
The Implications of Connexin 43 Deficiency during the Early Stages of Chemically Induced Mouse Colon Carcinogenesis

 , , and
, , and 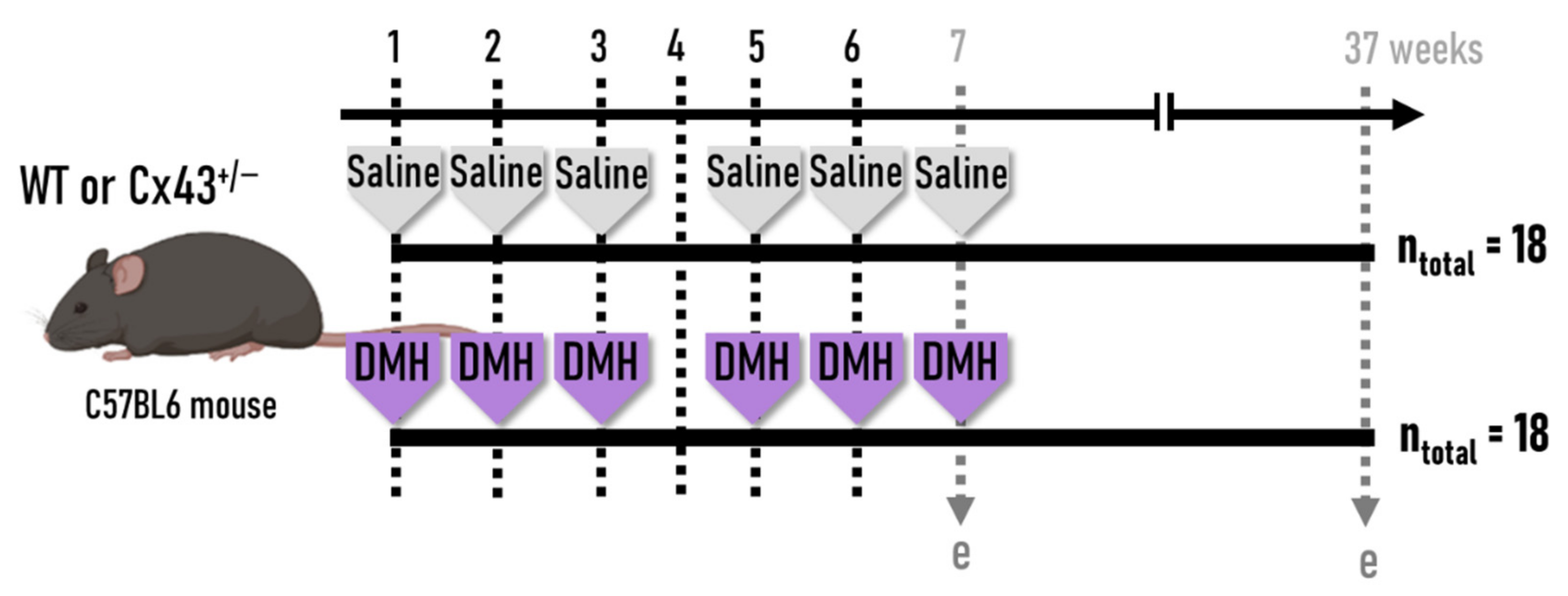{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Design and Sampling
2.3. Genotyping
2.4. Genotoxicity Assessment
2.5. ACF Screening and Tumor Analysis
2.6. Immunohistochemistry Analysis
2.7. Analysis of Antioxidant Enzymes
2.8. In Silico Analysis of Human CRA Samples
2.9. Statistical Analysis
3. Results
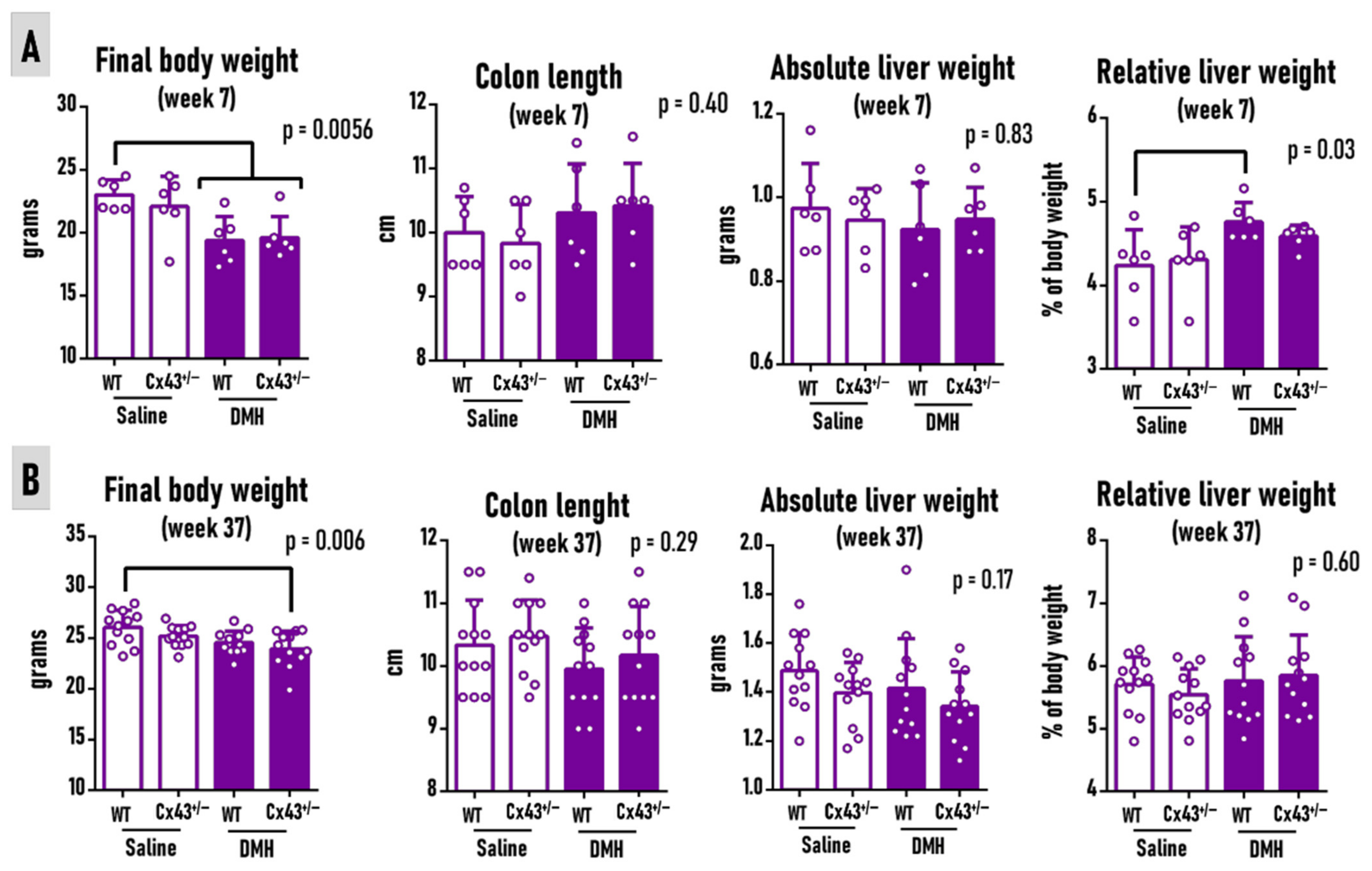
3.1. Cx43 Deficiency Does Not Alter DMH Effects on Body Weight, Liver Weights, or Colon Length at Weeks 7 and 37
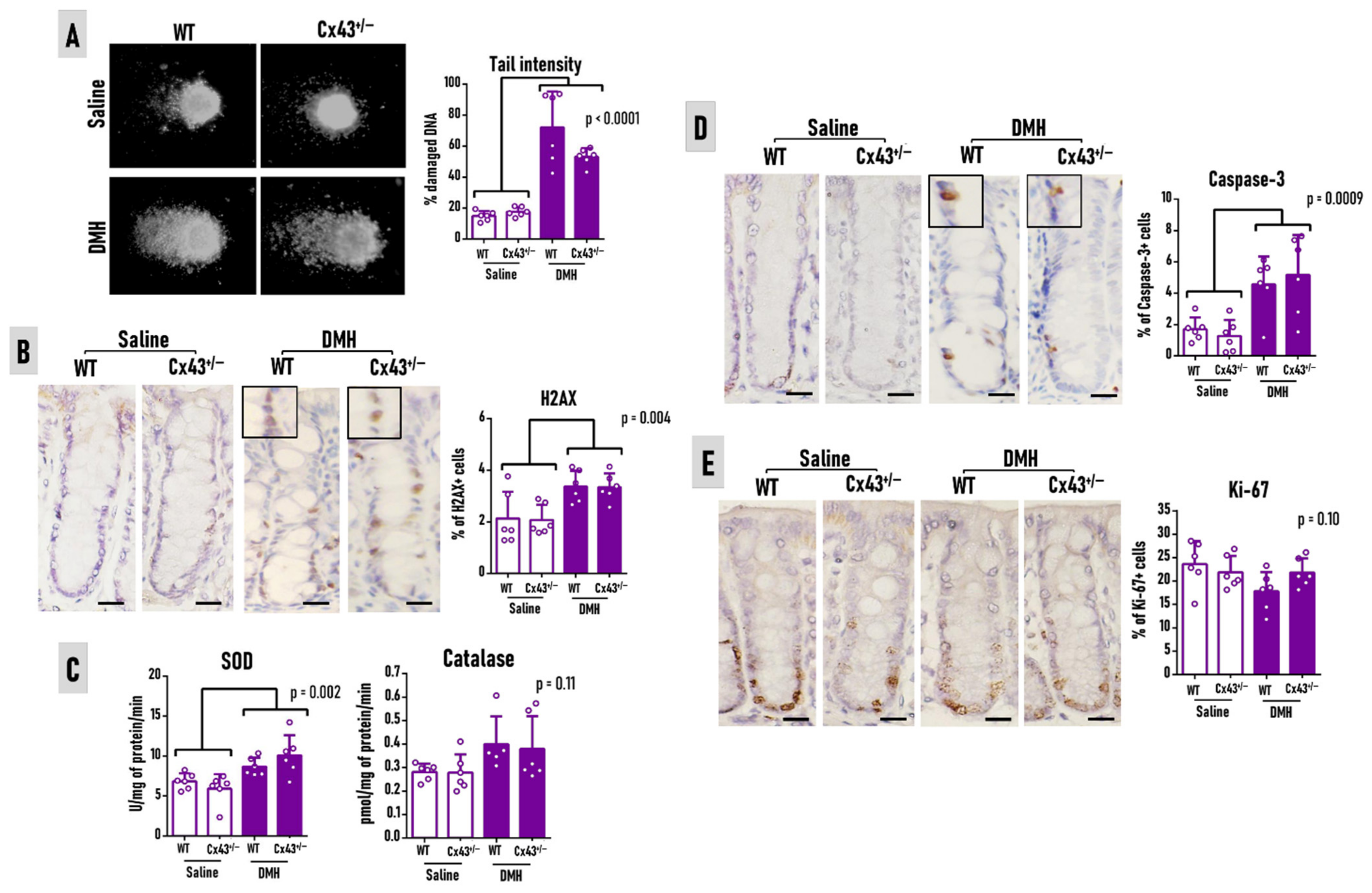
3.2. Cx43 Deficiency Does Not Alter Carcinogen-Induced Outcomes at Week 7
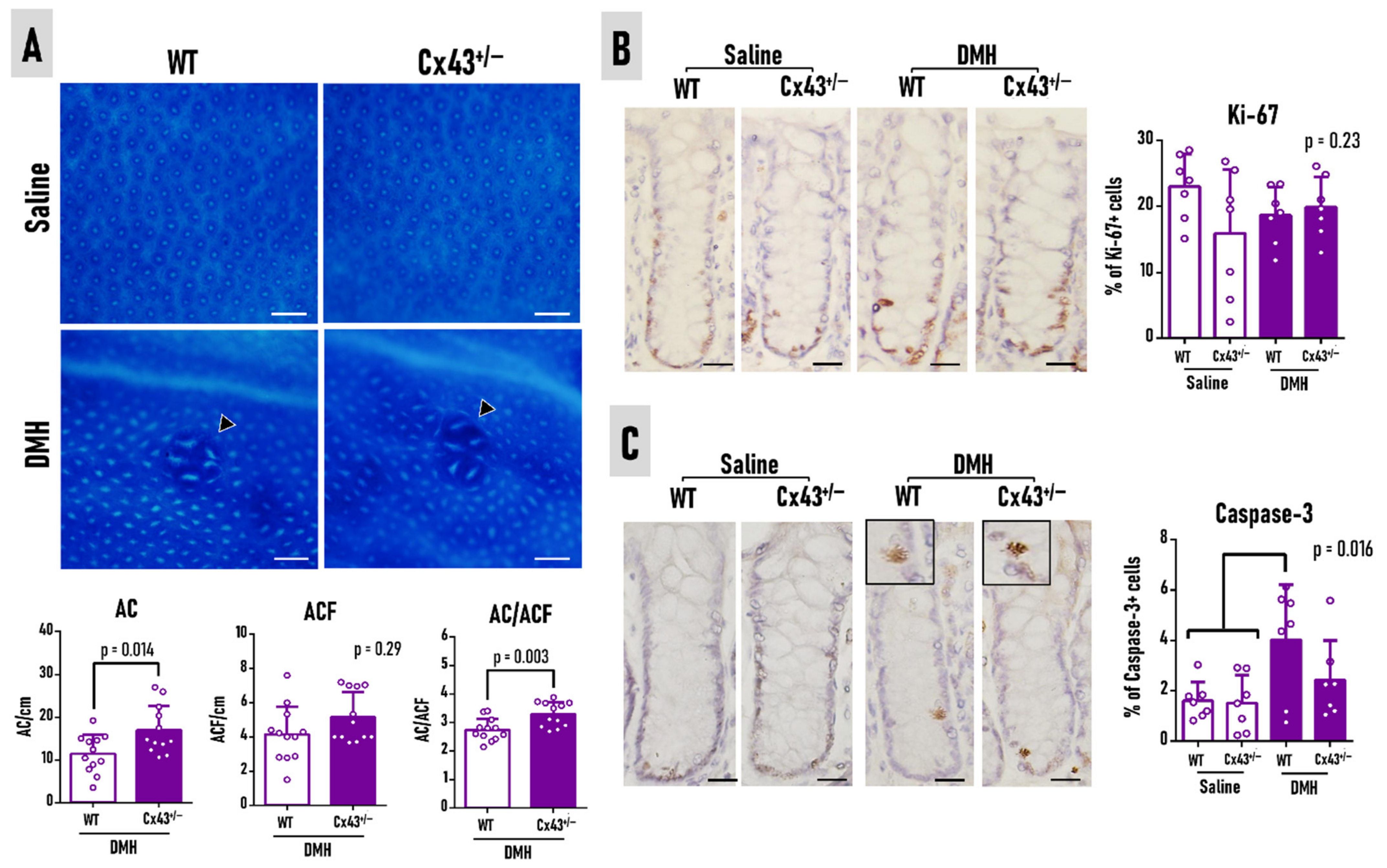
3.3. Cx43 Deficiency Slightly Attenuates Preneoplastic ACF Burden at Week 37
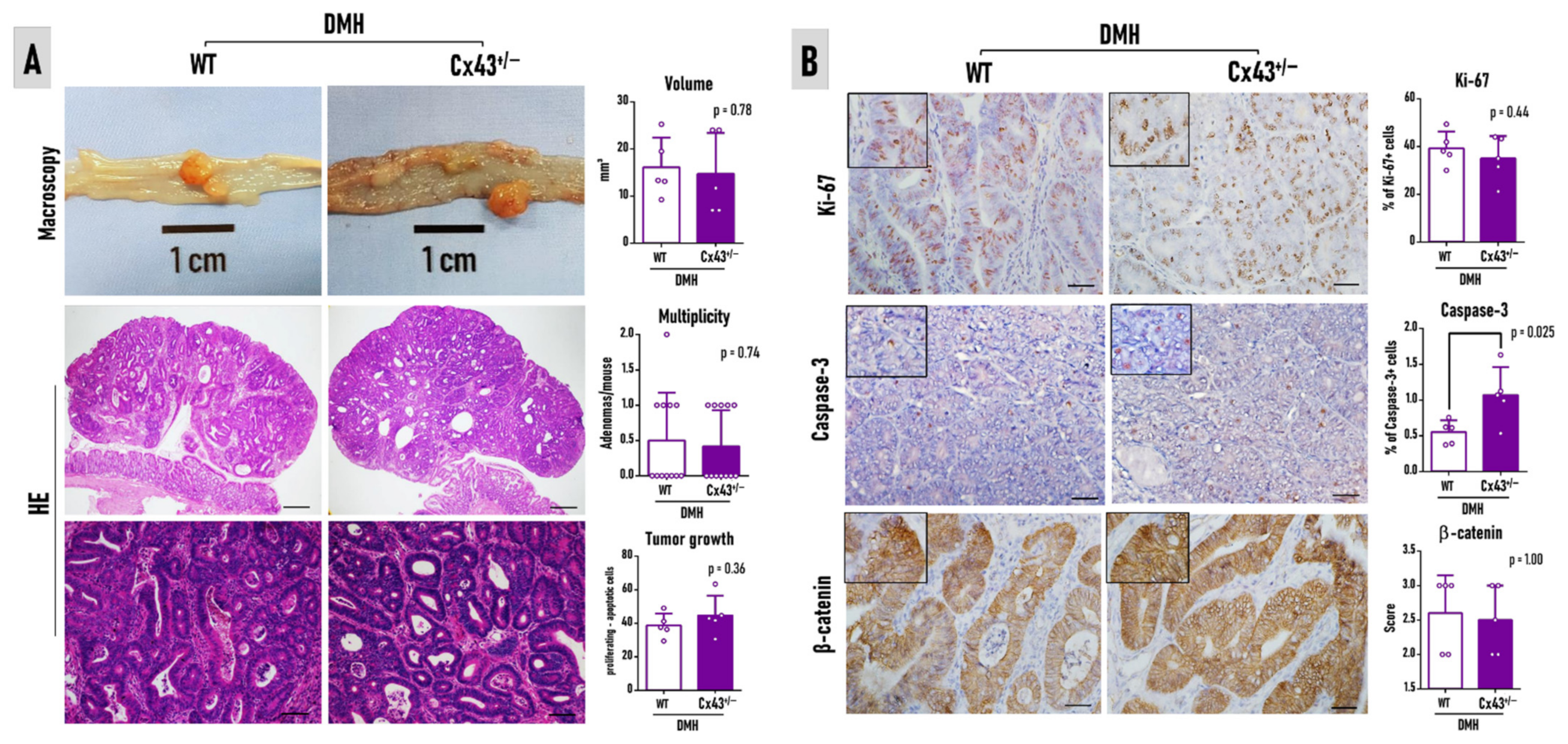
3.4. Cx43 Deficiency Does Not Alter Colorectal Adenoma Burden at Week 37
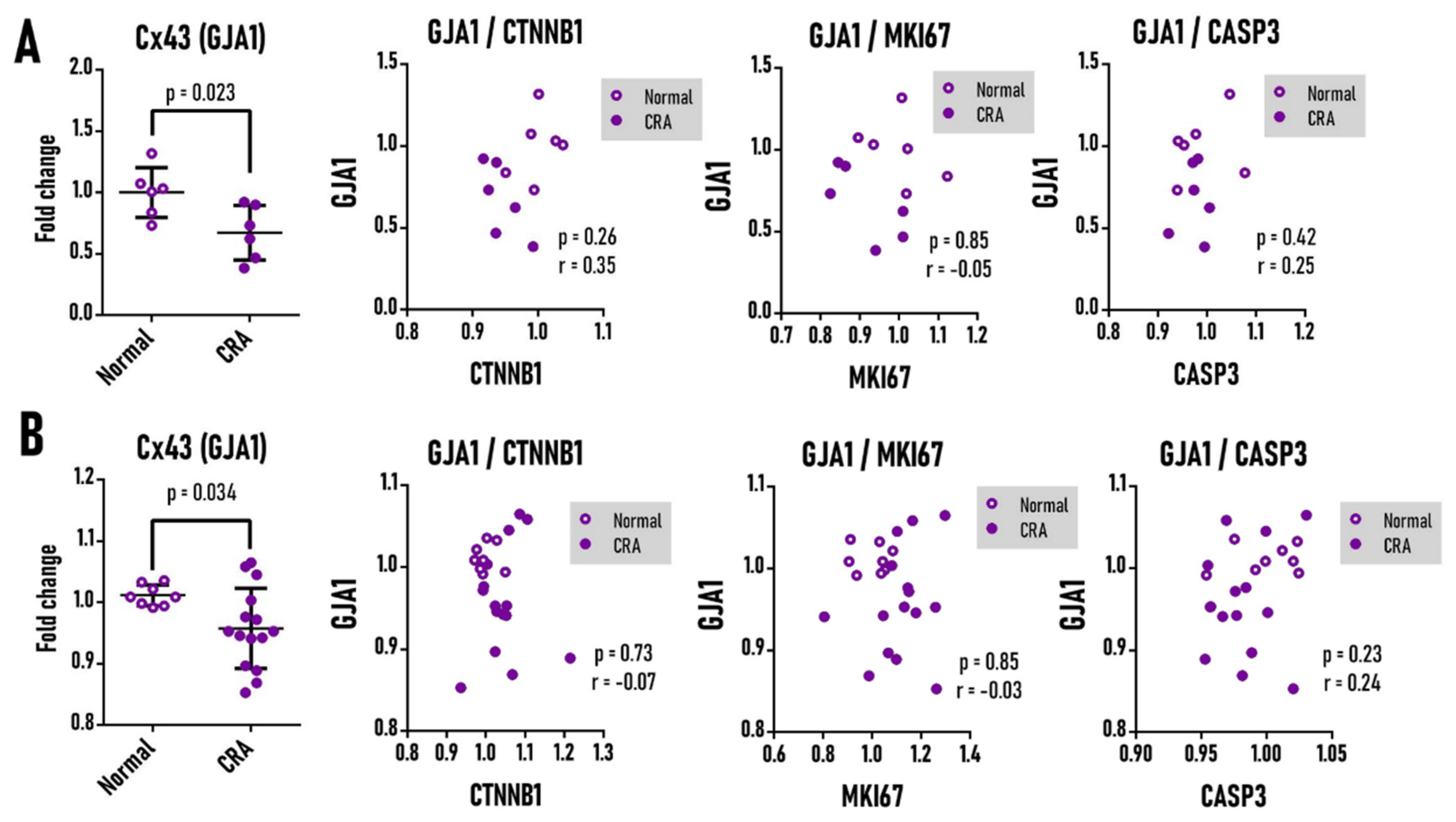
3.5. Cx43 mRNA Is Not Correlated to Ki-67, Caspase-3, and β-Catenin in Human Colorectal Adenoma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Leporrier, J.; Maurel, J.; Chiche, L.; Bara, S.; Segol, P.; Launoy, G. A population-based study of the incidence, management and prognosis of hepatic metastases from colorectal cancer. Br. J. Surg. 2006, 93, 465–474. [Google Scholar] [CrossRef] [PubMed]
- De Klerk, C.M.; Gupta, S.; Dekker, E.; Essink-Bot, M.L. Socioeconomic and ethnic inequities within organised colorectal cancer screening programmes worldwide. Gut 2018, 67, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, S.; Liu, Y.; Zhang, C.; Li, H.; Lai, B. Metastatic patterns and survival outcomes in patients with stage IV colon cancer: A population-based analysis. Cancer Med. 2020, 9, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.S.M.; Lau, R.; Aune, D.; Vieira, R.; Greenwood, D.C.; Kampman, E.; Norat, T. Red and processed meat and colorectal cancer incidence: Meta-analysis of prospective studies. PLoS ONE 2011, 6, e20456. [Google Scholar] [CrossRef] [Green Version]
- Vieira, A.R.; Abar, L.; Chan, D.S.M.; Vingeliene, S.; Polemiti, E.; Stevens, C.; Greenwood, D.; Norat, T. Foods and beverages and colorectal cancer risk: A systematic review and meta-analysis of cohort studies, an update of the evidence of the WCRF-AICR Continuous Update Project. Ann. Oncol. 2017, 28, 1788–1802. [Google Scholar] [CrossRef]
