


Alcohol Withdrawal Is an Oxidative Stress Challenge for the Brain: Does It Pave the Way toward Severe Alcohol-Related Cognitive Impairment?
 , , , ,
, , , ,  , and
, and
Abstract
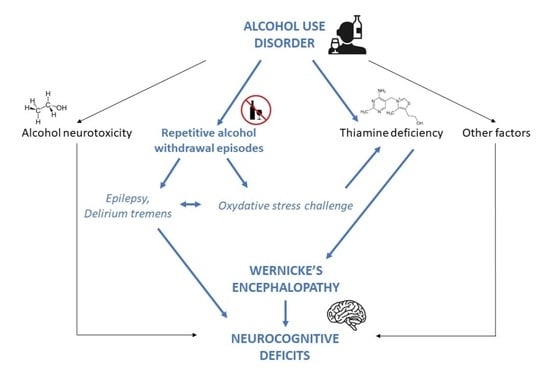
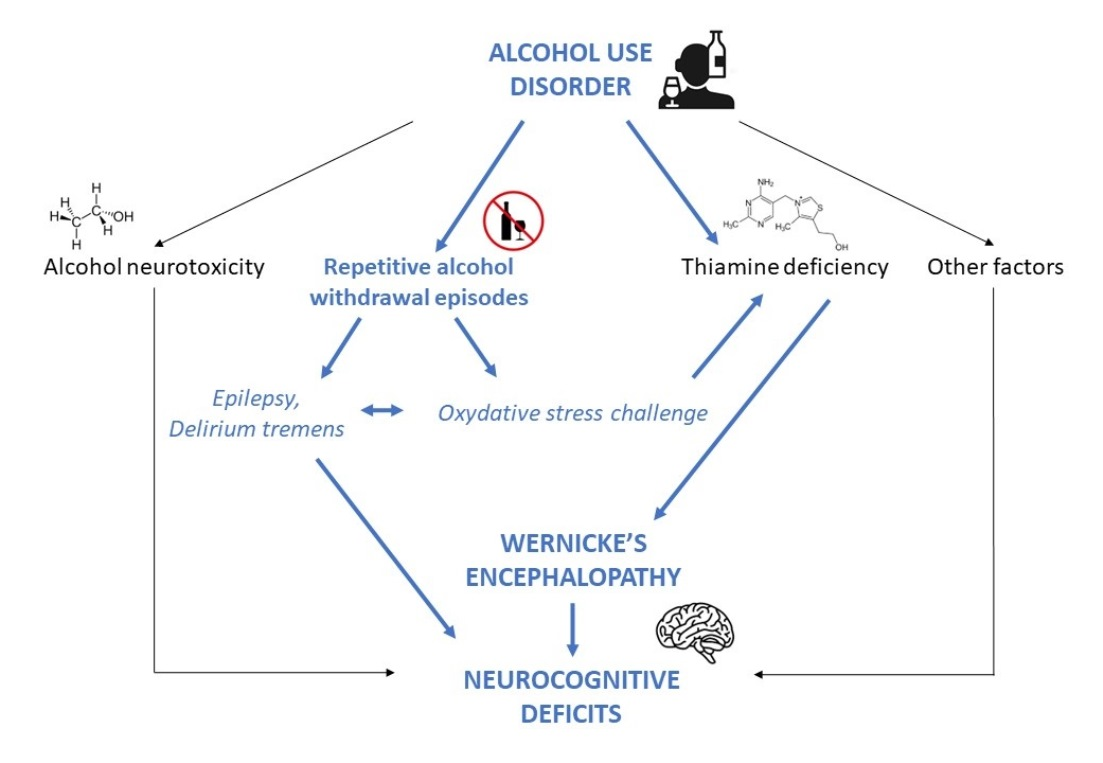
:
{kind=link}
{kind=link}
1. Introduction
1.1. Alcohol-Induced Brain Morbidity
1.2. Are All Heavy Alcohol Users at Risk of Brain Morbidity?
2. Alcohol Withdrawal: Definition
3. Alcohol Withdrawal as an Oxidative Stress Challenge for the Brain
3.1. Epilepsy, Delirium Tremens as AW Neurological Complications: The Role of Oxidative Stress
3.2. AW as a Vulnerability Period for Wernicke’s Encephalopathy: The Role of Oxidative Stress
3.2.1. Wernicke’s Encephalopathy Occurrence
3.2.2. Wernicke’s Encephalopathy Diagnosis
3.2.3. Systematic Thiamine Supplementation in AUD Patients
3.2.4. Wernicke’s Encephalopathy and Alcohol Withdrawal
3.2.5. Wernicke’s Encephalopathy and Oxidative Stress
4. What Makes the Alcohol Withdrawal Period at High Risk for Oxidative Brain Damage?
5. Alcohol Withdrawal and Severe Cognitive Impairments
6. Perspective
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Griswold, M.G.; Fullman, N.; Hawley, C.; Arian, N.; Zimsen, S.R.M.; Tymeson, H.D.; Venkateswaran, V.; Tapp, A.D.; Forouzanfar, M.H.; Salama, J.S.; et al. Alcohol use and burden for 195 countries and territories, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2018, 392, 1015–1035. [Google Scholar] [CrossRef] [Green Version]
- Sohi, I.; Franklin, A.; Chrystoja, B.; Wettlaufer, A.; Rehm, J.; Shield, K. The Global Impact of Alcohol Consumption on Premature Mortality and Health in 2016. Nutrients 2021, 13, 3145. [Google Scholar] [CrossRef] [PubMed]
- Guérin, S.; Laplanche, A.; Dunant, A.; Hill, C. Alcohol-attributable mortality in France. Eur. J. Public Health 2013, 23, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Shield, K.; Manthey, J.; Rylett, M.; Probst, C.; Wettlaufer, A.; Parry, C.D.H.; Rehm, J. National, regional, and global burdens of disease from 2000 to 2016 attributable to alcohol use: A comparative risk assessment study. Lancet Public Health 2020, 5, e51–e61. [Google Scholar] [CrossRef] [Green Version]
- Maillard, A.; Poussier, H.; Boudehent, C.; Lannuzel, C.; Vicente, A.; Vabret, F.; Cabe, N.; Pitel, A.-L. Short-term neuropsychological recovery in alcohol use disorder: A retrospective clinical study. Addict. Behav. 2020, 105, 106350. [Google Scholar] [CrossRef]
- Schwarzinger, M.; Pollock, B.G.; Hasan, O.S.M.; Dufouil, C.; Rehm, J. Contribution of alcohol use disorders to the burden of dementia in France 2008–13: A nationwide retrospective cohort study. Lancet Public Health 2018, 3, e124–e132. [Google Scholar] [CrossRef]
- Rehm, J.; Gmel, G. Alcohol consumption and total mortality/morbidity-definitions and methodological implications. Best Pract. Res. Clin. Gastroenterol. 2003, 17, 497–505. [Google Scholar] [CrossRef]
- Dawson, D.A. Drinking patterns among individuals with and without DSM-IV alcohol use disorders. J. Stud. Alcohol 2000, 61, 111–120. [Google Scholar] [CrossRef]
- John, U.; Rumpf, H.; Hanke, M.; Meyer, C. Severity of alcohol dependence and mortality after 20 years in an adult general population sample. Int. J. Methods Psychiatr. Res. 2022, 21, e1915. [Google Scholar] [CrossRef]
- Carvalho, A.F.; Heilig, M.; Perez, A.; Probst, C.; Rehm, J. Alcohol use disorders. Lancet 2019, 394, 781–792. [Google Scholar] [CrossRef]
