

Electroanalysis of Naringin at Electroactivated Pencil Graphite Electrode for the Assessment of Polyphenolics with Intermediate Antioxidant Power

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Instrumentation
2.2. Reagents and Solutions
2.3. Procedures
2.4. Real Samples Analysis
3. Results and Discussion
3.1. Establishing the Optimum Conditions for NG Voltammetric Analysis
3.1.1. The Influence of the Working Electrode Surface
3.1.2. The Influence of the pH and Nature of the Supporting Electrolyte
3.2. Voltammetric Behavior of NG at HB_PGE*
3.3. Quantitative Determination of NG at HB_PGE*
3.3.1. The Stability of NG Stock and Working Solutions
3.3.2. Linear Range, Limits of Detection and Quantification
3.3.3. Repeatability
3.4. Studies of Interferences on the Voltammetric Determination of NG at HB_PGE*
3.5. Analytical Application of Voltammetric NG Determination at HB_PGE*
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Csuti, A.; Sik, B.; Ajtony, Z. Measurement of naringin from citrus fruits by high-performance liquid chromatography—A review. Crit. Rev. Anal. Chem. 2022, 6, 1–14. [Google Scholar] [CrossRef]
- Alam, M.A.; Subhan, N.; Rahman, M.M.; Uddin, S.J.; Reza, H.M.; Sarker, S.D. Effect of citrus flavonoids, naringin and naringenin, on metabolic syndrome and their mechanisms of action. Adv. Nutr. 2014, 5, 404–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.K.; Das, S.; Sahu, P.P.; Mishra, P. Design and development of IDE sensor for naringin quantification in pomelo juice: An indicator of citrus maturity. Food Chem. 2022, 377, 131947. [Google Scholar] [CrossRef]
- Chen, R.; Qi, Q.-L.; Wang, M.-T.; Li, Q.-Y. Therapeutic potential of naringin: An overview. Pharm. Biol. 2016, 54, 3203–3210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Liu, S. Bioactivity of naringin and related mechanisms. Pharmazie 2021, 76, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.S.; Lima, K.C.M.S.; Botelho, C.N.; Pereira, N.M.; Fernandes, R.N.; Silva, G.G.; Damos, F.S.; Luz, R.C.S. Photoelectrochemical sensor for determination of naringin at low oxidation potential using a modified FTO electrode with cadmium sulfide and titanium dioxide sensitized with chloroprotoporphyrin IX iron(III). J. Solid State Electrochem. 2020, 24, 1715–1726. [Google Scholar] [CrossRef]
- Zhao, H.; Liu, M.; Liu, H.; Suo, R.; Lu, C. Naringin protects endothelial cells from apoptosis and inflammation by regulating the Hippo-YAP Pathway. Biosci. Rep. 2020, 40, BSR20193431. [Google Scholar] [CrossRef] [Green Version]
- Zhupanova, A.; Guss, E.; Ziyatdinova, G.; Budnikov, H. Simultaneous voltammetric determination of flavanones using an electrode based on functionalized single-walled carbon nanotubes and polyaluminon. Anal. Lett. 2020, 53, 2170–2189. [Google Scholar] [CrossRef]
- Ghanbari-Movahed, M.; Jackson, G.; Farzaei, M.H.; Bishayee, A. A systematic review of the preventive and therapeutic effects of naringin against human malignancies. Front. Pharmacol. 2021, 12, 639840. [Google Scholar] [CrossRef]