- Murphy, N.; Moreno, V.; Hughes, D.J.; Vodicka, L.; Vodicka, P.; Aglago, E.K.; Gunter, M.J.; Jenab, M. Lifestyle and dietary environmental factors in colorectal cancer susceptibility. Mol. Asp. Med. 2019, 69, 2–9. [Google Scholar] [CrossRef]
- Amitay, E.L.; Carr, P.R.; Jansen, L.; Roth, W.; Alwers, E.; Herpel, E.; Kloor, M.; Bläker, H.; Chang-Claude, J.; Brenner, H.; et al. Smoking, alcohol consumption and colorectal cancer risk by molecular pathological subtypes and pathways. Br. J. Cancer 2020, 122, 1604–1610. [Google Scholar] [CrossRef] [Green Version]
- Sawicki, T.; Ruszkowska, M.; Danielewicz, A.; Niedźwiedzka, E.; Arłukowicz, T.; Przybyłowicz, K.E. A review of colorectal cancer in terms of epidemiology, risk factors, development, symptoms and diagnosis. Cancers 2021, 13, 2025. [Google Scholar] [CrossRef]
- Leslie, A.; Carey, F.A.; Pratt, N.R.; Steele, R.J. The colorectal adenoma-carcinoma sequence. Br. J. Surg. 2002, 89, 845–860. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 6, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Bird, R.P. Observation and quantification of aberrant crypts in the murine colon treated with a colon carcinogen: Preliminary findings. Cancer Lett. 1987, 37, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Shpitz, B.; Bomstein, Y.; Mekori, Y.; Cohen, R.; Kaufman, Z.; Neufeld, D.; Galkin, M.; Bernheim, J. Aberrant crypt foci in human colons: Distribution and histomorphologic characteristics. Hum. Pathol. 1998, 29, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Takayama, T.; Katsuki, S.; Takahashi, Y.; Ohi, M.; Nojiri, S.; Sakamaki, S.; Kato, J.; Kogawa, K.; Miyake, H.; Niitsu, Y. Aberrant crypt foci of the colon as precursors of adenoma and cancer. N. Engl. J. Med. 1998, 339, 1277–1284. [Google Scholar] [CrossRef]
- Bird, R.P.; Good, C.K. The significance of aberrant crypt foci in understanding the pathogenesis of colon cancer. Toxicol. Lett. 2000, 112–113, 395–402. [Google Scholar] [CrossRef]
- Stopera, S.A.; Bird, R.P. Immunohistochemical demonstration of mutant p53 tumour suppressor gene product in aberrant crypt foci. Cytobios 1993, 73, 73–88. [Google Scholar]
