
Epitranscriptome Analysis of Oxidative Stressed Retinal Epithelial Cells Depicted a Possible RNA Editing Landscape of Retinal Degeneration
 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Photo-Oxidation Induction
2.2. MTT Assay
2.3. Total RNA Sequencing
2.4. RNA Editing Event Detection and High-Confidence Filtering
2.5. Comparison of RNA Editing between Treated Samples and Controls
2.6. Functional Enrichment of Differential Edited Genes and miRNAs
2.7. ADAR Expression Level Analysis
2.8. Data Validation by qRT-PCR
3. Results
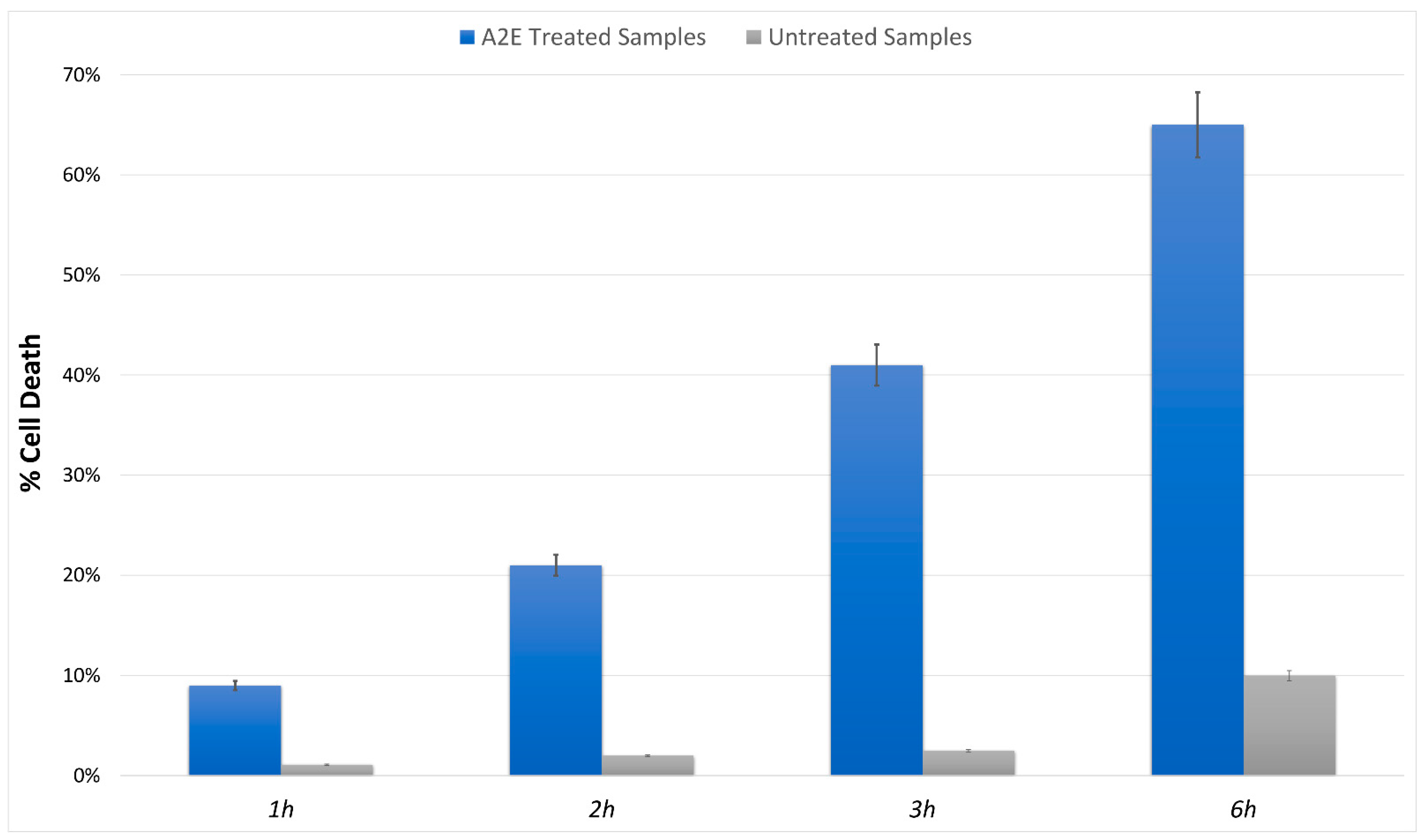
3.1. MTT Assay Showed Increased RPE Cell Death in A2E Treated Samples
3.2. Sequencing Analysis and Mapping Statistics
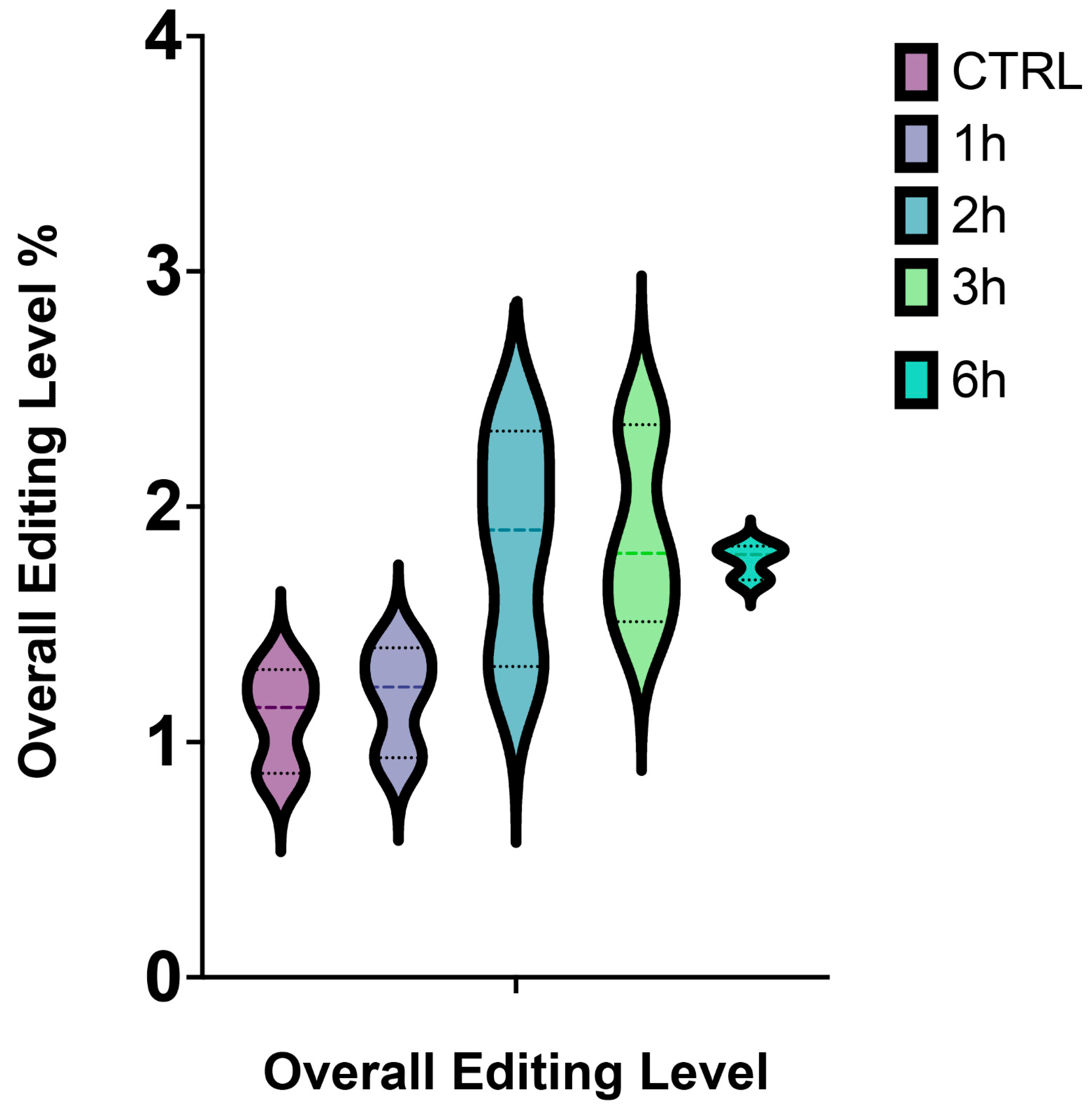
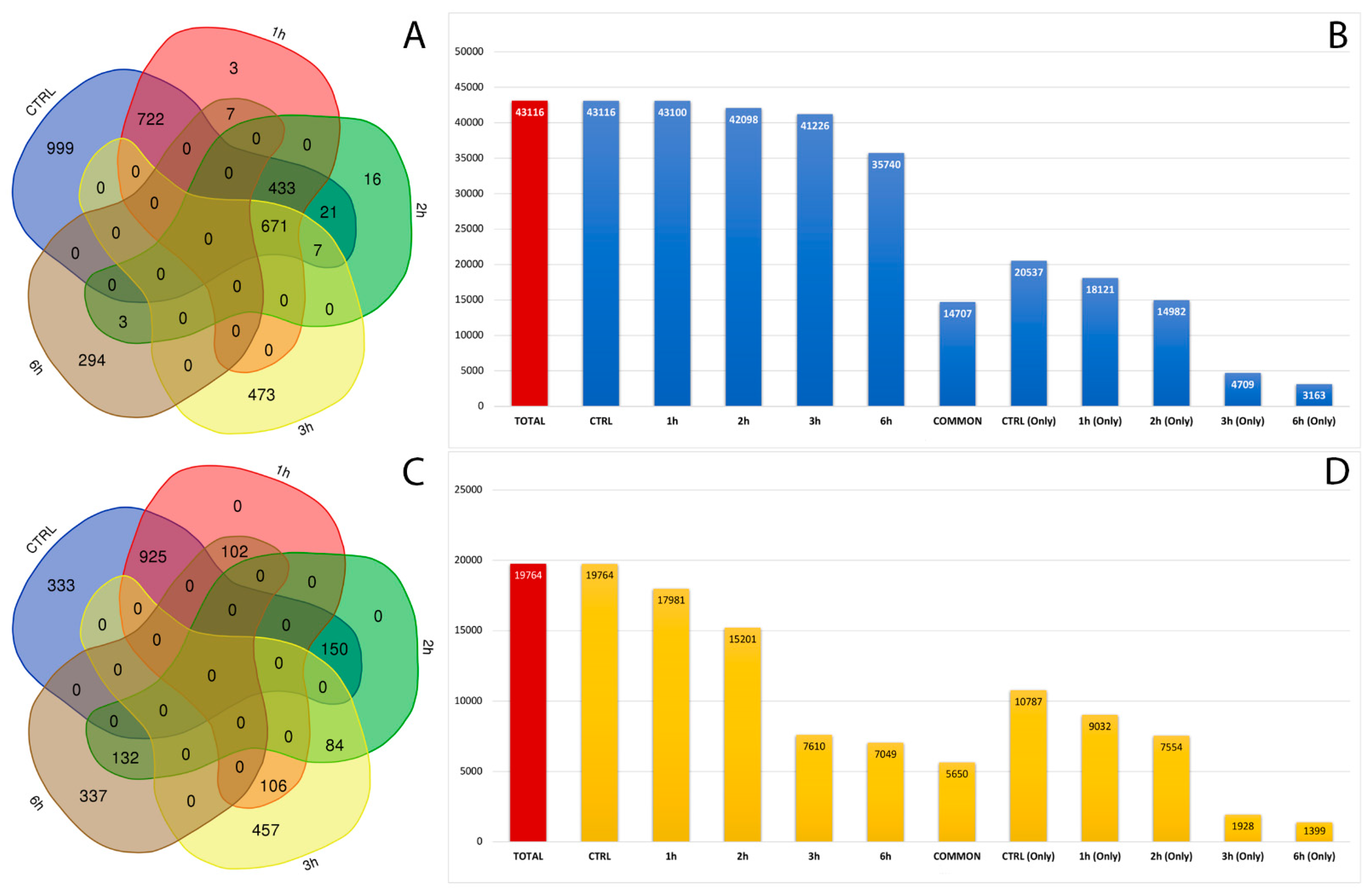
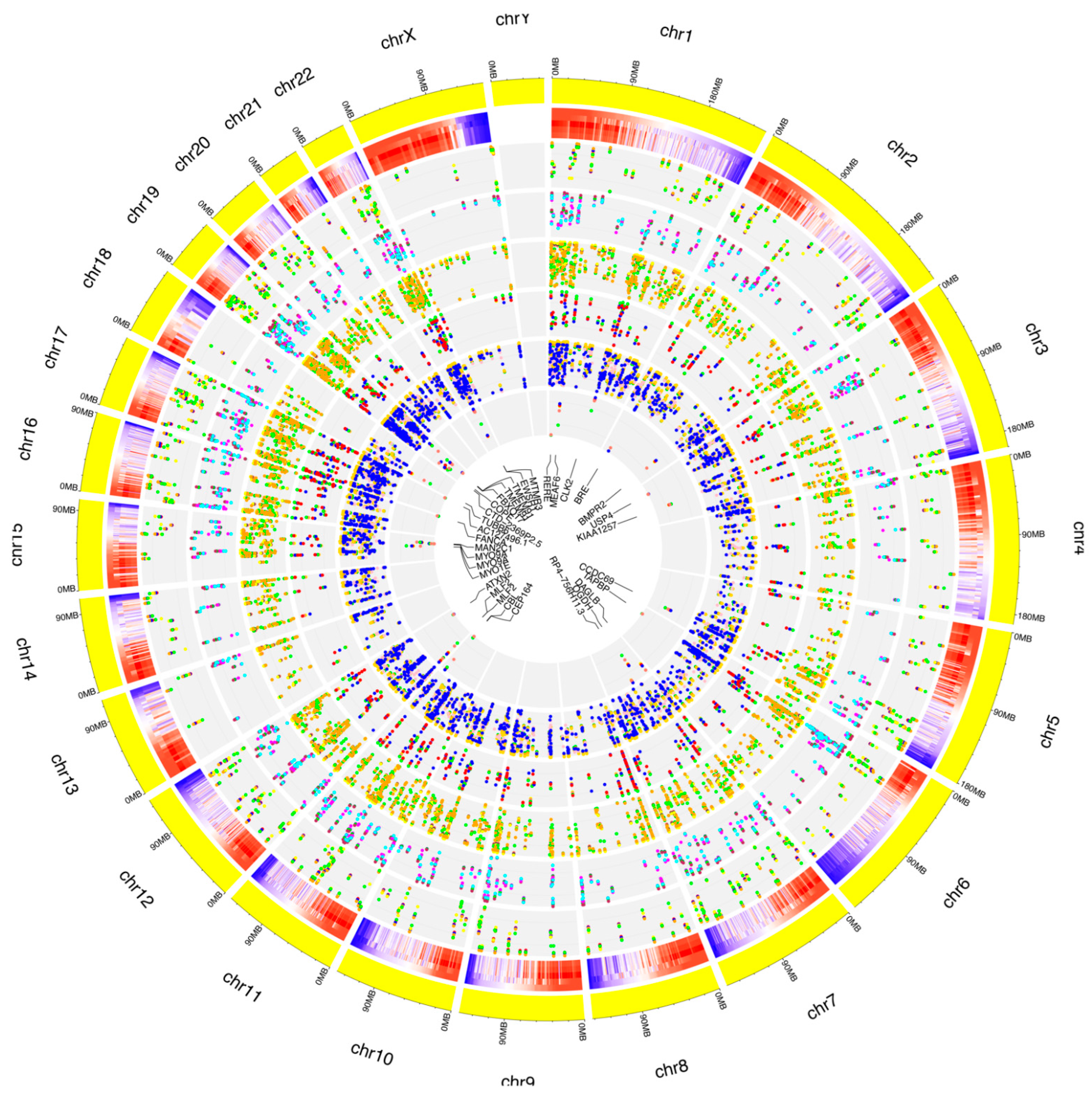
3.3. Identification of RNA Editing Sites in A2E Treated RPE Cells
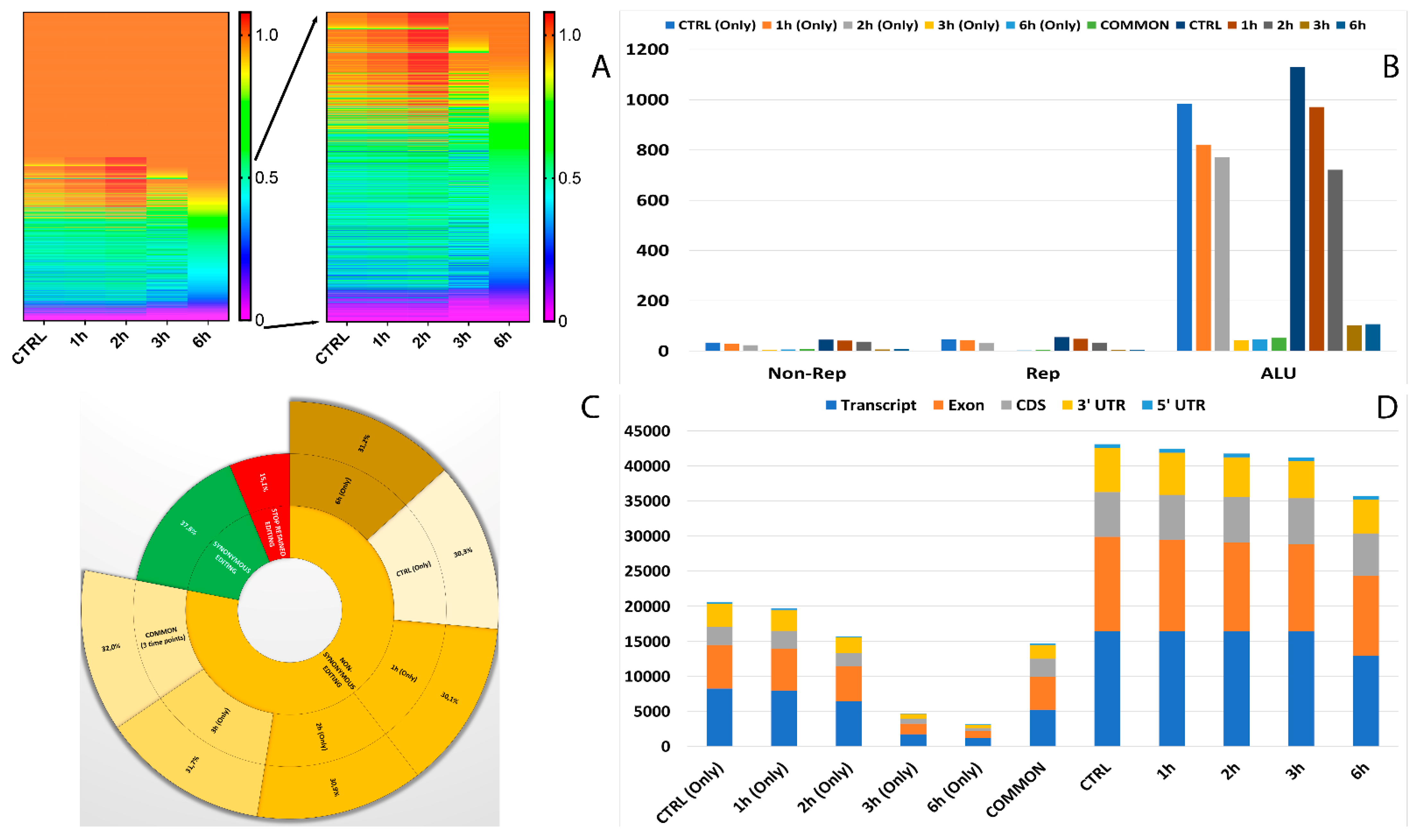
3.4. Characterization and Distribution of Known RNA Editing Sites across Different Genomic Regions
3.5. Characterization and Distribution of De Novo RNA Editing Sites across Different Genomic Regions
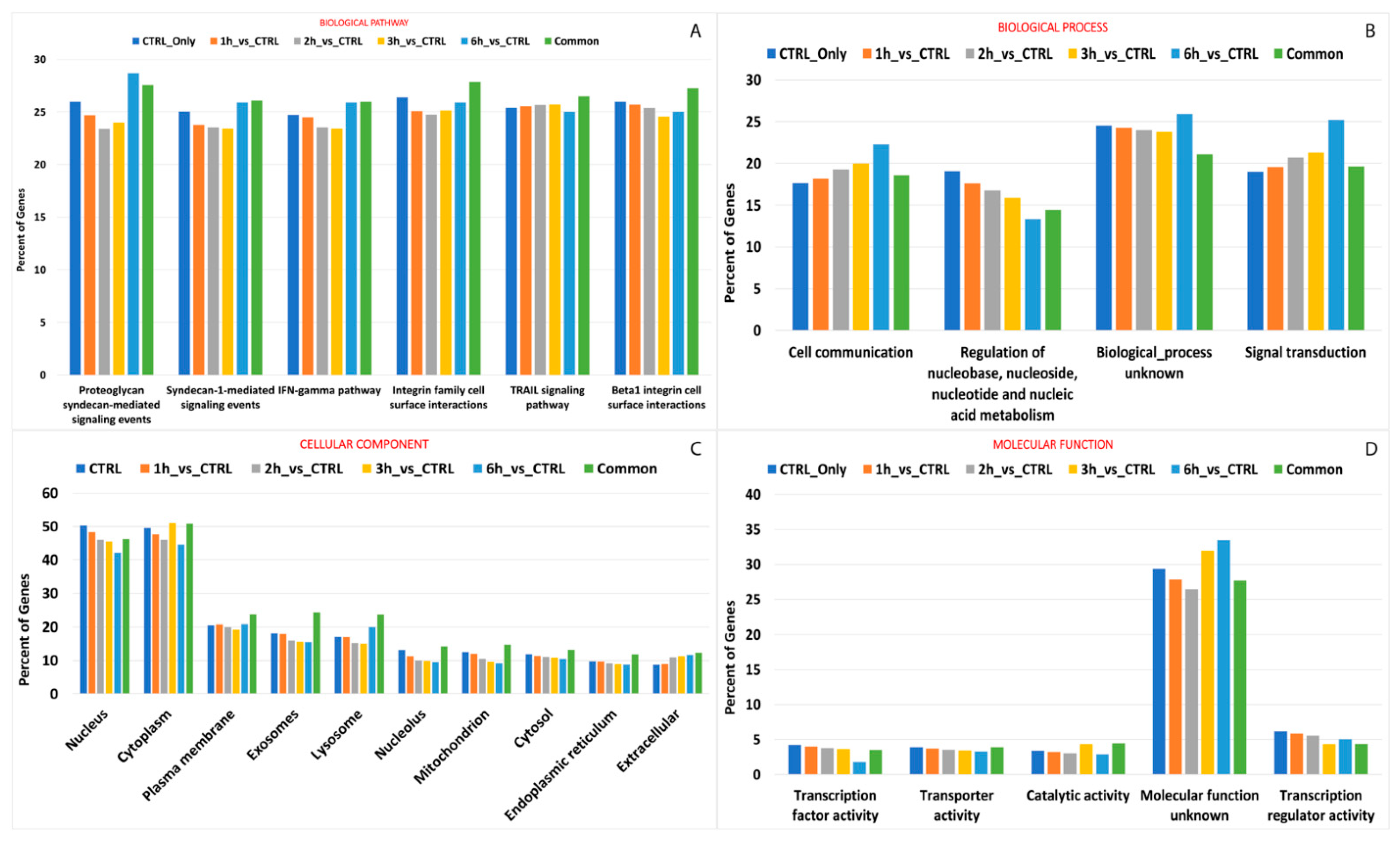
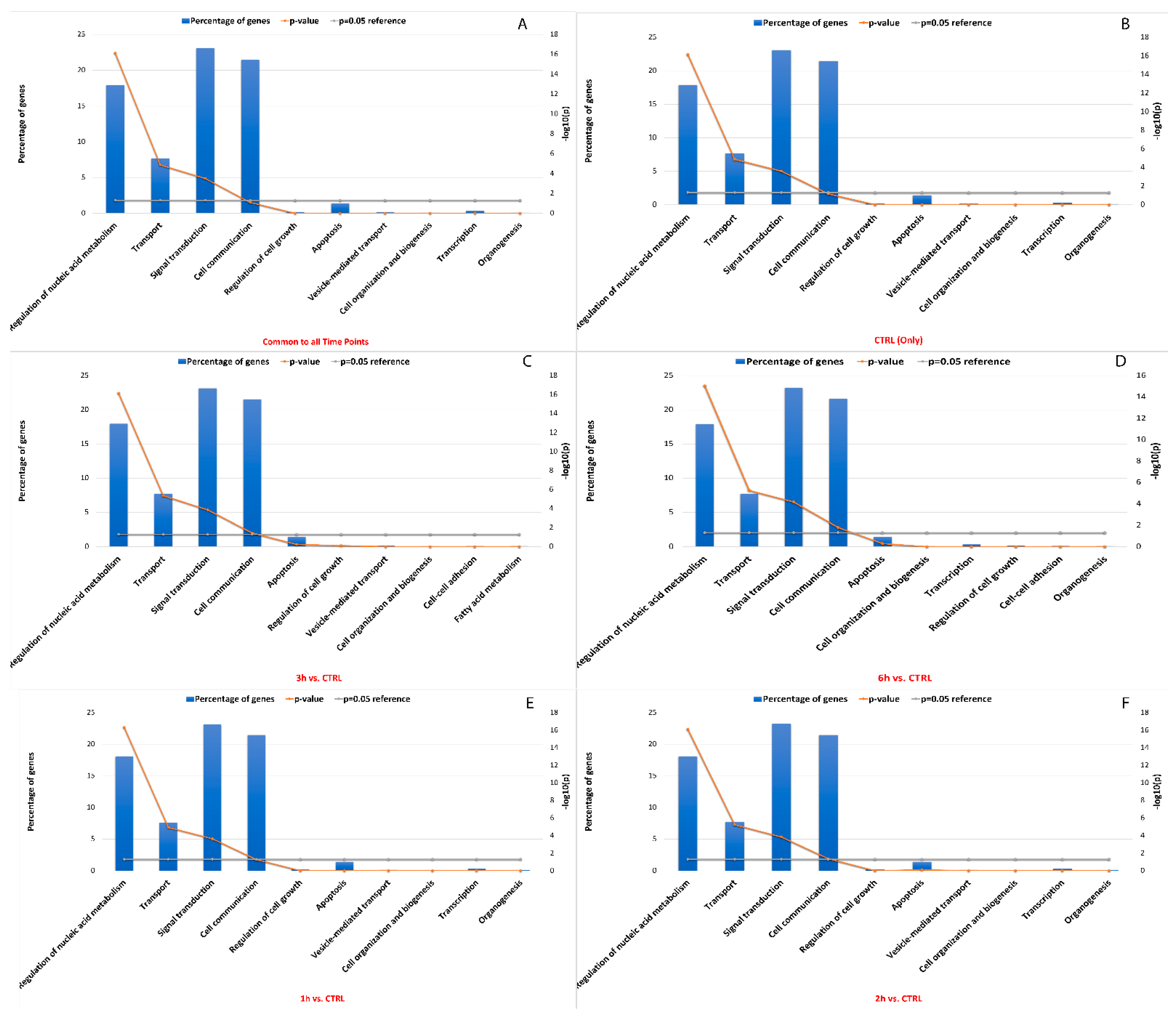
3.6. Editing Site Comparison between Control and Treated RPE Cells Highlighted Pathways Involving Surface Protein as Most Edited
3.7. Effects of RNA Editing Sites on miRNA–RNA Interactions
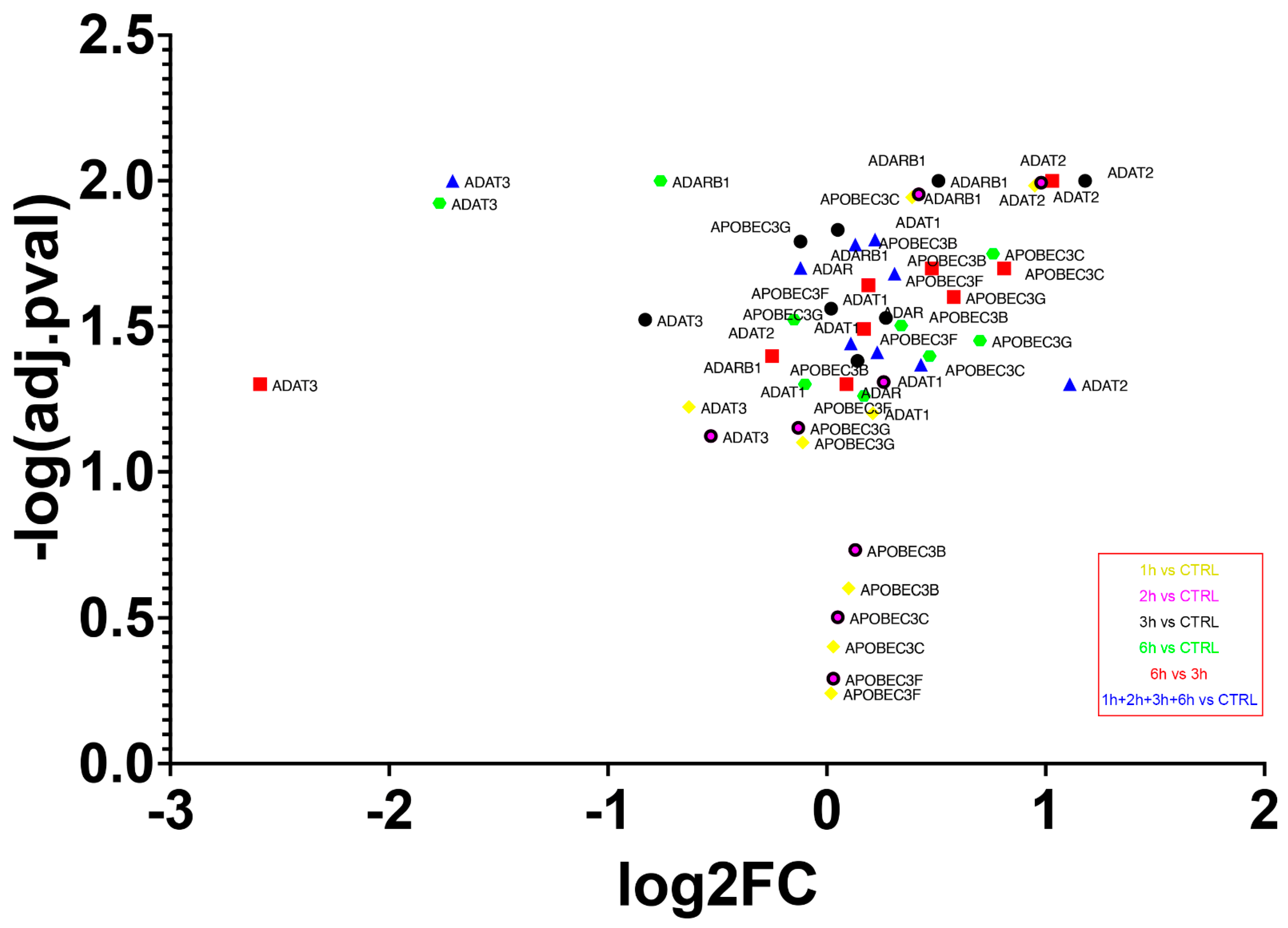
3.8. Deaminases Involved into Editing Showed a Global Up-Regulation of mRNAs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Christofi, T.; Zaravinos, A. RNA editing in the forefront of epitranscriptomics and human health. J. Transl. Med. 2019, 17, 319. [Google Scholar] [CrossRef]
- Ramaswami, G.; Li, J.B. Identification of human RNA editing sites: A historical perspective. Methods 2016, 107, 42–47. [Google Scholar] [CrossRef]
- Stepanova, V.V.; Gel’fand, M.S. RNA editing. The classical cases and the outlook of new technologies. Mol. Biol. 2014, 48, 15–21. [Google Scholar] [CrossRef]
- Daniel, C.; Lagergren, J.; Ohman, M. RNA editing of non-coding RNA and its role in gene regulation. Biochimie 2015, 117, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Gagnidze, K.; Rayon-Estrada, V.; Harroch, S.; Bulloch, K.; Papavasiliou, F.N. A New Chapter in Genetic Medicine: RNA Editing and its Role in Disease Pathogenesis. Trends Mol. Med. 2018, 24, 294–303. [Google Scholar] [CrossRef]