- Xu, C.; Huang, X.; Huang, Y.; Liu, X.; Wu, M.; Wang, J.; Duan, X. Naringin induces apoptosis of gastric carcinoma cells via blocking the PI3K/AKT pathway and activating pro-death autophagy. Mol. Med. Rep. 2021, 24, 772. [Google Scholar] [CrossRef]
- Deger, M.; Akdogan, N.; Izol, V.; Kaplan, H.M.; Pazarci, P.; Aridogan, I.A. Protective effect of naringin in rat model of renal ischemia reperfusion injury. Rev. Nefrol. Dial. Traspl. 2021, 41, 113–118. [Google Scholar]
- Amini, N.; Sarkaki, A.; Dianat, M.; Mard, S.A.; Ahangarpour, A.; Badavi, M. The renoprotective effects of naringin and trimetazidine on renal ischemia/reperfusion injury in rats through inhibition of apoptosis and downregulation of micoRNA-10a. Biomed. Pharmacoth. 2019, 112, 108568. [Google Scholar] [CrossRef] [PubMed]
- Ben-Azu, B.; Nwoke, E.E.; Aderibigbe, A.O.; Omogbiya, I.A.; Ajayi, A.M.; Olonode, E.T.; Umukoro, S.; Iwalewa, E.O. Possible neuroprotective mechanisms of action involved in the neurobehavioral property of naringin in mice. Biomed. Pharmacother. 2019, 109, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.E.; Alder, K.D.; Morris, M.T.; Munger, A.M.; Lee, I.; Cahill, S.V.; Kwon, H.-K.; Back, J.H.; Lee, F.Y. Re-appraising the potential of naringin for natural, novel orthopedic biotherapies. Ther. Adv. Musculoskel. Dis. 2020, 12, 1–21. [Google Scholar] [CrossRef]
- Mohanty, S.; Konkimalla, V.B.; Pal, A.; Sharma, T.; Si, S.C. Naringin as sustained delivery nanoparticles ameliorates the anti-inflammatory activity in a Freund’s complete adjuvant-induced arthritis model. ACS Omega 2021, 6, 28630–28641. [Google Scholar] [CrossRef]
- Kumari, S.D.; Chevala, N.T.; Jitta, S.R.; Kumar, L.; Verma, R.; Jose, J. Design and development of naringin-loaded proposomal gel for wound healing. J. Cosmet. Dermatol. 2022, 21, 5187–5202. [Google Scholar] [CrossRef]
- Chen, T.; Wu, H.; He, Y.; Pan, W.; Yan, Z.; Liao, Y.; Peng, W.; Gan, L.; Zhang, Y.; Su, W.; et al. Simultaneously quantitative analysis of naringin and its major human gut microbial metabolites naringenin and 3-(4′-hydroxyphenyl) propanoic acid via stable isotope deuterium-labeling coupled with RRLC-MS/MS method. Molecules 2019, 24, 4287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victor, M.M.; David, J.M.; Sakukuma, M.C.K.; França, E.L.; Nunes, A.V.J. A simple and efficient process for the extraction of naringin from grapefruit peel waste. Green Process. Synth. 2018, 7, 524–529. [Google Scholar] [CrossRef]
- Stabrauskiene, J.; Marksa, M.; Ivanauskas, L.; Bernatoniene, J. Optimization of naringin and naringenin extraction from Citrus × paradisi L. using hydrolysis and excipients as adsorbent. Pharmaceutics 2022, 14, 890. [Google Scholar] [CrossRef]