- Yamashita, N.; Minamoto, T.; Ochiai, A.; Onda, M.; Esumi, H. Frequent and characteristic K-ras activation in aberrant crypt foci of colon. Is there preference among K-ras mutants for malignant progression? Cancer 1995, 75, 1527–1533. [Google Scholar] [CrossRef]
- Yamada, Y.; Yoshimi, N.; Hirose, Y.; Kawabata, K.; Matsunaga, K.; Shimizu, M.; Hara, A.; Mori, H. Frequent beta-catenin gene mutations and accumulations of the protein in the putative preneoplastic lesions lacking macroscopic aberrant crypt foci appearance, in rat colon carcinogenesis. Cancer Res. 2000, 60, 3323–3327. [Google Scholar]
- Wargovich, M.J.; Brown, V.R.; Morris, J. Aberrant crypt foci: The case for inclusion as a biomarker for colon cancer. Cancers 2010, 2, 1705–1716. [Google Scholar] [CrossRef] [Green Version]
- Caetano, B.F.R.; Tablas, M.B.; Romualdo, G.R.; Rodrigues, M.A.M.; Barbisan, L.F. Early molecular events associated with liver and colon sub-acute responses to 1,2-dimethylhydrazine: Potential implications on preneoplastic and neoplastic lesion development. Toxicol. Lett. 2020, 329, 67–79. [Google Scholar] [CrossRef]
- Rosenberg, D.W.; Giardina, C.; Tanaka, T. Mouse models for the study of colon carcinogenesis. Carcinogenesis 2009, 30, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Perše, M.; Cerar, A. The dimethylhydrazine induced colorectal tumours in rat—Experimental colorectal carcinogenesis. Radiol. Oncol. 2005, 39, 61–70. [Google Scholar]
- Perše, M.; Cerar, A. Morphological and molecular alterations in 1,2 dimethylhydrazine and azoxymethane induced colon carcinogenesis in rats. J. Biomed. Biotechnol. 2011, 2011, 473964. [Google Scholar] [CrossRef] [PubMed]
- Wolter, S.; Frank, N. Metabolism of 1,2-dimethylhydrazine in isolated perfused rat liver. Chem. Biol. Interact. 1982, 42, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Wolter, S.; Schmähl, D.; Frank, N. Influence of diet on 1,2-dimethylhydrazine metabolism in rat liver. Nutr. Cancer 1984, 6, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Weisburger, J.H. Colon carcinogens: Their metabolism and mode of action. Cancer 1971, 28, 60–70. [Google Scholar] [CrossRef]
- Skrzydlewska, E.; Sulkowski, S.; Koda, M.; Zalewski, B.; Kanczuga-Koda, L.; Sulkowska, M. Lipid peroxidation and antioxidant status in colorectal cancer. World J. Gastroenterol. 2005, 11, 403–406. [Google Scholar] [CrossRef]