- Schuckit, M.A.; Smith, T.L. Endorsement of specific alcohol use disorder criterion items changes with age in individuals with persistent alcohol use disorders in 2 generations of the San Diego Prospective Study. Alcohol. Clin. Exp. Res. 2021, 45, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Clergue-Duval, V.; Sivapalan, R.; Hispard, E.; Azuar, J.; Bellivier, F.; Bloch, V.; Vorspan, F.; Naccache, F.; Questel, F. BNP worsens 12 days after alcohol cessation while other cardiovascular risk biomarkers improve: An observational study. Alcohol 2021, 90, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Vigouroux, A.; Garret, C.; Lascarrou, J.-B.; Martin, M.; Miailhe, A.-F.; Lemarié, J.; Dupeyrat, J.; Zambon, O.; Seguin, A.; Reignier, J.; et al. Alcohol withdrawal syndrome in ICU patients: Clinical features, management, and outcome predictors. PLoS ONE 2021, 16, e0261443. [Google Scholar] [CrossRef]
- Chernyavsky, S.; Dharapak, P.; Hui, J.; Laskova, V.; Merrill, E.; Pillay, K.; Siau, E.; Rizk, D. Alcohol and the Hospitalized Patient. Med. Clin. North Am. 2020, 104, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Haber, P.S.; Riordan, B.C.; Winter, D.T.; Barrett, L.; Saunders, J.; Hides, L.; Gullo, M.; Manning, V.; Day, C.A.; Bonomo, Y.; et al. New Australian guidelines for the treatment of alcohol problems: An overview of recommendations. Med. J. Aust. 2021, 215, S3–S32. [Google Scholar] [CrossRef]
- Lindsay, D.L.; Freedman, K.; Jarvis, M.; Lincoln, P.; Williams, J.; Nelson, L.S.; Safarian, T. Executive Summary of the American Society of Addiction Medicine (ASAM) Clinical Practice Guideline on Alcohol Withdrawal Management. J. Addict. Med. 2020, 14, 376–392. [Google Scholar] [CrossRef]
- Rolland, B.; Paille, F.; Gillet, C.; Rigaud, A.; Moirand, R.; Dano, C.; Dematteis, M.; Mann, K.; Aubin, H.-J. Pharmacotherapy for Alcohol Dependence: The 2015 Recommendations of the French Alcohol Society, Issued in Partnership with the European Federation of Addiction Societies. CNS Neurosci. Ther. 2016, 22, 25–37. [Google Scholar] [CrossRef] [Green Version]
- Cushman, P.; Forbes, R.; Lemer, W.; Stewart, M. Alcohol Withdrawal Syndromes: Clinical Management with Lofexidine. Alcohol. Clin. Exp. Res. 1985, 9, 103–108. [Google Scholar] [CrossRef]
- Pruckner, N.; Baumgartner, J.; Hinterbuchinger, B.; Glahn, A.; Vyssoki, S.; Vyssoki, B. Thiamine Substitution in Alcohol Use Disorder: A Narrative Review of Medical Guidelines. Eur. Addict. Res. 2019, 25, 103–110. [Google Scholar] [CrossRef]
- McLean, C.; Tapsell, L.; Grafenauer, S.; McMahon, A. Systematic review of nutritional interventions for people admitted to hospital for alcohol withdrawal. Nutr. Diet. 2020, 77, 76–89. [Google Scholar] [CrossRef]
- Becker, H.C.; Mulholland, P.J. Neurochemical mechanisms of alcohol withdrawal. Handb. Clin. Neurol. 2014, 125, 133–156. [Google Scholar] [PubMed]
- Jung, M.E.; Metzger, D.B. Alcohol Withdrawal and Brain Injuries: Beyond Classical Mechanisms. Molecules 2010, 15, 4984–5011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brousse, G.; Arnaud, B.; Vorspan, F.; Richard, D.; Dissard, A.; Dubois, M.; Pic, D.; Geneste, J.; Xavier, L.; Authier, N.; et al. Alteration of Glutamate/GABA Balance During Acute Alcohol Withdrawal in Emergency Department: A Prospective Analysis. Alcohol Alcohol. 2012, 47, 501–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- N’Gouemo, P. Voltage-Sensitive Calcium Channels in the Brain: Relevance to Alcohol Intoxication and Withdrawal. Handb. Exp. Pharm. 2018, 248, 263–280. [Google Scholar]