- Schaffer, A.A.; Levanon, E.Y. ALU A-to-I RNA Editing: Millions of Sites and Many Open Questions. Methods Mol. Biol. 2021, 2181, 149–162. [Google Scholar] [CrossRef]
- Kliuchnikova, A.A.; Kuznetsova, K.G.; Moshkovskii, S.A. ADAR-mediated messenger RNA editing: Analysis at the proteome level. Biomed. Khim. 2016, 62, 510–519. [Google Scholar] [CrossRef]
- Choi, E.H.; Suh, S.; Foik, A.T.; Leinonen, H.; Newby, G.A.; Gao, X.D.; Banskota, S.; Hoang, T.; Du, S.W.; Dong, Z.; et al. In vivo base editing rescues cone photoreceptors in a mouse model of early-onset inherited retinal degeneration. Nat. Commun. 2022, 13, 1830. [Google Scholar] [CrossRef]
- Nishikura, K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 2010, 79, 321–349. [Google Scholar] [CrossRef]
- Lerner, T.; Kluesner, M.; Tasakis, R.N.; Moriarity, B.S.; Papavasiliou, F.N.; Pecori, R. C-to-U RNA Editing: From Computational Detection to Experimental Validation. Methods Mol. Biol. 2021, 2181, 51–67. [Google Scholar] [CrossRef]
- Behm, M.; Ohman, M. RNA Editing: A Contributor to Neuronal Dynamics in the Mammalian Brain. Trends Genet. 2016, 32, 165–175. [Google Scholar] [CrossRef]
- Costa Cruz, P.H.; Kawahara, Y. RNA Editing in Neurological and Neurodegenerative Disorders. Methods Mol. Biol. 2021, 2181, 309–330. [Google Scholar] [CrossRef]
- Niavarani, A.; Currie, E.; Reyal, Y.; Anjos-Afonso, F.; Horswell, S.; Griessinger, E.; Luis Sardina, J.; Bonnet, D. APOBEC3A is implicated in a novel class of G-to-A mRNA editing in WT1 transcripts. PLoS ONE 2015, 10, e0120089. [Google Scholar] [CrossRef]
- Klimek-Tomczak, K.; Mikula, M.; Dzwonek, A.; Paziewska, A.; Karczmarski, J.; Hennig, E.; Bujnicki, J.M.; Bragoszewski, P.; Denisenko, O.; Bomsztyk, K.; et al. Editing of hnRNP K protein mRNA in colorectal adenocarcinoma and surrounding mucosa. Br. J. Cancer 2006, 94, 586–592. [Google Scholar] [CrossRef]
- Yu, H.; Zhao, S.; Ness, S.; Kang, H.; Sheng, Q.; Samuels, D.C.; Oyebamiji, O.; Zhao, Y.Y.; Guo, Y. Non-canonical RNA-DNA differences and other human genomic features are enriched within very short tandem repeats. PLoS Comput. Biol. 2020, 16, e1007968. [Google Scholar] [CrossRef] [PubMed]