- Clapper, M.L.; Chang, W.C.L.; Cooper, H.S. Dysplastic aberrant crypt foci: Biomarkers of early colorectal neoplasia and response to preventive intervention. Cancer Prev. Res. 2020, 13, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Willecke, K.; Jungbluth, S.; Dahl, E.; Hennemann, H.; Heynkes, R.; Grzeschik, K.H. Six genes of the human connexin gene family coding for gap junctional proteins are assigned to four different human chromosomes. Eur. J. Cell Biol. 1990, 53, 275–280. [Google Scholar]
- Werner, R.; Levine, E.; Rabadan-Diehl, C.; Dahl, G. Formation of hybrid cell-cell channels. Proc. Natl. Acad. Sci. USA 1989, 86, 5380–5384. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.; Goldberg, G. Transfer of Biologically Important Molecules Between Cells Through Gap Junction Channels. Curr. Med. Chem. 2003, 10, 2045–2058. [Google Scholar] [CrossRef] [PubMed]
- Dbouk, H.A.; Mroue, R.M.; El-Sabban, M.E.; Talhouk, R.S. Connexins: A myriad of functions extending beyond assembly of gap junction channels. Cell Commun. Signal. 2009, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanczuga-Koda, L.; Sulkowski, S.; Koda, M.; Sobaniec-Łotowska, M.; Sulkowska, M. Expression of connexins 26, 32 and 43 in the human colon—An immunohistochemical study. Folia Histochem. Cytobiol. 2004, 42, 203–207. [Google Scholar] [PubMed]
- Sirnes, S.; Lind, G.E.; Bruun, J.; Fykerud, T.A.; Mesnil, M.; Lothe, R.A.; Rivedal, E.; Kolberg, M.; Leithe, E. Connexins in colorectal cancer pathogenesis. Int. J. Cancer 2015, 137, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sirnes, S.; Bruun, J.; Kolberg, M.; Kjenseth, A.; Lind, G.E.; Svindland, A.; Brech, A.; Nesbakken, A.; Lothe, R.A.; Leithe, E.; et al. Connexin43 acts as a colorectal cancer tumor suppressor and predicts disease outcome. Int. J. Cancer 2012, 131, 570–581. [Google Scholar] [CrossRef]
- Ismail, R.; Rashid, R.; Andrabi, K.; Parray, F.Q.; Besina, S.; Shah, M.A.; Hussain, M.U. Pathological implications of Cx43 down-regulation in human colon cancer. Asian Pac. J. Cancer Prev. 2014, 15, 2987–2991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayad, E.; Eesa, A.N.; Radi, R.; Abdel-Salam, L.O.E.F. Expression of “connexin 43” in colorectal carcinomas: Histopathological and immunohistochemical study. Open Access Maced. J. Med. Sci. 2020, 8, 354–359. [Google Scholar] [CrossRef]