- Huang, M.-C.; Chen, C.-H.; Peng, F.-C.; Tang, S.-H.; Chen, C.-C. Alterations in Oxidative Stress Status During Early Alcohol Withdrawal in Alcoholic Patients. J. Med. Assoc. 2009, 108, 560–569. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.E.; Yan, L.-J.; Forster, M.J.; Simpkins, J.W. Ethanol withdrawal provokes mitochondrial injury in an estrogen preventable manner. J. Bioenerg. Biomembr. 2008, 40, 35–44. [Google Scholar] [CrossRef]
- Lebourgeois, S.; González-Marín, M.C.; Jeanblanc, J.; Naassila, M.; Vilpoux, C. Effect of N-acetylcysteine on motivation, seeking and relapse to ethanol self-administration: NAC reduces ethanol seeking and relapse in rats. Addict. Biol. 2018, 23, 643–652. [Google Scholar] [CrossRef]
- Lebourgeois, S.; González-Marín, M.C.; Antol, J.; Naassila, M.; Vilpoux, C. Evaluation of N-acetylcysteine on ethanol self-administration in ethanol-dependent rats. Neuropharmacology 2019, 150, 112–120. [Google Scholar] [CrossRef]
- Quintanilla, M.E.; Ezquer, F.; Morales, P.; Ezquer, M.; Olivares, B.; Santapau, D.; Herrera-Marschitz, M.; Israel, Y. N-Acetylcysteine and Acetylsalicylic Acid Inhibit Alcohol Consumption by Different Mechanisms: Combined Protection. Front. Behav. Neurosci. 2020, 14, 122. [Google Scholar] [CrossRef]
- Parsons, A.L.M.; Bucknor, E.M.V.; Castroflorio, E.; Soares, T.R.; Oliver, P.L.; Rial, D. The Interconnected Mechanisms of Oxidative Stress and Neuroinflammation in Epilepsy. Antioxidants 2022, 11, 157. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, M.; Ko, D.G.; Choi, B.Y.; Suh, S.W. The Role of NADPH Oxidase in Neuronal Death and Neurogenesis after Acute Neurological Disorders. Antioxidants 2021, 10, 739. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.-C.; Chen, C.-C.; Pan, C.-H.; Chen, C.-H. Comparison of Oxidative DNA Damage Between Alcohol-Dependent Patients with and Without Delirium Tremens. Alcohol. Clin. Exp. Res. 2014, 38, 2523–2528. [Google Scholar] [CrossRef] [PubMed]
- Abdou, E.; Hazell, A.S. Thiamine Deficiency: An Update of Pathophysiologic Mechanisms and Future Therapeutic Considerations. Neurochem. Res. 2015, 40, 353–361. [Google Scholar] [CrossRef]
- Arts, N.J.M.; Walvoort, S.J.; Kessels, R.P. Korsakoff’s syndrome: A critical review. Neuropsychiatr. Dis. Treat. 2017, 13, 2875–2890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arts, N.J.M.; Pitel, A.-L.; Kessels, R.P.C. The contribution of mamillary body damage to Wernicke’s encephalopathy and Korsakoff’s syndrome. Handb. Clin. Neurol. 2021, 180, 455–475. [Google Scholar]
- Chandrakumar, A.; Bhardwaj, A.; W’t Jong, G.W. Review of thiamine deficiency disorders: Wernicke encephalopathy and Korsakoff psychosis. J. Basic. Clin. Physiol. Pharm. 2019, 30, 153–162. [Google Scholar] [CrossRef]
- Coulbault, L.; Ritz, L.; Vabret, F.; Lannuzel, C.; Boudehent, C.; Nowoczyn, M.; Beaunieux, H.; Pitel, A.L. Thiamine and phosphate esters concentrations in whole blood and serum of patients with alcohol use disorder: A relation with cognitive deficits. Nutr. Neurosci. 2021, 24, 530–541. [Google Scholar] [CrossRef]
