
NOX as a Therapeutic Target in Liver Disease

Abstract
:1. Introduction
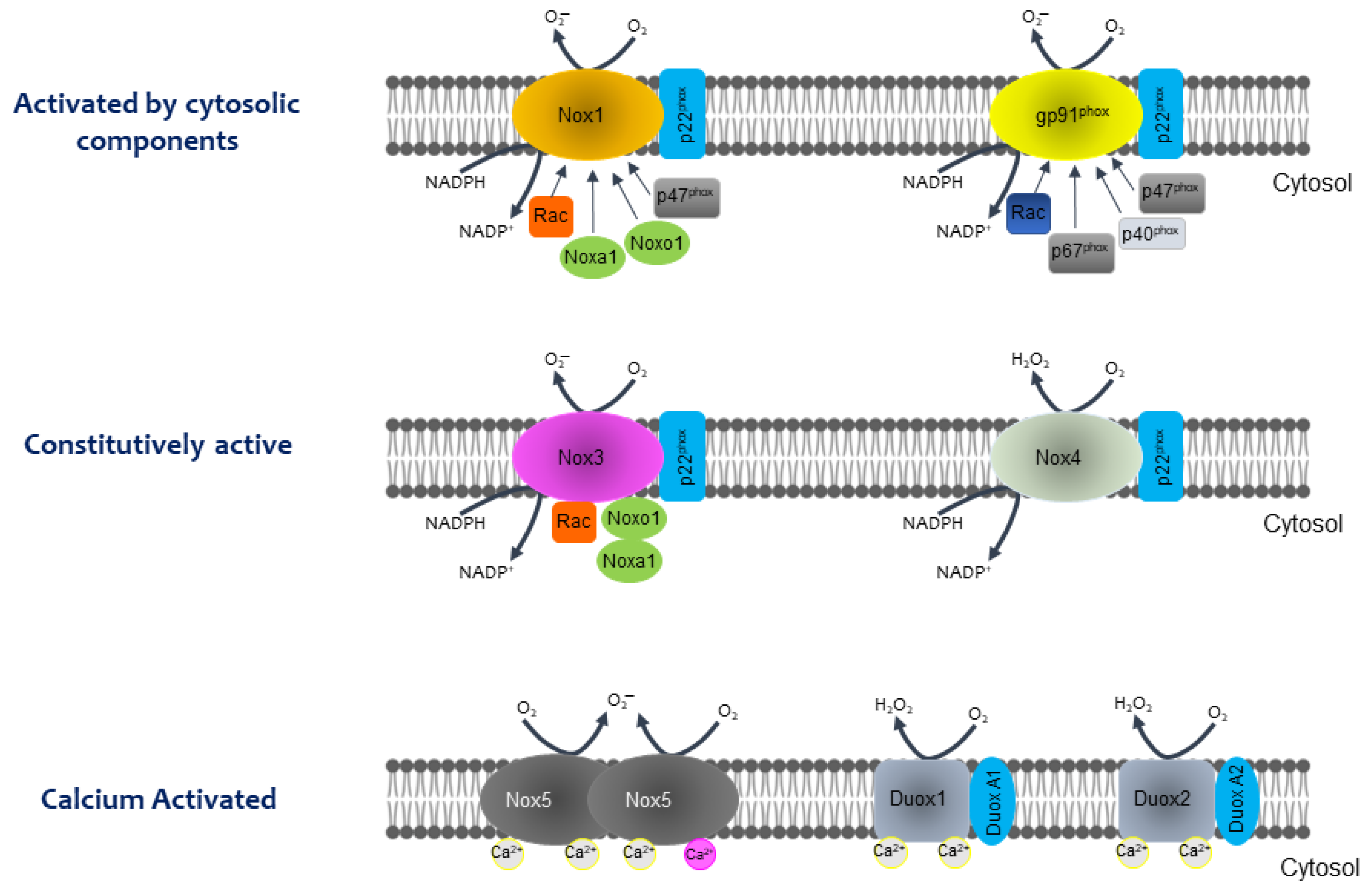
2. The NOX Family: From the Respiratory Burst to the Regulation of Metabolic Pathways
- They are dependent on NADPH;
- They are only found in membrane systems (plasma, mitochondrial, etc.);
- NOX consists of a catalytic subunit (gp96phox-like) linked to another subunit (p22phox), in most cases, and some subunits that regulate the activity of this enzyme system (p47phox, p67phox, p40phox, and Rac). The catalytic subunit has six transmembrane domains (seven for DUOX1 and 2), four hemes in transmembrane domains three and five, an NADPH-binding domain, and a FAD-binding domain at the C-terminus in the cytosolic region.
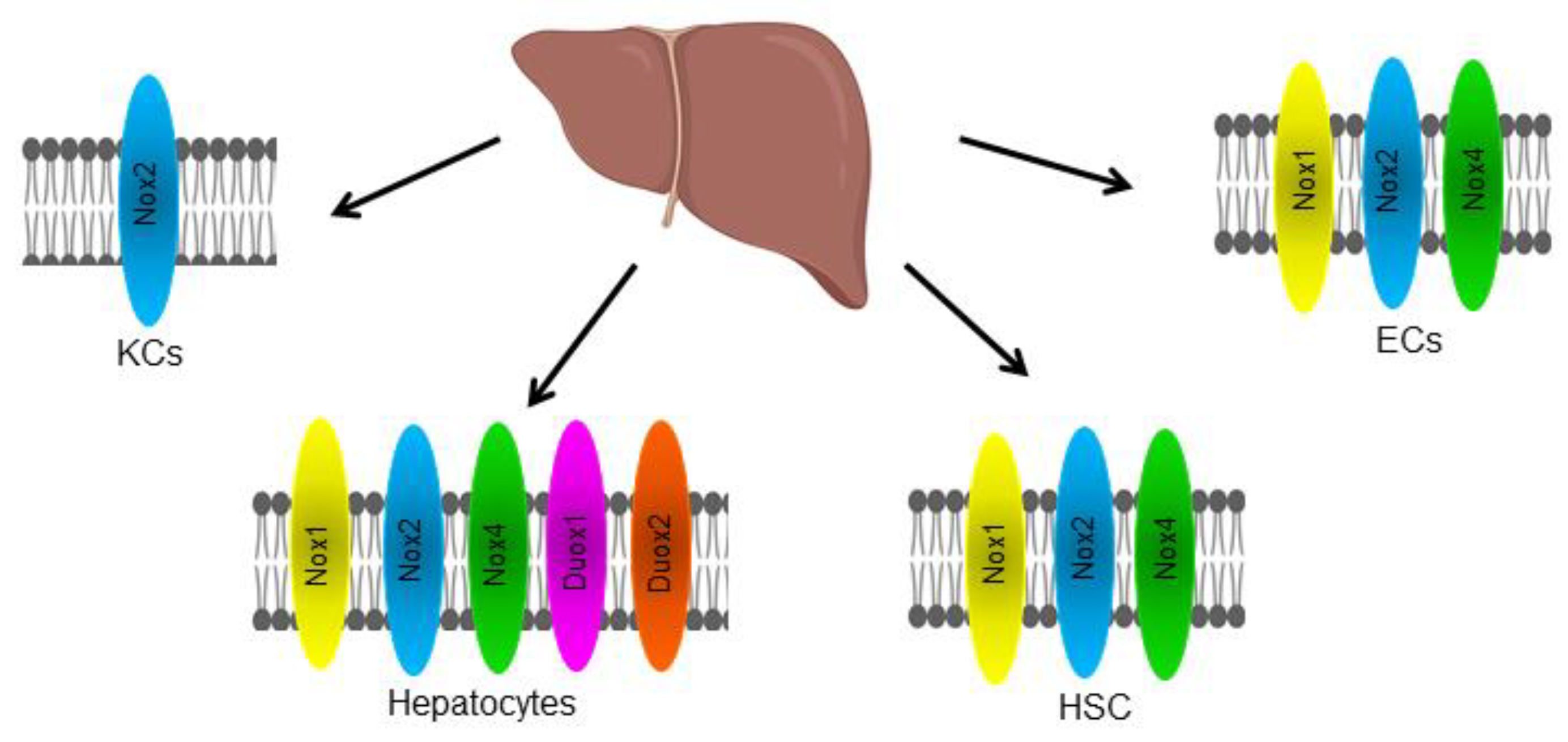
3. Cellular Distribution of NOX in the Liver
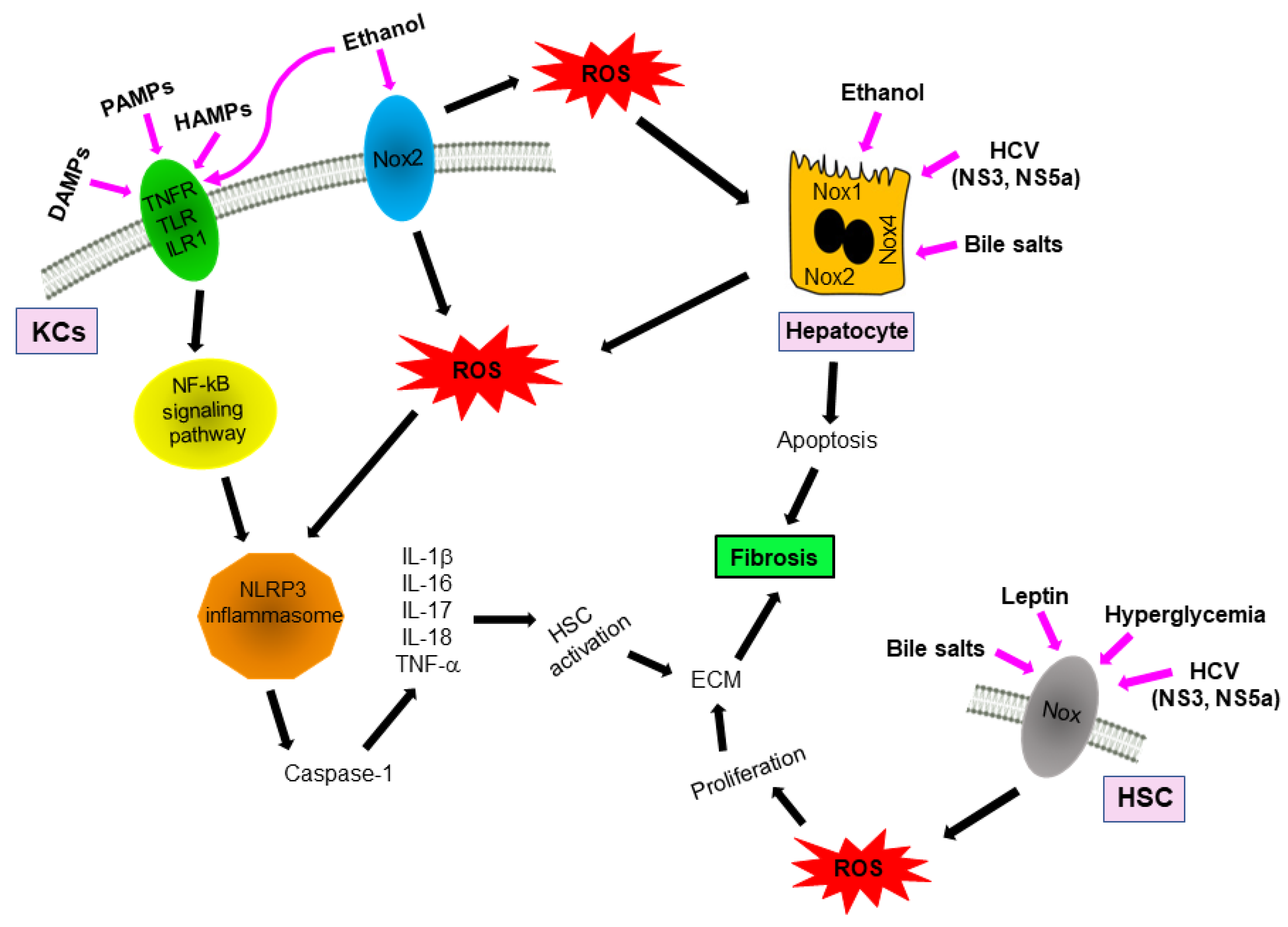
4. NOXs and Inflammasomes Activation
5. NOXs and Fibrosis
6. Alcohol and Non-Alcohol Associated Steatohepatitis
7. Hepatitis C Virus (HCV) Induced Hepatocellular Damage
8. Hydrophobic Bile Salts-Induced Liver Injury
9. Role of ROS in Acute and Chronic Liver Damage
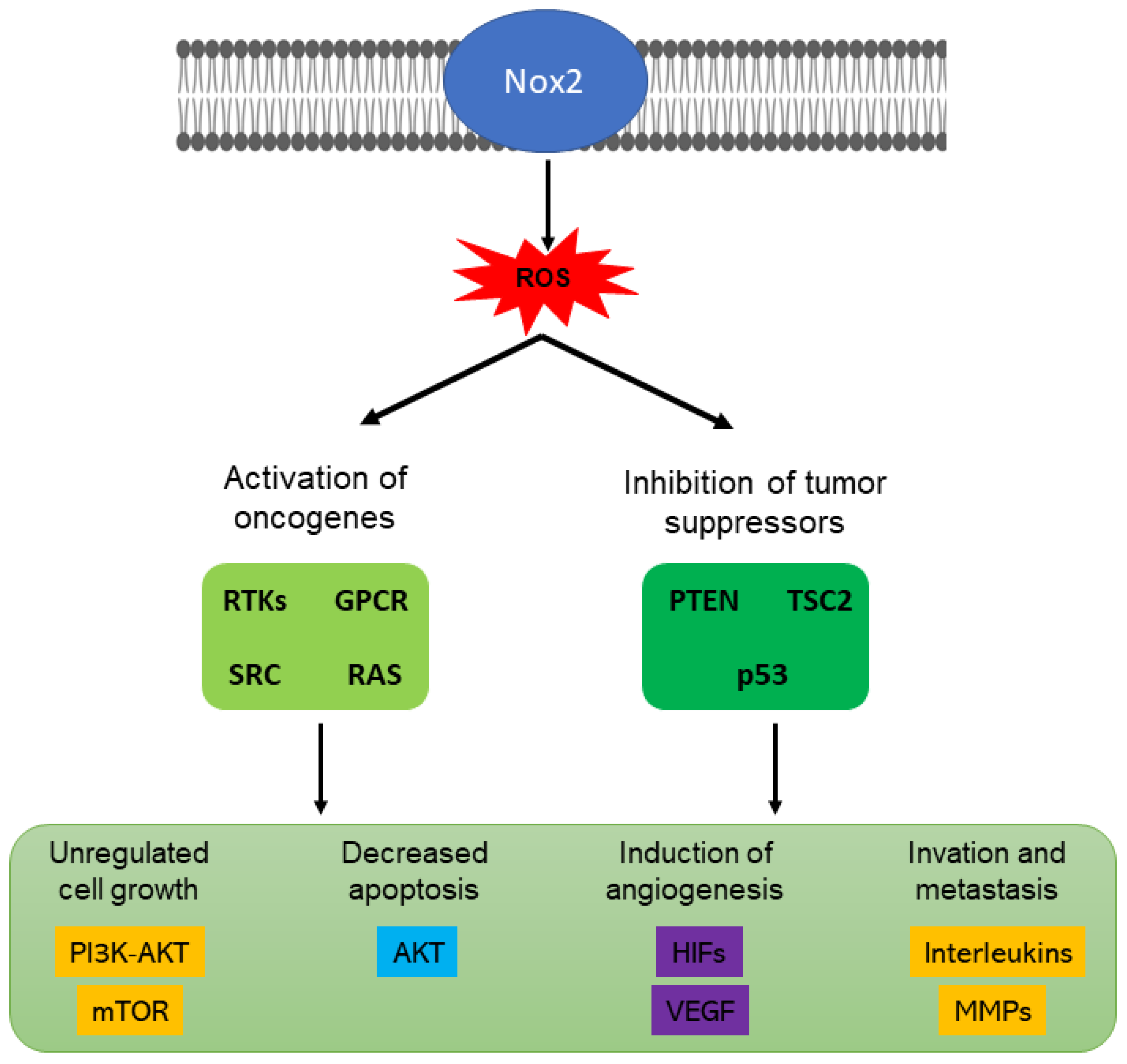
10. NOXs and Cancer
10.1. NOX1
10.2. NOX2
10.3. NOX4
10.4. DUOX1
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ROS | reactive oxygen species; |
| HCC | hepatic carcinogenesis; |
| HBV | hepatitis B virus; |
| HCV | hepatitis C virus; |
| ALD | alcohol-related liver disease; |
| NAFLD | nonalcoholic fatty liver disease; |
| NADPH | nicotinamide adenine dinucleotide phosphate hydrogen; |
| KC | Kupffer cells; |
| HSCs | hepatic stellate cells; |
| ECs | endothelial cells; |
| PRR | pattern recognition receptor; |
| DAMPs | damage-associated molecular patterns; |
| PAMPs | pathogen-associated molecular patterns; |
| HAMPs | homeostasis-altering molecular processes; |
| NOD | nucleotide-binding oligomerization domain; |
| NLRC | NOD-like receptor CARD domain containing; |
| NLRP | NOD-like receptor Pyrin domain containing; |
| TNFR | tumor necrosis factor receptor; |
| TLR | Toll-like receptors; |
| ECM | extracellular matrix; |
| TGF-β1 | transforming growth factor β1; |
| αSMA | α-smooth muscle actin; |
| BDL | bile duct ligation; |
| NASH | non-alcohol associated steatohepatitis; |
| HIF-1α | Hypoxia Inducible Factor 1α; |
| ET-1 | endothelin-1; |
| LSEC | liver sinusoidal endothelial cells; |
| DAAs | direct-acting antiviral agents; |
| HCC | hepatocellular carcinoma; |
| SVR | sustained virologic response; |
| FXR or NR1H4 | farnesoid receptor; |
| PKCf | Protein kinase C f; |
| ERK | Extracellular signal-regulated kinase; |
| VSMCs | Vascular Smooth Muscle Cells; |
| EGFR | epidermal growth factor receptor; |
| VEGF | vascular endothelial growth factor; |
| VEGFR2 | vascular endothelial growth factor receptor 2; |
| RTK | receptor tyrosine kinase; |
| GPCR | G protein-coupled receptors; |
| SRC | protooncogene tyrosine-protein kinase; |
| RAS | serine-threonine kinase; |
| PTEN | lipid tyrosine phosphatase; |
| PI3K | phosphoinositol 3-kinase; |
| mTOR | target of rapamycin; |
| HIF | hypoxia-inducible factors; |
| VEGF | vascular endothelial growth factor; |
| MMPs | matrix metalloproteinases; |
| HIFs | hypoxia-inducible transcription factors; |
| AMPK | AMP-activated protein kinase; |
| TSG | tumor-suppressor gene. |
References
- Moon, A.M.; Singal, A.G.; Tapper, E.B. Contemporary Epidemiology of Chronic Liver Disease and Cirrhosis. Clin. Gastroenterol. Hepatol. 2020, 18, 2650–2666. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Valle, V.; Chavez-Tapia, N.C.; Uribe, M.; Mendez-Sanchez, N. Role of oxidative stress and molecular changes in liver fibrosis: A review. Curr. Med. Chem. 2012, 19, 4850–4860. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef] [PubMed]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid Med. Cell Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luangmonkong, T.; Suriguga, S.; Mutsaers, H.A.M.; Groothuis, G.M.M.; Olinga, P.; Boersema, M. Targeting Oxidative Stress for the Treatment of Liver Fibrosis. Rev. Physiol. Biochem. Pharm. 2018, 175, 71–102. [Google Scholar] [CrossRef]
- Burton, R.H. Superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Superoxide Hydrog. Peroxide Relat. Mamm. Cell Prolif. 1995, 189, 775–794. [Google Scholar] [CrossRef]
- Matsuzaki, K. Modulation of TGF-beta signaling during progression of chronic liver diseases. Front. Biosci. 2009, 14, 2923–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, A.; Alvarez, A.M.; Benito, M.; Fabregat, I. Apoptosis induced by transforming growth factor-beta in fetal hepatocyte primary cultures: Involvement of reactive oxygen intermediates. J. Biol. Chem. 1996, 271, 7416–7422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedard, K.; Lardy, B.; Krause, K.H. NOX family NADPH oxidases: Not just in mammals. Biochimie 2007, 89, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Weissmann, N.; Schroder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free. Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Herranz-Iturbide, M.; Penuelas-Haro, I.; Espinosa-Sotelo, R.; Bertran, E.; Fabregat, I. The TGF-beta/NADPH Oxidases Axis in the Regulation of Liver Cell Biology in Health and Disease. Cells 2021, 10, 2312. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.C. The phagocyte respiratory burst: Historical perspectives and recent advances. Immunol. Lett. 2017, 192, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Vilchis-Landeros, M.M.; Matuz-Mares, D.; Vázquez-Meza, H. Regulation of Metabolic Processes by HydrogenPeroxide Generated by NADPH Oxidases. Processes 2020, 8, 1424–1440. [Google Scholar] [CrossRef]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masamune, A.; Watanabe, T.; Kikuta, K.; Satoh, K.; Shimosegawa, T. NADPH oxidase plays a crucial role in the activation of pancreatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G99–G108. [Google Scholar] [CrossRef]