- Dubina, M.V.; Iatckii, N.A.; Popov, D.E.; Vasil’ev, S.V.; Krutovskikh, V.A. Connexin 43, but not connexin 32, is mutated at advanced stages of human sporadic colon cancer. Oncogene 2002, 21, 4992–4996. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Zhang, P.J.; Chen, T.; Yum, S.W.; Pasha, T.; Furth, E.E. Connexin43 Expression Increases in the Epithelium and Stroma along the Colonic Neoplastic Progression Pathway: Implications for Its Oncogenic Role. Gastroenterol. Res. Pract 2011, 2011, 561719. [Google Scholar] [CrossRef] [Green Version]
- Gong, K.; Hong, Q.; Wu, H.; Wang, F.; Zhong, L.; Shen, L.; Xu, P.; Zhang, W.; Cao, H.; Zhan, Y.; et al. Gap junctions mediate glucose transfer to promote colon cancer growth in three-dimensional spheroid culture. Cancer Lett. 2022, 531, 27–38. [Google Scholar] [CrossRef]
- Kanczuga-Koda, L.; Koda, M.; Sulkowski, S.; Wincewicz, A.; Zalewski, B.; Sulkowska, M. Gradual loss of functional gap junction within progression of colorectal cancer—A shift from membranous CX32 and CX43 expression to cytoplasmic pattern during colorectal carcinogenesis. In Vivo 2010, 24, 101–108. [Google Scholar] [PubMed]
- Reaume, A.G.; De Sousa, P.A.; Kulkarni, S.; Langille, B.L.; Zhu, D.; Davies, T.C.; Juneja, S.C.; Kidder, G.M.; Rossant, J. Cardiac Malformation in Neonatal Mice Lacking Connexin 43. Science 1995, 267, 1831–1834. [Google Scholar] [CrossRef]
- Richards, T.C. Early changes in the dynamics of crypt cell populations in mouse colon following administration of 1,2-dimethylhydrazine. Cancer Res. 1977, 37, 1680–1685. [Google Scholar]
- James, J.T.; Shamsuddin, A.M.; Trump, B.F. Comparative study of the morphologic, histochemical, and proliferative changes induced in the large intestine of ICR/Ha and C57BL/Ha mice by 1,2-dimethylhydrazine. J. Natl. Cancer Inst. 1983, 71, 955–964. [Google Scholar] [PubMed]
- Ijiri, K. Apoptosis (cell death) induced in mouse bowel by 1,2-dimethylhydrazine, methylazoxymethanol acetate, and gamma-rays. Cancer Res. 1989, 49, 6342–6346. [Google Scholar] [PubMed]
- James, J.T.; Autrup, H. Methylated DNA adducts in the large intestine of ICR/Ha and C57BL/Ha mice given 1,2-dimethylhydrazine. J. Natl. Cancer Inst. 1983, 70, 541–546. [Google Scholar] [PubMed]