- Bertotti, S.; Fleming, I.; Camara, M.L.M.; Centeno Camean, C.; Carmona, S.J.; Aguero, F.; Balouz, V.; Zahn, A.; Di Noia, J.M.; Alfonzo, J.D.; et al. Characterization of ADAT2/3 molecules in Trypanosoma cruzi and regulation of mucin gene expression by tRNA editing. Biochem. J. 2022, 479, 561–580. [Google Scholar] [CrossRef]
- Correia de Sousa, M.; Gjorgjieva, M.; Dolicka, D.; Sobolewski, C.; Foti, M. Deciphering miRNAs’ Action through miRNA Editing. Int. J. Mol. Sci. 2019, 20, 6249. [Google Scholar] [CrossRef]
- Diroma, M.A.; Ciaccia, L.; Pesole, G.; Picardi, E. Bioinformatics Resources for RNA Editing. Methods Mol. Biol. 2021, 2181, 177–191. [Google Scholar] [CrossRef]
- Tran, S.S.; Zhou, Q.; Xiao, X. Statistical inference of differential RNA-editing sites from RNA-sequencing data by hierarchical modeling. Bioinformatics 2020, 36, 2796–2804. [Google Scholar] [CrossRef]
- Lee, J.H.; Ang, J.K.; Xiao, X. Analysis and design of RNA sequencing experiments for identifying RNA editing and other single-nucleotide variants. RNA 2013, 19, 725–732. [Google Scholar] [CrossRef] [Green Version]
- Donato, L.; Scimone, C.; Rinaldi, C.; D’Angelo, R.; Sidoti, A. New evaluation methods of read mapping by 17 aligners on simulated and empirical NGS data: An updated comparison of DNA- and RNA-Seq data from Illumina and Ion Torrent technologies. Neural Comput. Appl. 2021, 33, 15669–15692. [Google Scholar] [CrossRef]
- Tsoi, S.C.; Dyck, M.K. RNA amplification for pseudogene detection using RNA-seq. Methods Mol. Biol. 2014, 1167, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Boyer, N.P.; Thompson, D.A.; Koutalos, Y. Relative Contributions of All-Trans and 11-Cis Retinal to Formation of Lipofuscin and A2E Accumulating in Mouse Retinal Pigment Epithelium. Investig. Ophthalmol. Vis. Sci. 2021, 62, 1. [Google Scholar] [CrossRef]
- Boyer, N.P.; Higbee, D.; Currin, M.B.; Blakeley, L.R.; Chen, C.; Ablonczy, Z.; Crouch, R.K.; Koutalos, Y. Lipofuscin and N-retinylidene-N-retinylethanolamine (A2E) accumulate in retinal pigment epithelium in absence of light exposure: Their origin is 11-cis-retinal. J. Biol. Chem. 2012, 287, 22276–22286. [Google Scholar] [CrossRef]
- Donato, L.; Scimone, C.; Alibrandi, S.; Rinaldi, C.; Sidoti, A.; D’Angelo, R. Transcriptome Analyses of lncRNAs in A2E-Stressed Retinal Epithelial Cells Unveil Advanced Links between Metabolic Impairments Related to Oxidative Stress and Retinitis Pigmentosa. Antioxidants 2020, 9, 318. [Google Scholar] [CrossRef]