- Ota, Y.; Capizzano, A.A.; Moritani, T.; Naganawa, S.; Kurokawa, R.; Srinivasan, A. Comprehensive review of Wernicke encephalopathy: Pathophysiology, clinical symptoms and imaging findings. Jpn. J. Radiol. 2020, 38, 809–820. [Google Scholar] [CrossRef]
- Thomson, A.D.; Marshall, E.J. The natural history and pathophysiology of Wernicke’s Encephalopathy and Korsakoff’s Psychosis. Alcohol Alcohol. 2006, 41, 151–158. [Google Scholar] [CrossRef] [Green Version]
- Thomson, A.D.; Marshall, E.J. The treatment of patients at risk of developing wernicke’s encephalopathy in the community. Alcohol Alcohol. 2006, 41, 159–167. [Google Scholar] [CrossRef]
- Smith, T.J.; Johnson, C.R.; Koshy, R.; Hess, S.Y.; Qureshi, U.A.; Mynak, M.L.; Fischer, P.R. Thiamine deficiency disorders: A clinical perspective. Ann. N. Y. Acad. Sci. 2021, 1498, 9–28. [Google Scholar] [CrossRef] [PubMed]
- Gomes, F.; Bergeron, G.; Bourassa, M.W.; Fischer, P.R. Thiamine deficiency unrelated to alcohol consumption in high-income countries: A literature review. Ann. N. Y. Acad. Sci. 2021, 1498, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Oudman, E.; Wijnia, J.W.; Oey, M.J.; van Dam, M.; Postma, A. Wernicke-Korsakoff syndrome despite no alcohol abuse: A summary of systematic reports. J. Neurol. Sci. 2021, 426, 117482. [Google Scholar] [CrossRef] [PubMed]
- Scalzo, S.J.; Bowden, S.C.; Ambrose, M.L.; Whelan, G.; Cook, M.J. Wernicke-Korsakoff syndrome not related to alcohol use: A systematic review. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1362–1368. [Google Scholar] [CrossRef] [Green Version]
- Wijnia, J.W.; Oudman, E.; van Gool, W.A.; Wierdsma, A.I.; Bresser, E.L.; Bakker, J.; van de Wiel, A.; Mulder, C.L. Severe Infections are Common in Thiamine Deficiency and May be Related to Cognitive Outcomes: A Cohort Study of 68 Patients With Wernicke-Korsakoff Syndrome. Psychosomatics 2016, 57, 624–633. [Google Scholar] [CrossRef]
- Caine, D.; Halliday, G.M.; Kril, J.J.; Harper, C.G. Operational criteria for the classification of chronic alcoholics: Identification of Wernicke’s encephalopathy. J. Neurol. Neurosurg. Psychiatry 1997, 62, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Société Française d’Alcoologie. Mésusage de l’alcool: Dépistage, diagnostic et traitement. Recommandation de bonne pratique. Alcoologie Addictologie 2015, 37, 5–84. [Google Scholar]
- Whitfield, K.C.; Bourassa, M.W.; Adamolekun, B.; Bergeron, G.; Bettendorff, L.; Brown, K.H.; Cox, L.; Fattal-Valevski, A.; Fischer, P.R.; Frank, E.L.; et al. Thiamine deficiency disorders: Diagnosis, prevalence, and a roadmap for global control programs. Ann. N. Y. Acad. Sci. 2018, 1430, 3–43. [Google Scholar] [CrossRef]
- Jones, K.S.; Parkington, D.A.; Cox, L.J.; Koulman, A. Erythrocyte transketolase activity coefficient (ETKAC) assay protocol for the assessment of thiamine status. Ann. N. Y. Acad. Sci. 2021, 1498, 77–84. [Google Scholar] [CrossRef]
- Ganapathy, V.; Smith, S.B.; Prasad, P.D. SLC19: The folate/thiamine transporter family. Pflügers Arch. 2004, 447, 641–646. [Google Scholar] [CrossRef]