- Sasaki, Y.F.; Saga, A.; Akasaka, M.; Ishibashi, S. Organ-specific genotoxicity of the potent rodent colon carcinogen 1, 2-dimethylhydrazine and three hydrazine derivatives: Difference between intraperitoneal and oral administration. Mutat. Res. 1998, 415, 1–12. [Google Scholar] [CrossRef]
- Nolte, T.; Brander-Weber, P.; Dangler, C.; Deschl, U.; Elwell, M.R.; Greaves, P.; Hailey, R.; Leach, M.W.; Pandiri, A.R.; Rogers, A.; et al. Nonproliferative and Proliferative Lesions ofthe Gastrointestinal Tract, Pancreas and Salivary Glands of the Rat and Mouse. J. Toxicol. Pathol. 2016, 29, 1S–125S. [Google Scholar] [CrossRef] [Green Version]
- Marklund, S.; Marklund, G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur. J. Biochem. 1974, 47, 469–474. [Google Scholar] [CrossRef]
- Bergmeyer, H.U. Methods of Enzymatic Analysis, 2nd ed.; Academic Press: New York, NY, USA, 1974. [Google Scholar]
- Galamb, O.; Spisák, S.; Sipos, F.; Tóth, K.; Solymosi, N.; Wichmann, B.; Krenács, T.; Valcz, G.; Tulassay, Z.; Molnár, B. Reversal of gene expression changes in the colorectal normal-adenoma pathway by NS398 selective COX2 inhibitor. Br. J. Cancer 2010, 102, 765–773. [Google Scholar] [CrossRef]
- Galamb, O.; Wichmann, B.; Sipos, F.; Spisák, S.; Krenács, T.; Tóth, K.; Leiszter, K.; Kalmár, A.; Tulassay, Z.; Molnár, B. Dysplasia-carcinoma transition specific transcripts in colonic biopsy samples. PLoS ONE 2012, 7, e48547. [Google Scholar] [CrossRef] [PubMed]
- Schell, M.J.; Yang, M.; Teer, J.K.; Lo, F.Y.; Madan, A.; Coppola, D.; Monteiro, A.N.A.; Nebozhyn, M.V.; Yue, B.; Loboda, A.; et al. A multigene mutation classification of 468 colorectal cancers reveals a prognostic role for APC. Nat. Commun. 2016, 7, 11743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttman, J.A.; Lin, A.E.; Li, Y.; Bechberger, J.; Naus, C.C.; Vogl, A.W.; Finlay, B.B. Gap junction hemichannels contribute to the generation of diarrhoea during infectious enteric disease. Gut 2010, 59, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Clayton, J.A. Studying both sexes: A guiding principle for biomedicine. FASEB J. 2016, 30, 519–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amos-Landgraf, J.M.; Heijmans, J.; Wielenga, M.C.B.; Dunkin, E.; Krentz, K.J.; Clipson, L.; Ederveen, A.G.; Groothuis, P.G.; Mosselman, S.; Muncan, V.; et al. Sex disparity in colonic adenomagenesis involves promotion by male hormones, not protection