- Blasiak, J. Senescence in the pathogenesis of age-related macular degeneration. Cell. Mol. Life Sci. 2020, 77, 789–805. [Google Scholar] [CrossRef]
- Jin, H.L.; Choung, S.-Y.; Jeong, K.W. Protective mechanisms of polyphenol-enriched fraction of Vaccinium uliginosum L. Against blue light-induced cell death of human retinal pigmented epithelial cells. J. Funct. Foods 2017, 39, 28–36. [Google Scholar] [CrossRef]
- Kim, J.; Jin, H.L.; Jang, D.S.; Jeong, K.W.; Choung, S.Y. Quercetin-3-O-alpha-l-arabinopyranoside protects against retinal cell death via blue light-induced damage in human RPE cells and Balb-c mice. Food Funct. 2018, 9, 2171–2183. [Google Scholar] [CrossRef]
- Scimone, C.; Donato, L.; Alibrandi, S.; Vadala, M.; Giglia, G.; Sidoti, A.; D’Angelo, R. N-retinylidene-N-retinylethanolamine adduct induces expression of chronic inflammation cytokines in retinal pigment epithelium cells. Exp. Eye Res. 2021, 209, 108641. [Google Scholar] [CrossRef]
- Rinaldi, C.; Donato, L.; Alibrandi, S.; Scimone, C.; D’Angelo, R.; Sidoti, A. Oxidative Stress and the Neurovascular Unit. Life 2021, 11, 767. [Google Scholar] [CrossRef]
- Donato, L.; D’Angelo, R.; Alibrandi, S.; Rinaldi, C.; Sidoti, A.; Scimone, C. Effects of A2E-Induced Oxidative Stress on Retinal Epithelial Cells: New Insights on Differential Gene Response and Retinal Dystrophies. Antioxidants 2020, 9, 307. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Zhou, J.; Ben-Shabat, S.; Vollmer, H.; Itagaki, Y.; Nakanishi, K. Involvement of oxidative mechanisms in blue-light-induced damage to A2E-laden RPE. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1222–1227. [Google Scholar]
- Pham, T.N.M.; Shin, C.Y.; Park, S.H.; Lee, T.H.; Ryu, H.Y.; Kim, S.B.; Auh, K.; Jeong, K.W. Solanum melongena L. Extract Protects Retinal Pigment Epithelial Cells from Blue Light-Induced Phototoxicity in In Vitro and In Vivo Models. Nutrients 2021, 13, 359. [Google Scholar] [CrossRef]
- Jin, H.L.; Jeong, K.W. Transcriptome Analysis of Long-Term Exposure to Blue Light in Retinal Pigment Epithelial Cells. Biomol. Ther. 2022, 30, 291–297. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Conesa, A.; Garcia-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Vasimuddin, M.; Misra, S.; Li, H.; Aluru, S. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. In Proceedings of the 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS), Rio de Janeiro, Brazil, 20–24 May 2019; pp. 314–324. [Google Scholar]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Xiao, X. RASER: Reads aligner for SNPs and editing sites of RNA. Bioinformatics 2015, 31, 3906–3913. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, G.; Li, J.B. RADAR: A rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res. 2014, 42, D109–D113. [Google Scholar] [CrossRef]
- Lo Giudice, C.; Tangaro, M.A.; Pesole, G.; Picardi, E. Investigating RNA editing in deep transcriptome datasets with REDItools and REDIportal. Nat. Protoc. 2020, 15, 1098–1131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Lu, Y.; Yan, S.; Xing, Q.; Tian, W. SPRINT: An SNP-free toolkit for identifying RNA editing sites. Bioinformatics 2017, 33, 3538–3548. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef]
- IBM. IBM SPSS Statistics for Windows; Version 26.0; IBM: New York, NY, USA, 2020. [Google Scholar]
- Peng, Z.; Cheng, Y.; Tan, B.C.; Kang, L.; Tian, Z.; Zhu, Y.; Zhang, W.; Liang, Y.; Hu, X.; Tan, X.; et al. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat. Biotechnol. 2012, 30, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Fonseka, P.; Pathan, M.; Chitti, S.V.; Kang, T.; Mathivanan, S. FunRich enables enrichment analysis of OMICs datasets. J. Mol. Biol. 2021, 433, 166747. [Google Scholar] [CrossRef]
- The Gene Ontology, C. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res 2017, 45, D331–D338. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.Y.; Dosztanyi, Z.; El-Gebali, S.; Fraser, M.; et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R.; Jassal, B.; Jupe, S.; Korninger, F.; McKay, S.; et al. The Reactome pathway Knowledgebase. Nucleic Acids Res. 2016, 44, D481–D487. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct project--IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Aken, B.L.; Ayling, S.; Barrell, D.; Clarke, L.; Curwen, V.; Fairley, S.; Fernandez Banet, J.; Billis, K.; Garcia Giron, C.; Hourlier, T.; et al. The Ensembl gene annotation system. Database 2016, 2016, baw093. [Google Scholar] [CrossRef]
- Wain, H.M.; Bruford, E.A.; Lovering, R.C.; Lush, M.J.; Wright, M.W.; Povey, S. Guidelines for human gene nomenclature. Genomics 2002, 79, 464–470. [Google Scholar] [CrossRef]