- Bondi, C.D.; Manickam, N.; Lee, D.Y.; Block, K.; Gorin, Y.; Abboud, H.E.; Barnes, J.L. NAD(P)H oxidase mediates TGF-beta1-induced activation of kidney myofibroblasts. J. Am. Soc. Nephrol. 2010, 21, 93–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crosas-Molist, E.; Fabregat, I. Role of NADPH oxidases in the redox biology of liver fibrosis. Redox Biol. 2015, 6, 106–111. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Ruiz, I.; Blanes Ruiz, N.; Rada, P.; Pardo, V.; Ruiz, L.; Blas-Garcia, A.; Valdecantos, M.P.; Grau Sanz, M.; Solis Herruzo, J.A.; Valverde, A.M. Protein tyrosine phosphatase 1b deficiency protects against hepatic fibrosis by modulating nadph oxidases. Redox. Biol. 2019, 26, 101263. [Google Scholar] [CrossRef] [PubMed]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S.; Sorescu, D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef]
- Angeloni, C.; Prata, C.; Dalla Sega, F.V.; Piperno, R.; Hrelia, S. Traumatic brain injury and NADPH oxidase: A deep relationship. Oxid. Med. Cell Longev. 2015, 2015, 370312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, D.; Turner, M.; Munkonda, M.N.; Touyz, R.M. Endothelial Microparticle-Derived Reactive Oxygen Species: Role in Endothelial Signaling and Vascular Function. Oxid. Med. Cell Longev. 2016, 2016, 5047954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q. Human Three-Dimensional Hepatic Models: Cell Type Variety and Corresponding Applications. Front. Bioeng. Biotechnol. 2021, 9, 730008. [Google Scholar] [CrossRef] [PubMed]
- Manco, R.; Itzkovitz, S. Liver zonation. J. Hepatol. 2021, 74, 466–468. [Google Scholar] [CrossRef] [PubMed]
- Baumann, K. Endothelial cell diversity in the liver. Nat. Rev. Mol. Cell Biol. 2022, 23, 305. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Yang, Y.; Lai, S.; Jeong, J.; Jung, Y.; McConnell, M.; Utsumi, T.; Iwakiri, Y. Single-Cell Transcriptomics Reveals Zon Specific Alterations of Liver Sinusoidal Endothelial Cells in Cirrhosis. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 1139–1161. [Google Scholar] [CrossRef] [PubMed]
- Nuciforo, S.; Heim, M.H. Organoids to model liver disease. JHEP Rep. 2021, 3, 100198. [Google Scholar] [CrossRef] [PubMed]
- Lough, J.; Rosenthall, L.; Arzoumanian, A.; Goresky, C.A. Kupffer cell depletion associated with capillarization of liver sinusoids in carbon tetrachloride-induced rat liver cirrhosis. J. Hepatol. 1987, 5, 190–198. [Google Scholar] [CrossRef]
- Diaz-Cruz, A.; Guinzberg, R.; Guerra, R.; Vilchis, M.; Carrasco, D.; Garcia-Vazquez, F.J.; Pina, E. Adrenaline stimulates H2O2 generation in liver via NADPH oxidase. Free. Radic. Res. 2007, 41, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Cruz, A.; Vilchis-Landeros, M.M.; Guinzberg, R.; Villalobos-Molina, R.; Pina, E. NOX2 activated by alpha1-adrenoceptors modulates hepatic metabolic routes stimulated by beta-adrenoceptors. Free. Radic. Res. 2011, 45, 1366–1378. [Google Scholar] [CrossRef] [PubMed]
- Mortezaee, K. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) and liver fibrosis: A review. Cell Biochem. Funct. 2018, 36, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Suárez, R.; Buelvas, N. El inflamasoma: Mecanismos de activación. Investig. Clínica 2015, 56, 74–99. [Google Scholar]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montaño Estrada, L.F.; van der Goes, T.I.F.; Rendón Huerta, E.R. ¿Qué son los inflamosomas? El NLRP3 como por ejemplo. Rev. De La Fac. De Med. 2017, 60, 1–8. [Google Scholar]
- Ting, J.P.; Lovering, R.C.; Alnemri, E.S.; Bertin, J.; Boss, J.M.; Davis, B.K.; Flavell, R.A.; Girardin, S.E.; Godzik, A.; Harton, J.A.; et al. The NLR gene family: A standard nomenclature. Immunity 2008, 28, 285–287. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Huang, H.; Liu, B.; Zhang, Y.; Pan, X.; Yu, X.Y.; Shen, Z.; Song, Y.H. Inflammasomes as therapeutic targets in human diseases. Signal Transduct. Target. Ther. 2021, 6, 247. [Google Scholar] [CrossRef]
- Harijith, A.; Ebenezer, D.L.; Natarajan, V. Reactive oxygen species at the crossroads of inflammasome and inflammation. Front. Physiol. 2014, 5, 352. [Google Scholar] [CrossRef]
- Petagine, L.; Zariwala, M.G.; Patel, V.B. Alcoholic liver disease: Current insights into cellular mechanisms. World J. Biol. Chem. 2021, 12, 87–103. [Google Scholar] [CrossRef]
- Han, H.; Desert, R.; Das, S.; Song, Z.; Athavale, D.; Ge, X.; Nieto, N. Danger signals in liver injury and restoration of homeostasis. J. Hepatol. 2020, 73, 933–951. [Google Scholar] [CrossRef]