by female hormones. Proc. Natl. Acad. Sci. USA 2014, 111, 16514–16519. [Google Scholar] [CrossRef]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. γh2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Kerr, C.A.; Hines, B.M.; Shaw, J.M.; Dunne, R.; Bragg, L.M.; Clarke, J.; Lockett, T.; Head, R. Genomic homeostasis is dysregulated in favour of apoptosis in the colonic epithelium of the azoxymethane treated rat. BMC Physiol. 2013, 13, 2. [Google Scholar] [CrossRef] [Green Version]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Zhang, G.; Li, B.; Wu, Y.; Liu, X.; Tan, Y. Dioscin augments HSV-tk-mediated suicide gene therapy for melanoma by promoting connexin-based intercellular communication. Oncotarget 2017, 8, 798–807. [Google Scholar] [CrossRef] [Green Version]
- Goubaeva, F.; Mikami, M.; Giardina, S.; Ding, B.; Abe, J.; Yang, J. Cardiac mitochondrial connexin 43 regulates apoptosis. Biochem. Biophys. Res. Commun. 2007, 352, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zhao, X.; Yao, Y.; Qi, X.; Yuan, Y.; Hu, Y. Connexin 43 interacts with Bax to regulate apoptosis of pancreatic cancer through a gap junction-independent pathway. Int. J. Oncol. 2012, 41, 941–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, R.; Riquelme, M.A.; Werner, S.; Jiang, J.X. Connexin 43 channels protect osteocytes against oxidative stress-induced cell death. J. Bone Miner Res. 2013, 28, 1611–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecoraro, M.; Pala, B.; Di Marcantonio, M.C.; Muraro, R.; Marzocco, S.; Pinto, A.; Mincione, G.; Popolo, A. Doxorubicin-induced oxidative and nitrosative stress: Mitochondrial connexin 43 is at the crossroads. Int. J. Mol. Med. 2020, 46, 1197–1209. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Kim, P.; Kim, J.K.; Kim, Y.R.; Fukumura, D.; Yun, S.H. Longitudinal tracing of spontaneous regression and anti-angiogenic response of individual microadenomas during colon tumorigenesis. Theranostics 2015, 5, 724–732. [Google Scholar] [CrossRef] [Green Version]
- Schoen, R.E.; Mutch, M.; Rall, C.; Dry, S.M.; Seligson, D.; Umar, A.; Pinsky, P. The natural history of aberrant crypt foci. Gastrointest. Endosc. 2008, 67, 1097–1102. [Google Scholar] [CrossRef]