- Memon, A.F.; Solangi, A.R.; Memon, S.Q.; Mallah, A.; Memon, N.; Memon, A.A. Simultaneous determination of quercetin, rutin, naringin, and naringenin in different fruits by capillary zone electrophoresis. Food Anal. Methods 2017, 10, 83–91. [Google Scholar] [CrossRef]
- Ni, H.; Zhang, S.F.; Gao, Q.F.; Hu, Y.; Jiang, Z.D.; Chen, F. Development and evaluation of simultaneous quantification of naringin, prunin, naringenin, and limonin in citrus juice. Food Sci. Biotechnol. 2015, 24, 1239–1247. [Google Scholar] [CrossRef]
- Yao, Z.; Wu, S.; Zhang, H.; Feng, X.; Wang, Z.; Lin, M. Chiral determination of naringenin by ultra-performance liquid chromatography-tandem mass spectrometry and application in citrus peel and pulp. Front. Nutr. 2022, 9, 906859. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Guan, Y.; Ye, J. Determination of flavonoids and ascorbic acid in grapefruit peel and juice by capillary electrophoresis with electrochemical detection. Food Chem. 2007, 100, 1573–1579. [Google Scholar] [CrossRef]
- Ma, F.; Zhang, W.; Wang, J.; Zhang, L.; Chen, G. Fabrication of a carbon nanotube-polyurethane composite electrode by in situ polyaddition for use in amperometric detection in capillary electrophoresis. Microchim. Acta 2016, 183, 2579–2587. [Google Scholar] [CrossRef]
- Gupta, A.K.; Poonam Mishra, P.; Senapati, M.; Sahu, P.P. A novel electrochemical device for naringin quantification and removal from bitter variety of citrus fruits. J. Food Eng. 2021, 306, 110637. [Google Scholar] [CrossRef]
- Ziyatdinova, G.; Yakupova, E.; Ziganshina, E.; Budnikov, H. First order derivative voltammetry on the in situ surfactant modified electrode for naringin quantification. Electroanalysis 2019, 31, 2130–2137. [Google Scholar] [CrossRef]
- Ma, X.L.; Chen, R.Y.; Zheng, X.; Chen, X.; Chen, Z. Preparation and application of Naringin sensor based on molecularly imprinting technique. Chin. J. Anal. Chem. 2010, 38, 100–104. [Google Scholar] [CrossRef]
- Ziyatdinova, G.; Yakupova, E.; Guss, E.; Budnikov, H. The selective electrochemical sensing of naringin using electropolymerized ellagic acid film. J. Electrochem. Soc. 2020, 167, 107502. [Google Scholar] [CrossRef]
- Ensafi, A.A.; Karbalaei, S.; Heydari-Bafrooei, E.; Rezaei, B. Biosensing of naringin in marketed fruits and juices based on its interaction with DNA. J. Iran. Chem. Soc. 2016, 13, 19–27. [Google Scholar] [CrossRef]
- Reichart, E.; Obendorf, D. Determination of naringin in grapefruit juice by cathodic stripping differential pulse voltammetry at the hanging mercury drop electrode. Anal. Chim. Acta 1998, 360, 179–187. [Google Scholar] [CrossRef]
- Volikakis, G.J.; Efstathiou, C.E. Determination of rutin and other flavonoids by flow-injection/adsorptive stripping voltammetry using nujol-graphite and diphenylether-graphite paste electrodes. Talanta 2000, 51, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Zhang, S.; Li, Y.; Lin, Y. Excellent graphitic carbon nitride nanosheets-based photoelectrochemical platform motivated by Schottky barrier and LSPR effect and its sensing application. Analyst 2015, 140, 3514–3520. [Google Scholar] [CrossRef] [PubMed]