- Anders, H.J.; Schaefer, L. Beyond tissue injury-damage-associated molecular patterns, toll-like receptors, and inflammasomes also drive regeneration and fibrosis. J. Am. Soc Nephrol. 2014, 25, 1387–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguchi, A.; Yan, R.; Pan, S.Q.; Wu, R.; Kim, J.; Chen, Y.; Ansong, C.; Smith, R.D.; Tempaku, M.; Ohno-Machado, L.; et al. Comprehensive characterization of hepatocyte-derived extracellular vesicles identifies direct miRNA-based regulation of hepatic stellate cells and DAMP-based hepatic macrophage IL-1beta and IL-17 upregulation in alcoholic hepatitis mice. J. Mol. Med. 2020, 98, 1021–1034. [Google Scholar] [CrossRef] [PubMed]
- Piccinini, A.M.; Midwood, K.S. DAMPening inflammation by modulating TLR signalling. Mediat. Inflamm. 2010, 2010, 672395. [Google Scholar] [CrossRef] [Green Version]
- Paik, S.; Kim, J.K.; Silwal, P.; Sasakawa, C.; Jo, E.K. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol. Immunol. 2021, 18, 1141–1160. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619. [Google Scholar] [CrossRef]
- Cai, S.M.; Yang, R.Q.; Li, Y.; Ning, Z.W.; Zhang, L.L.; Zhou, G.S.; Luo, W.; Li, D.H.; Chen, Y.; Pan, M.X.; et al. Angiotensin-(1-7) Improves Liver Fibrosis by Regulating the NLRP3 Inflammasome via Redox Balance Modulation. Antioxid. Redox Signal 2016, 24, 795–812. [Google Scholar] [CrossRef]
- Hewinson, J.; Moore, S.F.; Glover, C.; Watts, A.G.; MacKenzie, A.B. A key role for redox signaling in rapid P2X7 receptor-induced IL-1 beta processing in human monocytes. J. Immunol. 2008, 180, 8410–8420. [Google Scholar] [CrossRef] [Green Version]
- Sekiyama, A.; Ueda, H.; Kashiwamura, S.; Sekiyama, R.; Takeda, M.; Rokutan, K.; Okamura, H. A stress-induced, superoxide-mediated caspase-1 activation pathway causes plasma IL-18 upregulation. Immunity 2005, 22, 669–677. [Google Scholar] [CrossRef] [Green Version]
- Knorr, J.; Wree, A.; Tacke, F.; Feldstein, A.E. The NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Semin. Liver Dis. 2020, 40, 298–306. [Google Scholar] [CrossRef]
- Wu, X.; Dong, L.; Lin, X.; Li, J. Relevance of the NLRP3 Inflammasome in the Pathogenesis of Chronic Liver Disease. Front. Immunol. 2017, 8, 1728. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L.; Roll, F.J.; Boyles, J.; Bissell, D.M. Hepatic lipocytes: The principal collagen-producing cells of normal rat liver. Proc. Natl. Acad. Sci. USA 1985, 82, 8681–8685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Kisseleva, T. The origin of fibrogenic myofibroblasts in fibrotic liver. Hepatology 2017, 65, 1039–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Kisseleva, T. Bone marrow-derived fibrocytes contribute to liver fibrosis. Exp. Biol. Med. 2015, 240, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Corder, N.L.; Koduru, B.; Wang, Y. Oxidative stress and hepatic Nox proteins in chronic hepatitis C and hepatocellular carcinoma. Free. Radic. Biol. Med. 2014, 72, 267–284. [Google Scholar] [CrossRef] [Green Version]
- Paik, Y.H.; Kim, J.; Aoyama, T.; De Minicis, S.; Bataller, R.; Brenner, D.A. Role of NADPH oxidases in liver fibrosis. Antioxid. Redox Signal 2014, 20, 2854–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, S.G.; Zilhao, R.; Thorsteinsdottir, S.; Carlos, A.R. Linking Oxidative Stress and DNA Damage to Changes in the Expression of Extracellular Matrix Components. Front. Genet. 2021, 12, 673002. [Google Scholar] [CrossRef]
- Garcia-Trevijano, E.R.; Iraburu, M.J.; Fontana, L.; Dominguez-Rosales, J.A.; Auster, A.; Covarrubias-Pinedo, A.; Rojkind, M. Transforming growth factor beta1 induces the expression of alpha1(I) procollagen mRNA by a hydrogen peroxide-C/EBPbeta-dependent mechanism in rat hepatic stellate cells. Hepatology 1999, 29, 960–970. [Google Scholar] [CrossRef]
- De Minicis, S.; Seki, E.; Paik, Y.H.; Osterreicher, C.H.; Kodama, Y.; Kluwe, J.; Torozzi, L.; Miyai, K.; Benedetti, A.; Schwabe, R.F.; et al. Role and cellular source of nicotinamide adenine dinucleotide phosphate oxidase in hepatic fibrosis. Hepatology 2010, 52, 1420–1430. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.X.; Venugopal, S.; Serizawa, N.; Chen, X.; Scott, F.; Li, Y.; Adamson, R.; Devaraj, S.; Shah, V.; Gershwin, M.E.; et al. Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology 2010, 139, 1375–1384. [Google Scholar] [CrossRef] [Green Version]
- Paik, Y.H.; Iwaisako, K.; Seki, E.; Inokuchi, S.; Schnabl, B.; Osterreicher, C.H.; Kisseleva, T.; Brenner, D.A. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology 2011, 53, 1730–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bataller, R.; Schwabe, R.F.; Choi, Y.H.; Yang, L.; Paik, Y.H.; Lindquist, J.; Qian, T.; Schoonhoven, R.; Hagedorn, C.H.; Lemasters, J.J.; et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J. Clin. Invest. 2003, 112, 1383–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herranz-Iturbide, M.; Lopez-Luque, J.; Gonzalez-Sanchez, E.; Caballero-Diaz, D.; Crosas-Molist, E.; Martin-Mur, B.; Gut, M.; Esteve-Codina, A.; Jaquet, V.; Jiang, J.X.; et al. NADPH oxidase 4 (Nox4) deletion accelerates liver regeneration in mice. Redox Biol. 2021, 40, 101841. [Google Scholar] [CrossRef]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef]
- Ma, Y.; Lee, G.; Heo, S.Y.; Roh, Y.S. Oxidative Stress Is a Key Modulator in the Development of Nonalcoholic Fatty Liver Disease. Antioxidants 2021, 11, 91. [Google Scholar] [CrossRef]