- Lockman, P.R.; McAfee, J.H.; Geldenhuys, W.J.; Allen, D.D. Cation Transport Specificity at the Blood-Brain Barrier. Neurochem. Res. 2004, 29, 2245–2250. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.D.; Guerrini, I.; Marshall, E.J. The Evolution and Treatment of Korsakoff’s Syndrome: Out of Sight, Out of Mind? Neuropsychol. Rev. 2012, 22, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvin, R.; Bråthen, G.; Ivashynka, A.; Hillbom, M.; Tanasescu, R.; Leone, M.A. EFNS guidelines for diagnosis, therapy and prevention of Wernicke encephalopathy: EFNS guidelines for Wernicke encephalopathy. Eur. J. Neurol. 2010, 17, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- Vetreno, R.P.; Ramos, R.L.; Anzalone, S.; Savage, L.M. Brain and behavioral pathology in an animal model of Wernicke’s encephalopathy and Wernicke-Korsakoff Syndrome. Brain Res. 2012, 1436, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Vedder, L.C.; Hall, J.M.; Jabrouin, K.R.; Savage, L.M. Interactions Between Chronic Ethanol Consumption and Thiamine Deficiency on Neural Plasticity, Spatial Memory, and Cognitive Flexibility. Alcohol. Clin. Exp. Res. 2015, 39, 2143–2153. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, R.M.; Langlais, P.J. An examination of the synergistic interaction of ethanol and thiamine deficiency in the development of neurological signs and long-term cognitive and memory impairments. Alcohol. Clin. Exp. Res. 2000, 24, 622–634. [Google Scholar] [CrossRef]
- Homewood, J.; Bond, N.W.; Mackenzie, A. The effects of single and repeated episodes of thiamin deficiency on memory in alcohol-consuming rats. Alcohol 1997, 14, 81–91. [Google Scholar] [CrossRef]
- Palencia, G.; Teixeira, F.; Ortiz, A.; Perez, R.; Rios, C.; Sotelo, J. Detrimental effects of malnutrition on the damage induced by alcoholism: A study of animal models that simulate chronic alcoholism and malnutrition of large human groups. J. Stud. Alcohol 1994, 55, 113–120. [Google Scholar] [CrossRef]
- Hazell, A.S. Astrocytes are a major target in thiamine deficiency and Wernicke’s encephalopathy. Neurochem. Int. 2009, 55, 129–135. [Google Scholar] [CrossRef]
- Hazell, A.S.; Sheedy, D.; Oanea, R.; Aghourian, M.; Sun, S.; Jung, J.Y.; Wang, D.; Wang, C. Loss of astrocytic glutamate transporters in Wernicke encephalopathy. Glia 2010, 58, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Jhala, S.S.; Hazell, A.S. Modeling neurodegenerative disease pathophysiology in thiamine deficiency: Consequences of impaired oxidative metabolism. Neurochem. Int. 2011, 58, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, F.; Pereira, S.; Pires, R.; Ferraz, V.; Romanosilva, M.; Oliveirasilva, I.; Ribeiro, A. Thiamine deficiency decreases glutamate uptake in the prefrontal cortex and impairs spatial memory performance in a water maze test. Pharmacol. Biochem. Behav. 2006, 83, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Clergue-Duval, V.; Azuar, J.; Fonsart, J.; Delage, C.; Rollet, D.; Amami, J.; Frapsauce, A.; Gautron, M.-A.; Hispard, E.; Bellivier, F.; et al. Ascorbic Acid Deficiency Prevalence and Associated Cognitive Impairment in Alcohol Detoxification Inpatients: A Pilot Study. Antioxidants 2021, 10, 1892. [Google Scholar] [CrossRef] [PubMed]