- Li, B.; Ruotti, V.; Stewart, R.M.; Thomson, J.A.; Dewey, C.N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 2010, 26, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Chatsirisupachai, K.; Palmer, D.; Ferreira, S.; de Magalhaes, J.P. A human tissue-specific transcriptomic analysis reveals a complex relationship between aging, cancer, and cellular senescence. Aging Cell 2019, 18, e13041. [Google Scholar] [CrossRef]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanfreau, G.F. Impact of RNA Modifications and RNA-Modifying Enzymes on Eukaryotic Ribonucleases. Enzymes 2017, 41, 299–329. [Google Scholar] [CrossRef]
- Bianchetti, G.; Clementi, M.E.; Sampaolese, B.; Serantoni, C.; Abeltino, A.; De Spirito, M.; Sasson, S.; Maulucci, G. Investigation of DHA-Induced Regulation of Redox Homeostasis in Retinal Pigment Epithelium Cells through the Combination of Metabolic Imaging and Molecular Biology. Antioxidants 2022, 11, 1072. [Google Scholar] [CrossRef]
- Clementi, M.E.; Maulucci, G.; Bianchetti, G.; Pizzoferrato, M.; Sampaolese, B.; Tringali, G. Cytoprotective Effects of Punicalagin on Hydrogen-Peroxide-Mediated Oxidative Stress and Mitochondrial Dysfunction in Retinal Pigment Epithelium Cells. Antioxidants 2021, 10, 192. [Google Scholar] [CrossRef]
- Gerasimov, E.S.; Gasparyan, A.A.; Afonin, D.A.; Zimmer, S.L.; Kraeva, N.; Lukes, J.; Yurchenko, V.; Kolesnikov, A. Complete minicircle genome of Leptomonas pyrrhocoris reveals sources of its non-canonical mitochondrial RNA editing events. Nucleic Acids Res. 2021, 49, 3354–3370. [Google Scholar] [CrossRef]
- Cayir, A. RNA A-to-I editing, environmental exposure, and human diseases. Crit. Rev. Toxicol. 2021, 51, 456–466. [Google Scholar] [CrossRef]
- Tan, M.H.; Li, Q.; Shanmugam, R.; Piskol, R.; Kohler, J.; Young, A.N.; Liu, K.I.; Zhang, R.; Ramaswami, G.; Ariyoshi, K.; et al. Dynamic landscape and regulation of RNA editing in mammals. Nature 2017, 550, 249–254. [Google Scholar] [CrossRef]
- Leonardi, A.; Rosani, U.; Brun, P. Ocular Surface Expression of SARS-CoV-2 Receptors. Ocul. Immunol. Inflamm. 2020, 28, 735–738. [Google Scholar] [CrossRef]
- Xiao, J.; Li, X.; Chen, L.; Han, X.; Zhao, W.; Li, L.; Chen, J.G. Apobec1 Promotes Neurotoxicity-Induced Dedifferentiation of Muller Glial Cells. Neurochem. Res. 2017, 42, 1151–1164. [Google Scholar] [CrossRef]
- Gussakovsky, D.; McKenna, S.A. Alu RNA and their roles in human disease states. RNA Biol. 2021, 18, 574–585. [Google Scholar] [CrossRef]