- Wheeler, M.D.; Kono, H.; Yin, M.; Nakagami, M.; Uesugi, T.; Arteel, G.E.; Gäbele, E.; Rusyn, I.; Yamashina, S.; Froh, M.; et al. The role of kupffer cell oxidant production in early ethanol-induced liver disease. Free. Radic. Biol. Med. 2001, 31, 1544–1549. [Google Scholar] [CrossRef]
- Albano, E. Alcohol, oxidative stress and free radical damage. Proc. Nutr. Soc. 2006, 65, 278–290. [Google Scholar] [CrossRef] [Green Version]
- Thakur, V.; Pritchard, M.T.; McMullen, M.R.; Wang, Q.; Nagy, L.E. Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: Role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-alpha production. J. Leukoc. Biol. 2006, 79, 1348–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, I.; Petrasek, J.; Szabo, G. The presence of p47phox in liver parenchymal cells is a key mediator in the pathogenesis of alcoholic liver steatosis. Alcohol. Clin. Exp. Res. 2012, 36, 1397–1406. [Google Scholar] [CrossRef] [Green Version]
- Yeligar, S.; Tsukamoto, H.; Kalra, V.K. Ethanol-induced expression of ET-1 and ET-BR in liver sinusoidal endothelial cells and human endothelial cells involves hypoxia-inducible factor-1alpha and microrNA-199. J. Immunol. 2009, 183, 5232–5243. [Google Scholar] [CrossRef] [Green Version]
- Kono, H.; Rusyn, I.; Yin, M.; Gabele, E.; Yamashina, S.; Dikalova, A.; Kadiiska, M.B.; Connor, H.D.; Mason, R.P.; Segal, B.H.; et al. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J. Clin. Invest. 2000, 106, 867–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colmenero, J.; Bataller, R.; Sancho-Bru, P.; Bellot, P.; Miquel, R.; Moreno, M.; Jares, P.; Bosch, J.; Arroyo, V.; Caballeria, J.; et al. Hepatic expression of candidate genes in patients with alcoholic hepatitis: Correlation with disease severity. Gastroenterology 2007, 132, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, R.; Enjoji, M.; Kohjima, M.; Tsuruta, S.; Fukushima, M.; Iwao, M.; Sonta, T.; Kotoh, K.; Inoguchi, T.; Nakamuta, M. High glucose stimulates hepatic stellate cells to proliferate and to produce collagen through free radical production and activation of mitogen-activated protein kinase. Liver Int. 2005, 25, 1018–1026. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Seki, E.; Oesterreicher, C.; Schnabl, B.; Schwabe, R.F.; Brenner, D.A. Reduced nicotinamide adenine dinucleotide phosphate oxidase mediates fibrotic and inflammatory effects of leptin on hepatic stellate cells. Hepatology 2008, 48, 2016–2026. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ruiz, I.; Gomez-Izquierdo, E.; Diaz-Sanjuan, T.; Grau, M.; Solis-Munoz, P.; Munoz-Yague, T.; Solis-Herruzo, J.A. Sp1 and Sp3 transcription factors mediate leptin-induced collagen alpha1(I) gene expression in primary culture of male rat hepatic stellate cells. Endocrinology 2012, 153, 5845–5856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlassara, H.; Uribarri, J. Advanced glycation end products (AGE) and diabetes: Cause, effect, or both? Curr. Diab. Rep. 2014, 14, 453. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.X.; Chen, X.; Serizawa, N.; Szyndralewiez, C.; Page, P.; Schroder, K.; Brandes, R.P.; Devaraj, S.; Torok, N.J. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free. Radic. Biol. Med. 2012, 53, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Li, H.; Wang, N.; Chen, H.; Jin, Q.; Zhang, R.; Wang, J.; Chen, Y. Polymorphism of rs1836882 in NOX4 gene modifies associations between dietary caloric intake and ROS levels in peripheral blood mononuclear cells. PLoS ONE 2013, 8, e85660. [Google Scholar] [CrossRef] [PubMed]
- Spengler, U. Direct antiviral agents (DAAs)-A new age in the treatment of hepatitis C virus infection. Pharmacol. Ther. 2018, 183, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Ou, J.H. Mechanisms of liver injury. III. Oxidative stress in the pathogenesis of hepatitis C virus. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G847–G851. [Google Scholar] [CrossRef] [PubMed]
- Bureau, C.; Bernad, J.; Chaouche, N.; Orfila, C.; Beraud, M.; Gonindard, C.; Alric, L.; Vinel, J.P.; Pipy, B. Nonstructural 3 protein of hepatitis C virus triggers an oxidative burst in human monocytes via activation of NADPH oxidase. J. Biol. Chem. 2001, 276, 23077–23083. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Mediavilla, M.V.; Sanchez-Campos, S.; Gonzalez-Perez, P.; Gomez-Gonzalo, M.; Majano, P.L.; Lopez-Cabrera, M.; Clemente, G.; Garcia-Monzon, C.; Gonzalez-Gallego, J. Differential contribution of hepatitis C virus NS5A and core proteins to the induction of oxidative and nitrosative stress in human hepatocyte-derived cells. J. Hepatol. 2005, 43, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Lerat, H.; Honda, M.; Beard, M.R.; Loesch, K.; Sun, J.; Yang, Y.; Okuda, M.; Gosert, R.; Xiao, S.Y.; Weinman, S.A.; et al. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology 2002, 122, 352–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona-Cuenca, I.; Roncero, C.; Sancho, P.; Caja, L.; Fausto, N.; Fernandez, M.; Fabregat, I. Upregulation of the NADPH oxidase NOX4 by TGF-beta in hepatocytes is required for its pro-apoptotic activity. J Hepatol. 2008, 49, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Mainez, J.; Crosas-Molist, E.; Roncero, C.; Fernandez-Rodriguez, C.M.; Pinedo, F.; Huber, H.; Eferl, R.; Mikulits, W.; Fabregat, I. NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS ONE 2012, 7, e45285. [Google Scholar] [CrossRef] [Green Version]