- Szczepaniak, O.; Ligaj, M.; Kobus-Cisowska, J.; Tichoniuk, M.; Dziedziński, M.; Przeor, M.; Szulc, P. The genoprotective role of naringin. Biomolecules 2020, 10, 700. [Google Scholar] [CrossRef] [PubMed]
- David, I.G.; Oancea, A.G.; Buleandra, M.; Popa, D.E.; Iorgulescu, E.E.; Ciobanu, A.M. Disposable pencil graphite electrode for diosmin voltammetric analysis. Micromachines 2021, 12, 351. [Google Scholar] [CrossRef] [PubMed]
- David, I.G.; Popa, D.E.; Buleandra, M. Pencil graphite electrodes: A versatile tool in electroanalysis. J. Anal. Methods Chem. 2017, 2017, 1905968. [Google Scholar] [CrossRef] [Green Version]
- David, I.G.; Buleandra, M.; Popa, D.E.; Cheregi, M.C.; David, V.; Iorgulescu, E.E.; Tartareanu, G.O. Recent developments in voltammetric analysis of pharmaceuticals using disposable pencil graphite electrodes. Processes 2022, 10, 472. [Google Scholar] [CrossRef]
- Temerk, Y.; Ibrahim, H.; Kotb, M.; Schuhmann, W. Renewable pencil electrodes for highly sensitive anodic stripping voltammetric determination of 3-hydroxyflavone and morin in bulk form and in biological fluids. Electroanalysis 2013, 25, 1381–1387. [Google Scholar] [CrossRef]
- Temerk, Y.; Ibrahim, H.; Schuhmann, W. Simultaneous anodic adsorptive stripping voltammetric determination of luteolin and 3-hydroxyflavone in biological fluids using renewable pencil graphite electrodes. Electroanalysis 2019, 31, 1095–1103. [Google Scholar] [CrossRef]
- Šafranko, S.; Stanković, A.; Asserghine, A.; Jakovljević, M.; Hajra, S.; Nundy, S.; Medvidović-Kosanović, M.; Jokić, S. Electroactivated disposable pencil graphite electrode—New, cost-effective, and sensitive electrochemical detection of bioflavonoid hesperidin. Electroanalysis 2020, 33, 1063–1071. [Google Scholar] [CrossRef]
- David, I.G.; Litescu, S.C.; Popa, D.E.; Buleandra, M.; Iordache, L.; Albu, C.; Alecu, A.; Penu, R.L. Voltammetric analysis of naringenin at a disposable pencil graphite electrode—Application to polyphenol content determination in citrus juice. Anal. Methods 2018, 10, 5763–5772. [Google Scholar] [CrossRef]
- David, I.G.; Numan, N.; Buleandră, M.; Popa, D.-E.; Lițescu, S.C.; Riga, S.; Ciobanu, A.M. Rapid voltammetric screening method for the assessment of bioflavonoid content using the disposable bare pencil graphite electrode. Chemosensors 2021, 9, 323. [Google Scholar] [CrossRef]
- Sariga, A.G.; Rajeev, R.; Thadathil, D.A.; Varghese, A. A Comprehensive Review on the Electrochemical Sensing of Flavonoids. Crit. Rev. Anal. Chem. 2022. [Google Scholar] [CrossRef]
- Blasco, A.J.; Rogerio, M.; González, M.; Escarpa, A. “Electrochemical Index” as a screening method to determine “total polyphenolics” in foods: A proposal. Anal. Chim. Acta 2005, 539, 237–244. [Google Scholar] [CrossRef]