- Gautron, M.-A.; Questel, F.; Lejoyeux, M.; Bellivier, F.; Vorspan, F. Nutritional Status During Inpatient Alcohol Detoxification. Alcohol Alcohol. 2018, 53, 64–70. [Google Scholar] [CrossRef]
- Girre, C.; Hispard, E.; Therond, P.; Guedj, S.; Bourdon, R.; Dally, S. Effect of Abstinence from Alcohol on the Depression of Glutathione Peroxidase Activity and Selenium and Vitamin E Levels in Chronic Alcoholic Patients. Alcohol. Clin. Exp. Res. 1990, 14, 909–912. [Google Scholar] [CrossRef]
- O’Brien, N.L.; Quadri, G.; Lightley, I.; Sharp, S.I.; Guerrini, I.; Smith, I.; Heydtmann, M.; Morgan, M.Y.; Thomson, A.D.; Bass, N.J.; et al. SLC19A1 Genetic Variation Leads to Altered Thiamine Diphosphate Transport: Implications for the Risk of Developing Wernicke-Korsakoff’s Syndrome. Alcohol Alcohol. 2022, 57, 581–588. [Google Scholar] [CrossRef]
- Airagnes, G.; Ducoutumany, G.; Laffy-Beaufils, B.; Le Faou, A.-L.; Limosin, F. Alcohol withdrawal syndrome management: Is there anything new? Rev. Med. Interne 2019, 40, 373–379. [Google Scholar] [CrossRef]
- Maguire, D.; Burns, A.; Talwar, D.; Catchpole, A.; Stefanowicz, F.; Ross, D.P.; Galloway, P.; Ireland, A.; Robson, G.; Adamson, M.; et al. Randomised trial of intravenous thiamine and/or magnesium sulphate administration on erythrocyte transketolase activity, lactate concentrations and alcohol withdrawal scores. Sci. Rep. 2022, 12, 6941. [Google Scholar] [CrossRef]
- Sarai, M.; Tejani, A.M.; Chan, A.H.W.; Kuo, I.F.; Li, J. Magnesium for alcohol withdrawal. Cochrane Database Syst. Rev. 2013, CD008358. [Google Scholar] [CrossRef]
- Mahajan, V.R.; Elvig, S.K.; Vendruscolo, L.F.; Koob, G.F.; Darcey, V.L.; King, M.T.; Kranzler, H.R.; Volkow, N.D.; Wiers, C.E. Nutritional Ketosis as a Potential Treatment for Alcohol Use Disorder. Front. Psychiatry 2021, 12, 781668. [Google Scholar] [CrossRef]
- Shi, Z.; Xie, Y.; Ren, H.; He, B.; Wang, M.; Wan, J.; Yuan, T.; Yao, X.; Su, H. Fish oil treatment reduces chronic alcohol exposure induced synaptic changes. Addict. Biol. 2019, 24, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Duka, T.; Townshend, J.M.; Collier, K.; Stephens, D.N. Kindling of withdrawal: A study of craving and anxiety after multiple detoxifications in alcoholic inpatients. Alcohol. Clin. Exp. Res. 2002, 26, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Duka, T.; Townshend, J.M.; Collier, K.; Stephens, D.N. Impairment in Cognitive Functions After Multiple Detoxifications in Alcoholic Inpatients. Alcohol. Clin. Exp. Res. 2003, 27, 1563–1572. [Google Scholar] [CrossRef] [PubMed]
- Laniepce, A.; Cabé, N.; André, C.; Bertran, F.; Boudehent, C.; Lahbairi, N.; Maillard, A.; Mary, A.; Segobin, S.; Vabret, F.; et al. The effect of alcohol withdrawal syndrome severity on sleep, brain and cognition. Brain Commun. 2020, 2, fcaa123. [Google Scholar] [CrossRef]
- Frey, B.N.; Zunta-Soares, G.B.; Caetano, S.C.; Nicoletti, M.A.; Hatch, J.P.; Brambilla, P.; Mallinger, A.G.; Soares, J.C. Illness duration and total brain gray matter in bipolar disorder: Evidence for neurodegeneration? Eur. Neuropsychopharmacol. 2008, 18, 717–722. [Google Scholar] [CrossRef]