- Boudreau, H.E.; Emerson, S.U.; Korzeniowska, A.; Jendrysik, M.A.; Leto, T.L. Hepatitis C virus (HCV) proteins induce NADPH oxidase 4 expression in a transforming growth factor beta-dependent manner: A new contributor to HCV-induced oxidative stress. J. Virol. 2009, 83, 12934–12946. [Google Scholar] [CrossRef] [Green Version]
- Bataller, R.; Paik, Y.H.; Lindquist, J.N.; Lemasters, J.J.; Brenner, D.A. Hepatitis C virus core and nonstructural proteins induce fibrogenic effects in hepatic stellate cells. Gastroenterology 2004, 126, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Lohmann, V.; Penin, F. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat. Rev. Microbiol. 2013, 11, 482–496. [Google Scholar] [CrossRef] [Green Version]
- Zayas, M.; Long, G.; Madan, V.; Bartenschlager, R. Coordination of Hepatitis C Virus Assembly by Distinct Regulatory Regions in Nonstructural Protein 5A. PLoS Pathog. 2016, 12, e1005376. [Google Scholar] [CrossRef] [Green Version]
- Sarrazin, C.; Dvory-Sobol, H.; Svarovskaia, E.S.; Doehle, B.P.; Pang, P.S.; Chuang, S.M.; Ma, J.; Ding, X.; Afdhal, N.H.; Kowdley, K.V.; et al. Prevalence of Resistance-Associated Substitutions in HCV NS5A, NS5B, or NS3 and Outcomes of Treatment with Ledipasvir and Sofosbuvir. Gastroenterology 2016, 151, 501–512.e501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoofnagle, J.H.; Sherker, A.H. Hepatitis C: Down but Not Out. Ann. Intern. Med. 2017, 166, 675–676. [Google Scholar] [CrossRef] [PubMed]
- Geddawy, A.; Ibrahim, Y.F.; Elbahie, N.M.; Ibrahim, M.A. Direct Acting Anti-Hepatitis C Virus Drugs: Clinical Pharmacology and Future Direction. J. Transl. Int. Med. 2017, 5, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Copple, B.L.; Li, T. Pharmacology of bile acid receptors: Evolution of bile acids from simple detergents to complex signaling molecules. Pharm. Res. 2016, 104, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Morita, S.Y.; Ikeda, N.; Horikami, M.; Soda, K.; Ishihara, K.; Teraoka, R.; Terada, T.; Kitagawa, S. Effects of phosphatidylethanolamine N-methyltransferase on phospholipid composition, microvillus formation and bile salt resistance in LLC-PK1 cells. FEBS J. 2011, 278, 4768–4781. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.J.; Briz, O. Bile-acid-induced cell injury and protection. World J. Gastroenterol. 2009, 15, 1677–1689. [Google Scholar] [CrossRef]
- Houten, S.M.; Watanabe, M.; Auwerx, J. Endocrine functions of bile acids. EMBO J. 2006, 25, 1419–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Gioiello, A.; Costantino, G. Aplicaciones terapéuticas potenciales de los moduladores del receptor farnesoide X (FXR). Opinión Expert. Pat. 2006, 16, 333–341. [Google Scholar] [CrossRef]
- Chawla, A.; Saez, E.; Evans, R.M. Don’t know much bile-ology. Cell 2000, 103, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, L.; Han, S.I.; Fang, Y.; Park, J.S.; Gupta, S.; Gilfor, D.; Amorino, G.; Valerie, K.; Sealy, L.; Engelhardt, J.F.; et al. Bile acid regulation of C/EBPbeta, CREB, and c-Jun function, via the extracellular signal-regulated kinase and c-Jun NH2-terminal kinase pathways, modulates the apoptotic response of hepatocytes. Mol. Cell Biol. 2003, 23, 3052–3066. [Google Scholar] [CrossRef] [PubMed]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Mencarelli, A.; Renga, B.; Migliorati, M.; Cipriani, S.; Distrutti, E.; Santucci, L.; Fiorucci, S. The bile acid sensor farnesoid X receptor is a modulator of liver immunity in a rodent model of acute hepatitis. J. Immunol. 2009, 183, 6657–6666. [Google Scholar] [CrossRef] [Green Version]
- Kalaany, N.Y.; Mangelsdorf, D.J. LXRS and FXR: The yin and yang of cholesterol and fat metabolism. Annu. Rev. Physiol. 2006, 68, 159–191. [Google Scholar] [CrossRef]
- Li, G.; Guo, G.L. Farnesoid X receptor, the bile acid sensing nuclear receptor, in liver regeneration. Acta Pharm. Sin. B 2015, 5, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Li, F.; Guo, G.L. Tissue-specific function of farnesoid X receptor in liver and intestine. Pharm. Res. 2011, 63, 259–265. [Google Scholar] [CrossRef] [Green Version]
- Guo, G.L.; Lambert, G.; Negishi, M.; Ward, J.M.; Brewer, H.B., Jr.; Kliewer, S.A.; Gonzalez, F.J.; Sinal, C.J. Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J. Biol. Chem. 2003, 278, 45062–45071. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Ma, K.; Zhang, J.; Qatanani, M.; Cuvillier, J.; Liu, J.; Dong, B.; Huang, X.; Moore, D.D. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science 2006, 312, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Antonelli, E.; Rizzo, G.; Renga, B.; Mencarelli, A.; Riccardi, L.; Orlandi, S.; Pellicciari, R.; Morelli, A. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology 2004, 127, 1497–1512. [Google Scholar] [CrossRef]