- Karaboduk, K. Development of a voltammetric method for the determination of rapamycin in pharmaceutical samples at pretreated pencil graphite electrode. J. Chin. Chem. Soc. 2021, 68, 1722–1730. [Google Scholar] [CrossRef]
- Buleandră, M.; Popa, D.E.; David, I.G.; Ciucu, A.A. A simple and efficient cyclic square wave voltammetric method for simultaneous determination of epinephrine and norepinephrine using an activated pencil graphite electrode. Microchem. J. 2021, 160, 105621. [Google Scholar] [CrossRef]
- Koyun, O.; Gorduk, S.; Arvas, M.B.; Sahin, Y. Electrochemically treated pencil graphite electrodes prepared in one step for the electrochemical determination of paracetamol. Russ. J. Electrochem. 2018, 54, 796–808. [Google Scholar] [CrossRef]
- Gosser, D.K. Cyclic Voltammetry: Simulation and Analysis of Reaction Mechanisms; Wiley-VCH: New York, NY, USA, 1993. [Google Scholar]
- Laviron, E. General expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems. J. Electroanal. Chem. 1979, 101, 19–28. [Google Scholar] [CrossRef]
- Yunhua, W.; Xiaobo, J.; Shengshui, H. Studies on electrochemical oxidation of azithromycin and its interaction with bovine serum albumin. Bioelectrochem. 2004, 64, 91–97. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; Wiley: New York, NY, USA, 2001. [Google Scholar]
- Brodowska, K. Naringenin complexes with copper ions: Potentiometric studies. Biotechnol. Food Sci. 2013, 77, 45–53. [Google Scholar]
- Milicevic, A.; Novak Jovanovic, I. The relationship between the first oxidation potential and changes in electronic structures upon the electrochemical oxidation of flavonoids: Approach to O-glycosyl, galloyl and methoxy substituents. J. Mol. Liq. 2021, 335, 116223. [Google Scholar] [CrossRef]
- Miller, J.N.; Miller, J.C. Statistics and Chemometrics for Analytical Chemistry, 5th ed.; Pearson Prentice Hall: Edinburgh, UK, 2005. [Google Scholar]
- AOAC International. Guidelines for Standard Method Performance Requirements; Appendix F; AOAC International: Rockville, MD, USA, 2012; pp. 1–17. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electroactivation Technique | CV (−0.500 to 2.000 V; n = 10; v = 0.500 V/s) | E = ct. (2.000 V; 60 s) | None | |
|---|---|---|---|---|
| Medium | Ip (A) | |||
| BRB pH 2.21 | 2.42 × 10−6 | 4.47 × 10−6 | 4.70 × 10−6 | |
| ABS pH 4.00 | 4.78 × 10−6 | 3.75 × 10−6 | ||
| PBS pH 7.00 | 4.84 × 10−6 | 6.33 × 10−6 | ||
| M NaOH | 6.34 × 10−6 | 4.85 × 10−6 | ||
| Technique | Regression Equation | x/n |
|---|---|---|
| CV | Ep = −0.055 × pH + 1.160 (R2 = 0.9848) | 0.93 |
| DPV | Ep = −0.050 × pH + 1.046 (R2 = 0.9848) | 0.85 |
| Ip = f(v) | Ip = f(v1/2) | log Ip = f(log v) | Ep = f(log v) | Ep = f(v) |
|---|---|---|---|---|
| Ip = 1.00 ×10−4 × v + 2.00 × 10−6 R2 = 0.9838 | Ip = 7.00 × 10−5 × v1/2 − 8.00 × 10−6 R2 = 0.9899 | log Ip = 0.8345 × log v − 4.0614 R2 = 0.9790 | log Ep = 0.0938 × log v + 0.9204 R2 = 0.9806 | Ep = 0.7722 × v + 0.7544 R2 = 0.9768 |
| Technique | Electrode | Linear Range (mol/L) | Limit of Detection (mol/L) | Sample | Ref. |