- Ambati, M.; Apicella, I.; Wang, S.B.; Narendran, S.; Leung, H.; Pereira, F.; Nagasaka, Y.; Huang, P.; Varshney, A.; Baker, K.L.; et al. Identification of fluoxetine as a direct NLRP3 inhibitor to treat atrophic macular degeneration. Proc. Natl. Acad. Sci. USA 2021, 118, e2102975118. [Google Scholar] [CrossRef]
- Hwang, Y.E.; Baek, Y.M.; Baek, A.; Kim, D.E. Oxidative stress causes Alu RNA accumulation via PIWIL4 sequestration into stress granules. BMB Rep. 2019, 52, 196–201. [Google Scholar] [CrossRef]
- Scimone, C.; Alibrandi, S.; Scalinci, S.Z.; Trovato Battagliola, E.; D’Angelo, R.; Sidoti, A.; Donato, L. Expression of Pro-Angiogenic Markers Is Enhanced by Blue Light in Human RPE Cells. Antioxidants 2020, 9, 1154. [Google Scholar] [CrossRef]
- Kechagia, J.Z.; Ivaska, J.; Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell. Biol. 2019, 20, 457–473. [Google Scholar] [CrossRef]
- Mrugacz, M.; Bryl, A.; Falkowski, M.; Zorena, K. Integrins: An Important Link between Angiogenesis, Inflammation and Eye Diseases. Cells 2021, 10, 1703. [Google Scholar] [CrossRef]
- Park, H.; Yamamoto, H.; Mohn, L.; Ambuhl, L.; Kanai, K.; Schmidt, I.; Kim, K.P.; Fraccaroli, A.; Feil, S.; Junge, H.J.; et al. Integrin-linked kinase controls retinal angiogenesis and is linked to Wnt signaling and exudative vitreoretinopathy. Nat. Commun. 2019, 10, 5243. [Google Scholar] [CrossRef]
- Hubert, K.E.; Davies, M.H.; Stempel, A.J.; Griffith, T.S.; Powers, M.R. TRAIL-deficient mice exhibit delayed regression of retinal neovascularization. Am. J. Pathol. 2009, 175, 2697–2708. [Google Scholar] [CrossRef]
- Corti, F.; Wang, Y.; Rhodes, J.M.; Atri, D.; Archer-Hartmann, S.; Zhang, J.; Zhuang, Z.W.; Chen, D.; Wang, T.; Wang, Z.; et al. N-terminal syndecan-2 domain selectively enhances 6-O heparan sulfate chains sulfation and promotes VEGFA165-dependent neovascularization. Nat. Commun. 2019, 10, 1562. [Google Scholar] [CrossRef]
- Elfenbein, A.; Simons, M. Syndecan-4 signaling at a glance. J. Cell Sci. 2013, 126, 3799–3804. [Google Scholar] [CrossRef]
- Lipphardt, M.; Song, J.W.; Ratliff, B.B.; Dihazi, H.; Muller, G.A.; Goligorsky, M.S. Endothelial dysfunction is a superinducer of syndecan-4: Fibrogenic role of its ectodomain. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H484–H496. [Google Scholar] [CrossRef]