- Liu, Y.; Binz, J.; Numerick, M.J.; Dennis, S.; Luo, G.; Desai, B.; MacKenzie, K.I.; Mansfield, T.A.; Kliewer, S.A.; Goodwin, B.; et al. Hepatoprotection by the farnesoid X receptor agonist GW4064 in rat models of intra- and extrahepatic cholestasis. J. Clin. Invest. 2003, 112, 1678–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.D.; Chen, W.D.; Wang, M.; Yu, D.; Forman, B.M.; Huang, W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 2008, 48, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Degirolamo, C.; Modica, S.; Vacca, M.; Di Tullio, G.; Morgano, A.; D’Orazio, A.; Kannisto, K.; Parini, P.; Moschetta, A. Prevention of spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice by intestinal-specific farnesoid X receptor reactivation. Hepatology 2015, 61, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, A.; Thomas, A.; Edwards, G.; Jaseja, R.; Guo, G.L.; Apte, U. Increased activation of the Wnt/beta-catenin pathway in spontaneous hepatocellular carcinoma observed in farnesoid X receptor knockout mice. J. Pharm. Exp. 2011, 338, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Kong, B.; Zhu, Y.; Li, G.; Williams, J.A.; Buckley, K.; Tawfik, O.; Luyendyk, J.P.; Guo, G.L. Mice with hepatocyte-specific FXR deficiency are resistant to spontaneous but susceptible to cholic acid-induced hepatocarcinogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G295–G302. [Google Scholar] [CrossRef] [Green Version]
- Lozano, E.; Sanchez-Vicente, L.; Monte, M.J.; Herraez, E.; Briz, O.; Banales, J.M.; Marin, J.J.; Macias, R.I. Cocarcinogenic effects of intrahepatic bile acid accumulation in cholangiocarcinoma development. Mol. Cancer Res. 2014, 12, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Huang, X.; Yi, T.; Yen, Y.; Moore, D.D.; Huang, W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 2007, 67, 863–867. [Google Scholar] [CrossRef] [Green Version]
- Becker, S.; Reinehr, R.; Graf, D.; vom Dahl, S.; Haussinger, D. Hydrophobic bile salts induce hepatocyte shrinkage via NADPH oxidase activation. Cell Physiol. Biochem. 2007, 19, 89–98. [Google Scholar] [CrossRef]
- Becker, S.; Reinehr, R.; Grether-Beck, S.; Eberle, A.; Haussinger, D. Hydrophobic bile salts trigger ceramide formation through endosomal acidification. Biol. Chem. 2007, 388, 185–196. [Google Scholar] [CrossRef]
- Reinehr, R.; Becker, S.; Keitel, V.; Eberle, A.; Grether-Beck, S.; Haussinger, D. Bile salt-induced apoptosis involves NADPH oxidase isoform activation. Gastroenterology 2005, 129, 2009–2031. [Google Scholar] [CrossRef]
- Sommerfeld, A.; Reinehr, R.; Haussinger, D. Bile acid-induced epidermal growth factor receptor activation in quiescent rat hepatic stellate cells can trigger both proliferation and apoptosis. J. Biol. Chem. 2009, 284, 22173–22183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinehr, R.; Becker, S.; Eberle, A.; Grether-Beck, S.; Haussinger, D. Involvement of NADPH oxidase isoforms and Src family kinases in CD95-dependent hepatocyte apoptosis. J. Biol. Chem. 2005, 280, 27179–27194. [Google Scholar] [CrossRef] [PubMed]
- Svegliati-Baroni, G.; Ridolfi, F.; Hannivoort, R.; Saccomanno, S.; Homan, M.; De Minicis, S.; Jansen, P.L.; Candelaresi, C.; Benedetti, A.; Moshage, H. Bile acids induce hepatic stellate cell proliferation via activation of the epidermal growth factor receptor. Gastroenterology 2005, 128, 1042–1055. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Invest. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K. Liver regeneration. J. Cell Physiol. 2007, 213, 286–300. [Google Scholar] [CrossRef]
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat. Commun. 2019, 10, 1076. [Google Scholar] [CrossRef] [Green Version]
- Bai, H.; Zhang, W.; Qin, X.J.; Zhang, T.; Wu, H.; Liu, J.Z.; Hai, C.X. Hydrogen peroxide modulates the proliferation/quiescence switch in the liver during embryonic development and post hepatectomy regeneration. Antioxid. Redox Signal 2015, 22, 921–937. [Google Scholar] [CrossRef]
- Michalopoulos, G.K.; Bhushan, B. Liver regeneration: Biological and pathological mechanisms and implications. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 40–55. [Google Scholar] [CrossRef]
- Cao, L.; Quan, X.B.; Zeng, W.J.; Yang, X.O.; Wang, M.J. Mechanism of Hepatocyte Apoptosis. J. Cell Death 2016, 9, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Tang, P.; Dang, H.; Huang, J.; Xu, T.; Yuan, P.; Hu, J.; Sheng, J.F. NADPH oxidase NOX4 is a glycolytic regulator through mROS-HIF1alpha axis in thyroid carcinomas. Sci. Rep. 2018, 8, 15897. [Google Scholar] [CrossRef] [Green Version]
- Aykin-Burns, N.; Ahmad, I.M.; Zhu, Y.; Oberley, L.W.; Spitz, D.R. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem. J. 2009, 418, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Chetram, M.A.; Hinton, C.V. ROS-mediated regulation of CXCR4 in cancer. Front. Biol. 2013, 8, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Osorio Alves, J.; Matta Pereira, L.; Cabral Coutinho do Rego Monteiro, I.; Pontes Dos Santos, L.H.; Soares Marreiros Ferraz, A.; Carneiro Loureiro, A.C.; Calado Lima, C.; Leal-Cardoso, J.H.; Pires Carvalho, D.; Soares Fortunato, R.; et al. Strenuous Acute Exercise Induces Slow and Fast Twitch-Dependent NADPH Oxidase Expression in Rat Skeletal Muscle. Antioxidants 2020, 9, 57. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Shi, Q.; Deng, W.H.; Yu, J.; Zuo, T.; Mei, F.C.; Wang, W.X. Relationship between expression of NADPH oxidase 2 and invasion and prognosis of human gastric cancer. World J. Gastroenterol. 2015, 21, 6271–6279. [Google Scholar] [CrossRef]