- Femia, A.P.; Bendinelli, B.; Giannini, A.; Salvadori, M.; Pinzani, P.; Dolara, P.; Caderni, G. Mucin-depleted foci have β-catenin gene mutations, altered expression of its protein, and are dose- and time-dependent in the colon of 1,2-dimethylhydrazine-treated rats. Int. J. Cancer 2005, 116, 9–15. [Google Scholar] [CrossRef]
- Femia, A.P.; Dolara, P.; Giannini, A.; Salvadori, M.; Biggeri, A.; Caderni, G. Frequent mutation of Apc gene in rat colon tumors and mucin-depleted foci, preneoplastic lesions in experimental colon carcinogenesis. Cancer Res. 2007, 67, 445–449. [Google Scholar] [CrossRef] [Green Version]
- Lüchtenborg, M.; Weijenberg, M.P.; Roemen, G.M.J.M.; de Bruïne, A.P.; van den Brandt, P.A.; Lentjes, M.H.F.M.; Brink, M.; van Engeland, M.; Goldbohm, R.A.; de Goeij, A.F.P.M. APC mutations in sporadic coloretal carcinomas from The Netherlands Cohort Study. Carcinogenesis 2004, 25, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Blum, C.A.; Tanaka, T.; Zhong, X.; Li, Q.; Dashwood, W.M.; Pereira, C.; Xu, M.; Dashwood, R.H. Mutational analysis of Ctnnb1 and Apc in tumors from rats given 1,2-dimethylhydrazine or 2-amino-3-methylimidazo[4,5-f]quinoline: Mutational “hotspots” and the relative expression of β-catenin and c-jun. Mol. Carcinog. 2003, 36, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Turovsky, E.A.; Varlamova, E.G. Mechanism of Ca2+-dependent pro-apoptotic action of selenium nanoparticles, mediated by activation of Cx43 hemichannels. Biology 2021, 10, 743. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santo, S.G.E.; da Silva, T.C.; Vinken, M.; Cogliati, B.; Barbisan, L.F.; Romualdo, G.R. The Implications of Connexin 43 Deficiency during the Early Stages of Chemically Induced Mouse Colon Carcinogenesis. Antioxidants 2022, 11, 2368. https://doi.org/10.3390/antiox11122368
Santo SGE, da Silva TC, Vinken M, Cogliati B, Barbisan LF, Romualdo GR. The Implications of Connexin 43 Deficiency during the Early Stages of Chemically Induced Mouse Colon Carcinogenesis. Antioxidants. 2022; 11(12):2368. https://doi.org/10.3390/antiox11122368
Chicago/Turabian StyleSanto, Sara Gomes Espírito, Tereza Cristina da Silva, Mathieu Vinken, Bruno Cogliati, Luís Fernando Barbisan, and Guilherme Ribeiro Romualdo. 2022. "The Implications of Connexin 43 Deficiency during the Early Stages of Chemically Induced Mouse Colon Carcinogenesis" Antioxidants 11, no. 12: 2368. https://doi.org/10.3390/antiox11122368