]
- Candelise, N.; Scaricamazza, S.; Salvatori, I.; Ferri, A.; Valle, C.; Manganelli, V.; Garofalo, T.; Sorice, M.; Msasi, R. Protein aggregation landscape in neurodegenerative diseases: Clinical relevance and future applications. Int. J. Mol. Sci. 2019, 22, 6016. [Google Scholar] [CrossRef]
- Zbinden, A.; Perez-Berlanga, M.; De Rossi, P.; Polymenidou, M. Phase separation and neurodegenerative diseases: A disturbance in the force. Dev. Cell 2020, 55, 45–68. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Eckmann, C.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Jülicher, F.; Hyman, A.A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef]
- Riederer, B.M.; Leuba, G.; Vernay, A.; Riederer, I.M. The role of the ubiquitin proteasome system in Alzheimer’s disease. Exp. Biol. Med. 2011, 236, 268–276. [Google Scholar] [CrossRef]
- Benaroudj, N.; Zwickl, P.; Seemülle, E.; Baumeister, W.; Goldberg, A.L. ATP hydrolysis by the proteasome regulatory complex PAN serves multiple functions in protein degradation. Mol. Cell 2003, 11, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Grune, T.; Jung, T.; Merker, K.; Davies, K.J. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroidand ‘aggresomes’ during oxidative stress, aging and disease. Int. J. Biochem. Cell Biol. 2004, 36, 2519–2530. [Google Scholar] [CrossRef] [PubMed]
- Tseng, B.P.; Green, K.N.; Chan, J.L.; Blurton-Jones, M.; Laferla, F.M. Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol. Aging 2007, 29, 1607–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, C.L.; Bimonte-Nelson, H.A.; Nelson, M.; Eckman, C.B.; Granholm, A.C. Behavioral and neurobiological markers of Alzheimer’s disease in Ts65Dn mice: Effects of estrogen. Neurobiol. Aging 2004, 25, 873–884. [Google Scholar] [CrossRef]
- Krohne, T.U.; Kaemmerer, E.; Holz, F.G.; Kopitz, J. Lipid peroxidation products reduce lysosomal protease activities in human retinal pigment epithelial cells via two different mechanisms of action. Exp. Eye Res. 2010, 90, 261–266. [Google Scholar] [CrossRef]
- Di Domenico, F.; Tramutola, A.; Perluigi, M. Cathepsin D as a therapeutic target in Alzheimer’s disease. Expert Opin. Ther. Targets 2016, 20, 1393–1395. [Google Scholar] [CrossRef] [Green Version]
- Kurz, T.; Terman, A.; Gustafsson, B.; Brunk, U.T. Lysosomes and oxidative stress in aging and apoptosis. Biochim. Biophys. Acta 2008, 1780, 1291–1303. [Google Scholar] [CrossRef]
- Jung, T.; Badder, N.; Grune, T. Lipofuscin. Ann. N. Y. Acad. Sci. 2007, 1119, 97–111. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puente-Bedia, A.; Berciano, M.T.; Martínez-Cué, C.; Lafarga, M.; Rueda, N. Oxidative-Stress-Associated Proteostasis Disturbances and Increased DNA Damage in the Hippocampal Granule Cells of the Ts65Dn Model of Down Syndrome. Antioxidants 2022, 11, 2438. https://doi.org/10.3390/antiox11122438
Puente-Bedia A, Berciano MT, Martínez-Cué C, Lafarga M, Rueda N. Oxidative-Stress-Associated Proteostasis Disturbances and Increased DNA Damage in the Hippocampal Granule Cells of the Ts65Dn Model of Down Syndrome. Antioxidants. 2022; 11(12):2438. https://doi.org/10.3390/antiox11122438
Chicago/Turabian StylePuente-Bedia, Alba, María T. Berciano, Carmen Martínez-Cué, Miguel Lafarga, and Noemí Rueda. 2022. "Oxidative-Stress-Associated Proteostasis Disturbances and Increased DNA Damage in the Hippocampal Granule Cells of the Ts65Dn Model of Down Syndrome" Antioxidants 11, no. 12: 2438. https://doi.org/10.3390/antiox11122438