- Block, K.; Gorin, Y. Aiding and abetting roles of NOX oxidases in cellular transformation. Nat. Rev. Cancer 2012, 12, 627–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambeth, J.D.; Kawahara, T.; Diebold, B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic. Biol. Med. 2007, 43, 319–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, K.; Wu, Y.; Meitzler, J.L.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Antony, S.; Doroshow, J.H. NADPH oxidases and cancer. Clin. Sci. 2015, 128, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Buvelot, H.; Jaquet, V.; Krause, K.H. Mammalian NADPH Oxidases. Methods Mol. Biol. 2019, 1982, 17–36. [Google Scholar] [CrossRef] [PubMed]
- Helmcke, I.; Heumuller, S.; Tikkanen, R.; Schroder, K.; Brandes, R.P. Identification of structural elements in Nox1 and Nox4 controlling localization and activity. Antioxid. Redox Signal 2009, 11, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Caceres, J.; Mainez, J.; Mayoral, R.; Martin-Sanz, P.; Egea, G.; Fabregat, I. Caveolin-1-dependent activation of the metalloprotease TACE/ADAM17 by TGF-beta in hepatocytes requires activation of Src and the NADPH oxidase NOX1. FEBS J. 2016, 283, 1300–1310. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Zhang, Y.; Marden, J.J.; Banfi, B.; Engelhardt, J.F. Endosomal NADPH oxidase regulates c-Src activation following hypoxia/reoxygenation injury. Biochem. J. 2008, 411, 531–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, N.Y.; Zhou, W.; Li, Q.; Zhang, Y.; Luo, M.; Yan, Z.; Lynch, T.J.; Abbott, D.; Banfi, B.; Engelhardt, J.F. Hepatocytes produce TNF-alpha following hypoxia-reoxygenation and liver ischemia-reperfusion in a NADPH oxidase- and c-Src-dependent manner. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G84–G94. [Google Scholar] [CrossRef] [PubMed]
- Juhasz, A.; Markel, S.; Gaur, S.; Liu, H.; Lu, J.; Jiang, G.; Wu, X.; Antony, S.; Wu, Y.; Melillo, G.; et al. NADPH oxidase 1 supports proliferation of colon cancer cells by modulating reactive oxygen species-dependent signal transduction. J. Biol. Chem. 2017, 292, 7866–7887. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.P.; Wang, X.; Gong, L.F.; Chen, W.J.; Hao, Z.; Feng, S.W.; Wu, Y.B.; Ye, T.; Cai, Y.K. Nox1 promotes colon cancer cell metastasis via activation of the ADAM17 pathway. Eur. Rev. Med. Pharm. Sci. 2016, 20, 4474–4481. [Google Scholar]
- Grauers Wiktorin, H.; Aydin, E.; Hellstrand, K.; Martner, A. NOX2-Derived Reactive Oxygen Species in Cancer. Oxid. Med. Cell Longev. 2020, 2020, 7095902. [Google Scholar] [CrossRef] [PubMed]
- Adane, B.; Ye, H.; Khan, N.; Pei, S.; Minhajuddin, M.; Stevens, B.M.; Jones, C.L.; D’Alessandro, A.; Reisz, J.A.; Zaberezhnyy, V.; et al. The Hematopoietic Oxidase NOX2 Regulates Self-Renewal of Leukemic Stem Cells. Cell Rep. 2019, 27, 238–254.e236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aurelius, J.; Thoren, F.B.; Akhiani, A.A.; Brune, M.; Palmqvist, L.; Hansson, M.; Hellstrand, K.; Martner, A. Monocytic AML cells inactivate antileukemic lymphocytes: Role of NADPH oxidase/gp91(phox) expression and the PARP-1/PAR pathway of apoptosis. Blood 2012, 119, 5832–5837. [Google Scholar] [CrossRef] [Green Version]
- Mellqvist, U.H.; Hansson, M.; Brune, M.; Dahlgren, C.; Hermodsson, S.; Hellstrand, K. Natural killer cell dysfunction and apoptosis induced by chronic myelogenous leukemia cells: Role of reactive oxygen species and regulation by histamine. Blood 2000, 96, 1961–1968. [Google Scholar] [CrossRef]
- Kim, S.M.; Hur, D.Y.; Hong, S.W.; Kim, J.H. EBV-encoded EBNA1 regulates cell viability by modulating miR34a-NOX2-ROS signaling in gastric cancer cells. Biochem. Biophys. Res. Commun. 2017, 494, 550–555. [Google Scholar] [CrossRef]
- Loffredo, L.; Zicari, A.M.; Perri, L.; Carnevale, R.; Nocella, C.; Angelico, F.; Del Ben, M.; Mosca, A.; Zaffina, S.; Panera, N.; et al. Does Nox2 Overactivate in Children with Nonalcoholic Fatty Liver Disease? Antioxid. Redox Signal 2019, 30, 1325–1330. [Google Scholar] [CrossRef]
- Wang, X.; Zeldin, S.; Shi, H.; Zhu, C.; Saito, Y.; Corey, K.E.; Osganian, S.A.; Remotti, H.E.; Verna, E.C.; Pajvani, U.B.; et al. TAZ-induced Cybb contributes to liver tumor formation in non-alcoholic steatohepatitis. J. Hepatol. 2022, 76, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Ogboo, B.C.; Grabovyy, U.V.; Maini, A.; Scouten, S.; van der Vliet, A.; Mattevi, A.; Heppner, D.E. Architecture of the NADPH oxidase family of enzymes. Redox Biol. 2022, 52, 102298. [Google Scholar] [CrossRef]
- Schroder, K. NADPH oxidase-derived reactive oxygen species: Dosis facit venenum. Exp. Physiol. 2019, 104, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Serrander, L.; Cartier, L.; Bedard, K.; Banfi, B.; Lardy, B.; Plastre, O.; Sienkiewicz, A.; Forro, L.; Schlegel, W.; Krause, K.H. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem. J. 2007, 406, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Szanto, I. NADPH Oxidase 4 (NOX4) in Cancer: Linking Redox Signals to Oncogenic Metabolic Adaptation. Int. J. Mol. Sci. 2022, 23, 2702. [Google Scholar] [CrossRef] [PubMed]
- Meitzler, J.L.; Makhlouf, H.R.; Antony, S.; Wu, Y.; Butcher, D.; Jiang, G.; Juhasz, A.; Lu, J.; Dahan, I.; Jansen-Durr, P.; et al. Decoding NADPH oxidase 4 expression in human tumors. Redox Biol. 2017, 13, 182–195. [Google Scholar] [CrossRef]
- Weyemi, U.; Lagente-Chevallier, O.; Boufraqech, M.; Prenois, F.; Courtin, F.; Caillou, B.; Talbot, M.; Dardalhon, M.; Al Ghuzlan, A.; Bidart, J.M.; et al. ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene 2012, 31, 1117–1129. [Google Scholar] [CrossRef] [Green Version]