- Multhaupt, H.A.; Yoneda, A.; Whiteford, J.R.; Oh, E.S.; Lee, W.; Couchman, J.R. Syndecan signaling: When, where and why? J. Physiol. Pharmacol. 2009, 60 (Suppl. S4), 31–38. [Google Scholar]
- Donato, L.; Bramanti, P.; Scimone, C.; Rinaldi, C.; D’Angelo, R.; Sidoti, A. miRNAexpression profile of retinal pigment epithelial cells under oxidative stress conditions. FEBS Open Bio. 2018, 8, 219–233. [Google Scholar] [CrossRef] [Green Version]
- Shi, B.; Zhang, X.; Chao, L.; Zheng, Y.; Tan, Y.; Wang, L.; Zhang, W. Comprehensive analysis of key genes, microRNAs and long non-coding RNAs in hepatocellular carcinoma. FEBS Open Bio. 2018, 8, 1424–1436. [Google Scholar] [CrossRef]
- Talib, M.; van Schooneveld, M.J.; van Duuren, R.J.G.; Van Cauwenbergh, C.; Ten Brink, J.B.; De Baere, E.; Florijn, R.J.; Schalij-Delfos, N.E.; Leroy, B.P.; Bergen, A.A.; et al. Long-Term Follow-Up of Retinal Degenerations Associated With LRAT Mutations and Their Comparability to Phenotypes Associated with RPE65 Mutations. Transl. Vis. Sci. Technol. 2019, 8, 24. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ALIGNER | COMMAND LINE USED |
|---|---|
| BWA-MEM |
|
| CLC GENOMICS WORKBENCH |
|
| HISAT2 |
|
| STAR |
--genomeFastaFiles Genome_data/reference.tar.gz
--genomeLoad LoadAndKeep\ --genomeDir /path/to/genomeFasta/\ --runThreadN 4\ --outStd SAM > alignment.sam |
| RASER |
|
| TOPHAT2 |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donato, L.; Scimone, C.; Alibrandi, S.; Scalinci, S.Z.; Rinaldi, C.; D’Angelo, R.; Sidoti, A. Epitranscriptome Analysis of Oxidative Stressed Retinal Epithelial Cells Depicted a Possible RNA Editing Landscape of Retinal Degeneration. Antioxidants 2022, 11, 1967. https://doi.org/10.3390/antiox11101967
Donato L, Scimone C, Alibrandi S, Scalinci SZ, Rinaldi C, D’Angelo R, Sidoti A. Epitranscriptome Analysis of Oxidative Stressed Retinal Epithelial Cells Depicted a Possible RNA Editing Landscape of Retinal Degeneration. Antioxidants. 2022; 11(10):1967. https://doi.org/10.3390/antiox11101967
Chicago/Turabian StyleDonato, Luigi, Concetta Scimone, Simona Alibrandi, Sergio Zaccaria Scalinci, Carmela Rinaldi, Rosalia D’Angelo, and Antonina Sidoti. 2022. "Epitranscriptome Analysis of Oxidative Stressed Retinal Epithelial Cells Depicted a Possible RNA Editing Landscape of Retinal Degeneration" Antioxidants 11, no. 10: 1967. https://doi.org/10.3390/antiox11101967