|---|---|---|---|---|---|
| DPV | Poly-o-aminophenol MIP/graphite electrode | 6.00 × 10−5–1.40 × 10−4 | 1.60 × 10−5 | [27] | |
| 1-DLSV | SDS in situ MWCNTs-COOH/GCE | 7.50 × 10−7–1.00 × 10−4 | 1.40 × 10−7 | Grapefruit juice | [26] |
| DPV | Polyaluminon/f-SWCNTs/GCE | 1.00 × 10−7–2.50 × 10−5 | 2.00 × 10−8 | Orange and grapefruit juice | [8] |
| DPV | PolyEA/MWCNTs/GCE | 5.00 × 10−8–1.00 × 10−4 | 1.40 × 10−9 | Grapefruit juice | [28] |
| DPV | dsDNA/PDDA-MWCNTs/PGE | 1.00 × 10−7–1.00 × 10−3 | 1.72 × 10−8 | Orange, lemon, grapefruit juice | [29] |
| CSDPV | HMDE | 1.72 × 10−7–6.88 × 10−5 | 5.50 × 10−8 | Grapefruit juice | [30] |
| Flow injection/ AdSDPV | Nujol based CPE | - | d: 1.40 × 10−8 m.e: 9.00 × 10−9 | - | [31] |
| Diphenylether based CPE | d: 1.90 × 10−8 m.e: 1.70 × 10−8 | ||||
| PE (Amp) | CPPI-TiO2/CdS/FTO | 1.00 × 10−6–3.32 × 10−4 | 3.00 × 10−8 | Orange, limon, tangerine juice | [6] |
| PE (CV) | Au NRs/g-C3N4/Cys/GCE | 1.00 × 10−10–1.00 × 10−4 | 3.00 × 10−11 | [32] | |
| DPV | PGE | 1.40 × 10−6–2.00 × 10−5 2.00 × 10−5–1.40 × 10−4 | 6.02 × 10−7 | Grapefruit peel and fresh juice | This work |
| AdSDPV | 6.00 × 10−7–8.00 × 10−6 | 1.35 × 10−7 |
| Technique | AdSDPV | DPV | ||||
|---|---|---|---|---|---|---|
| NG concentration (mol/L) | 6.00 × 10−7 | 2.00 × 10−6 | 8.00 × 10−6 | 2.00 × 10−6 | 8.00 × 10−6 | 5.00 × 10−5 |
| RSD% | 9.32 | 4.52 | 2.12 | 7.62 | 5.47 | 2.13 |
| Sample | NG Concentration (mol/L) | NGN Concentration (mol/L) | ||
|---|---|---|---|---|
| DPV at HB_PGE* | HPLC-DAD-MS | HPLC-DAD-MS | ||
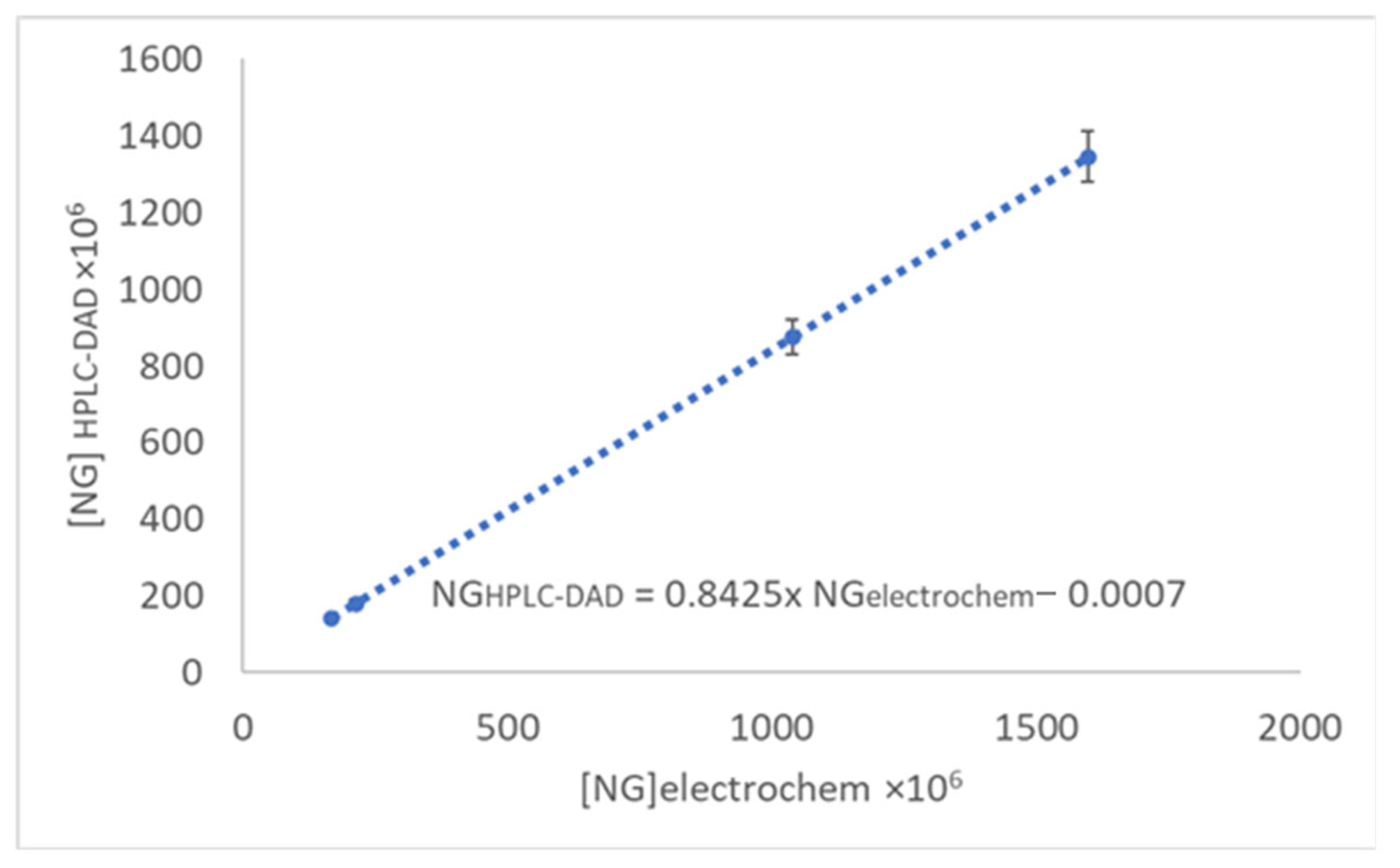
| Pink grapefruit | Peel | 1038.68 × 10−6 | 875.05 × 10−6 | 7.50 × 10−7 |
| Fresh juice | 214.05 × 10−6 | 180.33 × 10−6 | N.D. | |
| White grapefruit | Peel | 1597.18 × 10−6 | 1345.56 × 10−6 | 5.16 × 10−7 |
| Fresh juice | 166.73 × 10−6 | 140.46 × 10−6 | 4.41 × 10−7 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
David, I.G.; Litescu, S.C.; Moraru, R.; Albu, C.; Buleandra, M.; Popa, D.E.; Riga, S.; Ciobanu, A.M.; Noor, H. Electroanalysis of Naringin at Electroactivated Pencil Graphite Electrode for the Assessment of Polyphenolics with Intermediate Antioxidant Power. Antioxidants 2022, 11, 2306. https://doi.org/10.3390/antiox11122306
David IG, Litescu SC, Moraru R, Albu C, Buleandra M, Popa DE, Riga S, Ciobanu AM, Noor H. Electroanalysis of Naringin at Electroactivated Pencil Graphite Electrode for the Assessment of Polyphenolics with Intermediate Antioxidant Power. Antioxidants. 2022; 11(12):2306. https://doi.org/10.3390/antiox11122306
Chicago/Turabian StyleDavid, Iulia Gabriela, Simona Carmen Litescu, Raluca Moraru, Camelia Albu, Mihaela Buleandra, Dana Elena Popa, Sorin Riga, Adela Magdalena Ciobanu, and Hassan Noor. 2022. "Electroanalysis of Naringin at Electroactivated Pencil Graphite Electrode for the Assessment of Polyphenolics with Intermediate Antioxidant Power" Antioxidants 11, no. 12: 2306. https://doi.org/10.3390/antiox11122306