- Kim, H.K.; Nunes, P.V.; Oliveira, K.C.; Young, L.T.; Lafer, B. Neuropathological relationship between major depression and dementia: A hypothetical model and review. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 67, 51–57. [Google Scholar] [CrossRef]
- Meyer, J.H. Neuroprogression and Immune Activation in Major Depressive Disorder. Mod. Trends Pharm. 2017, 31, 27–36. [Google Scholar]
- Serafini, G.; Pardini, M.; Monacelli, F.; Orso, B.; Girtler, N.; Brugnolo, A.; Amore, M.; Nobili, F.; Disease Management Team on Dementia of The Irccs Ospedale Policlinico San Martino. Neuroprogression as an Illness Trajectory in Bipolar Disorder: A Selective Review of the Current Literature. Brain Sci. 2021, 11, 276. [Google Scholar] [CrossRef]
- Hugon, J.; Hourregue, C.; Cognat, E.; Lilamand, M.; Porte, B.; Mouton-Liger, F.; Dumurgier, J.; Paquet, C. Chronic traumatic encephalopathy. Neurochirurgie 2021, 67, 290–294. [Google Scholar] [CrossRef]
- McKee, A.C.; Cantu, R.C.; Nowinski, C.J.; Hedley-Whyte, E.T.; Gavett, B.E.; Budson, A.E.; Santini, V.E.; Lee, H.-S.; Kubilus, C.A.; Stern, R.A. Chronic traumatic encephalopathy in athletes: Progressive tauopathy after repetitive head injury. J. Neuropathol. Exp. Neurol. 2009, 68, 709–735. [Google Scholar] [CrossRef]
- McKee, A.C.; Stern, R.A.; Nowinski, C.J.; Stein, T.D.; Alvarez, V.E.; Daneshvar, D.H.; Lee, H.-S.; Wojtowicz, S.M.; Hall, G.; Baugh, C.M.; et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013, 136, 43–64. [Google Scholar] [CrossRef] [PubMed]
- Azuar, J.; Bouaziz-Amar, E.; Cognat, E.; Dumurgier, J.; Clergue-Duval, V.; Barré, T.; Amami, J.; Hispard, E.; Bellivier, F.; Paquet, C.; et al. Cerebrospinal Fluid Biomarkers in Patients with Alcohol Use Disorder and Persistent Cognitive Impairment. Alcohol. Clin. Exp. Res. 2021, 45, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Clergue-Duval, V.; Vrillon, A.; Jeanblanc, J.; Questel, F.; Azuar, J.; Fouquet, G.; Mouton-Liger, F.; Rollet, D.; Hispard, E.; Bouaziz-Amar, E.; et al. Plasma tau, NfL, GFAP and UCHL1 as candidate biomarkers of alcohol withdrawal-associated brain damage: A pilot study. Addict. Biol. 2022, 27, e13232. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clergue-Duval, V.; Coulbault, L.; Questel, F.; Cabé, N.; Laniepce, A.; Delage, C.; Boudehent, C.; Bloch, V.; Segobin, S.; Naassila, M.; et al. Alcohol Withdrawal Is an Oxidative Stress Challenge for the Brain: Does It Pave the Way toward Severe Alcohol-Related Cognitive Impairment? Antioxidants 2022, 11, 2078. https://doi.org/10.3390/antiox11102078
Clergue-Duval V, Coulbault L, Questel F, Cabé N, Laniepce A, Delage C, Boudehent C, Bloch V, Segobin S, Naassila M, et al. Alcohol Withdrawal Is an Oxidative Stress Challenge for the Brain: Does It Pave the Way toward Severe Alcohol-Related Cognitive Impairment? Antioxidants. 2022; 11(10):2078. https://doi.org/10.3390/antiox11102078
Chicago/Turabian StyleClergue-Duval, Virgile, Laurent Coulbault, Frank Questel, Nicolas Cabé, Alice Laniepce, Clément Delage, Céline Boudehent, Vanessa Bloch, Shailendra Segobin, Mickael Naassila, and et al. 2022. "Alcohol Withdrawal Is an Oxidative Stress Challenge for the Brain: Does It Pave the Way toward Severe Alcohol-Related Cognitive Impairment?" Antioxidants 11, no. 10: 2078. https://doi.org/10.3390/antiox11102078