- Soni, S.; Padwad, Y.S. HIF-1 in cancer therapy: Two decade long story of a transcription factor. Acta Oncol. 2017, 56, 503–515. [Google Scholar] [CrossRef] [Green Version]
- De Deken, X.; Wang, D.; Many, M.C.; Costagliola, S.; Libert, F.; Vassart, G.; Dumont, J.E.; Miot, F. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J. Biol. Chem. 2000, 275, 23227–23233. [Google Scholar] [CrossRef] [Green Version]
- Fischer, H. Mechanisms and function of DUOX in epithelia of the lung. Antioxid. Redox Signal 2009, 11, 2453–2465. [Google Scholar] [CrossRef]
- Luxen, S.; Belinsky, S.A.; Knaus, U.G. Silencing of DUOX NADPH oxidases by promoter hypermethylation in lung cancer. Cancer Res. 2008, 68, 1037–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulcrano, M.; Boukheris, H.; Talbot, M.; Caillou, B.; Dupuy, C.; Virion, A.; De Vathaire, F.; Schlumberger, M. Poorly differentiated follicular thyroid carcinoma: Prognostic factors and relevance of histological classification. Thyroid 2007, 17, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Amara, N.; Cooper, M.P.; Voronkova, M.A.; Webb, B.A.; Lynch, E.M.; Kollman, J.M.; Ma, T.; Yu, K.; Lai, Z.; Sangaraju, D.; et al. Selective activation of PFKL suppresses the phagocytic oxidative burst. Cell 2021, 184, 4480–4494.e4415. [Google Scholar] [CrossRef]
- Lan, T.; Kisseleva, T.; Brenner, D.A. Deficiency of NOX1 or NOX4 Prevents Liver Inflammation and Fibrosis in Mice through Inhibition of Hepatic Stellate Cell Activation. PLoS ONE 2015, 10, e0129743. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, T.; Paik, Y.H.; Watanabe, S.; Laleu, B.; Gaggini, F.; Fioraso-Cartier, L.; Molango, S.; Heitz, F.; Merlot, C.; Szyndralewiez, C.; et al. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology 2012, 56, 2316–2327. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Shi, A.; Dong, L.; Lu, X.; Wang, Y.; Zhao, J.; Dai, F.; Guo, X. Chlorogenic acid protects against liver fibrosis in vivo and in vitro through inhibition of oxidative stress. Clin. Nutr. 2016, 35, 1366–1373. [Google Scholar] [CrossRef]
- Colmenero, J.; Bataller, R.; Sancho-Bru, P.; Dominguez, M.; Moreno, M.; Forns, X.; Bruguera, M.; Arroyo, V.; Brenner, D.A.; Gines, P. Effects of losartan on hepatic expression of nonphagocytic NADPH oxidase and fibrogenic genes in patients with chronic hepatitis C. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G726–G734. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Wang, C.; Jin, Y.; Meng, Q.; Liu, Q.; Liu, Z.; Liu, K.; Sun, H. Catalpol ameliorates hepatic insulin resistance in type 2 diabetes through acting on AMPK/NOX4/PI3K/AKT pathway. Pharm. Res. 2018, 130, 466–480. [Google Scholar] [CrossRef]
- Fayed, M.R.; El-Naga, R.N.; Akool, E.S.; El-Demerdash, E. The potential antifibrotic impact of apocynin and alpha-lipoic acid in concanavalin A-induced liver fibrosis in rats: Role of NADPH oxidases 1 and 4. Drug Discov. 2018, 12, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Wagner, A.H.; Kohler, T.; Ruckschloss, U.; Just, I.; Hecker, M. Improvement of nitric oxide-dependent vasodilatation by HMG-CoA reductase inhibitors through attenuation of endothelial superoxide anion formation. Arter. Thromb. Vasc. Biol. 2000, 20, 61–69. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Nox Inhibited | Pathway Affected | Disease | Study Model | Reference |
|---|---|---|---|---|---|
| LDC7559 (NA-11) (Indirect action) | NOX2 | Respiratory burst (neutrophils) | Viral infection | Human blood | [163] |
| GKT137831 (Setanaxib) (Direct action) | NOX1/NOX4 | Suppressed chemokine production, inhibited hepatic stellate cell (HSC) activation | Hepatic fibrosis | Mouse | [164] |
| GKT137831 (Setanaxib) (Direct action) | NOX1/NOX4 | Bile duct ligation-induced hepatic fibrosis (BDL) | Hepatic fibrosis and hepatocyte apoptosis | Mouse | [77] |
| GKT137831 (Setanaxib) (Direct action) | NOX1/NOX4 | Decrease in oxidative stress and inflammation | Hepatic fibrosis | Mouse | [165] |
| Chlorogenic acid (Indirect action) | NOX | Upregulation of NFE2L2, a transcription factor that regulates the expression of antioxidant enzymes | Hepatic fibrosis | Rats | [166] |
| Losartan (Indirect action) | Non-specific inhibition of different NOX | The expression of profibrogenic and NOX genes was reduced | Hypertension and heart failure | Human | [167] |
| Catalpol (Indirect action) | NOX4 | AMPK/NOX4/PI3K/AKT | Hepatic insulin resistance in type 2 diabetes | Mouse | [168] |
| Apocinin (Direct action) | NOX2 | Inhibits the binding of p47phox to gp91phox | Inflammation and aging | Rats | [169] |
| Statinas (Direct action) | NOX1/NOX2 | Inhibit Rac binding to gp91phox | Hepatic fibrosis | Rats | [170] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matuz-Mares, D.; Vázquez-Meza, H.; Vilchis-Landeros, M.M. NOX as a Therapeutic Target in Liver Disease. Antioxidants 2022, 11, 2038. https://doi.org/10.3390/antiox11102038
Matuz-Mares D, Vázquez-Meza H, Vilchis-Landeros MM. NOX as a Therapeutic Target in Liver Disease. Antioxidants. 2022; 11(10):2038. https://doi.org/10.3390/antiox11102038
Chicago/Turabian StyleMatuz-Mares, Deyamira, Héctor Vázquez-Meza, and María Magdalena Vilchis-Landeros. 2022. "NOX as a Therapeutic Target in Liver Disease" Antioxidants 11, no. 10: 2038. https://doi.org/10.3390/antiox11102038