
Design and Synthesis of (Z)-5-(Substituted benzylidene)-3-cyclohexyl-2-thioxothiazolidin-4-one Analogues as Anti-Tyrosinase and Antioxidant Compounds: In Vitro and In Silico Insights
 , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. General Methods
2.1.1. Synthesis of N-Cyclohexylrhodanine (9)
2.1.2. General Procedure for the Syntheses of (Z)-5-(Substituted benzylidene)-3-cyclohexyl-2-thioxothiazolidin-4-one Analogues 1–8
(Z)-3-Cyclohexyl-5-(4-hydroxybenzylidene)-2-thioxothiazolidin-4-one (1)
(Z)-3-Cyclohexyl-5-(3,4-dihydroxybenzylidene)-2-thioxothiazolidin-4-one (2)
(Z)-3-Cyclohexyl-5-(2,4-dihydroxybenzylidene)-2-thioxothiazolidin-4-one (3)
(Z)-3-Cyclohexyl-5-(4-hydroxy-3-methoxybenzylidene)-2-thioxothiazolidin-4-one (4)
(Z)-3-Cyclohexyl-5-(3-hydroxy-4-methoxybenzylidene)-2-thioxothiazolidin-4-one (5)
(Z)-3-Cyclohexyl-5-(4-hydroxy-3,5-dimethoxybenzylidene)-2-thioxothiazolidin-4-one (6)
(Z)-5-(3-Bromo-4-hydroxybenzylidene)-3-cyclohexyl-2-thioxothiazolidin-4-one (7)
(Z)-3-Cyclohexyl-5-(3,5-dibromo-4-hydroxybenzylidene)-2-thioxothiazolidin-4-one (8)
2.2. Mushroom Tyrosinase Activity Assay
2.3. In Silico Docking Simulations of Analogues 2, 3, and Kojic Acid with Mushroom Tyrosinase Using Schrödinger Suite Software
2.4. In Silico Docking Simulations of Analog 3 and Cinnamic Acid with Mushroom Tyrosinase Using AutoDock Vina Software
2.5. In Silico Docking Simulations of Analogues 2 and 3 and Kojic Acid with a Human Tyrosinase Homology Model
2.6. Kinetic Studies on Mushroom Tyrosinase Inhibition by Analogues 2 and 3
2.7. Cell Culture
2.8. Viabilities of B16F10 Murine Melanoma Cells Treated with Analogues 2 or 3
2.9. Cellular Tyrosinase Activity Assay
2.10. Melanin Content Analysis
2.11. DPPH Radical Scavenging Activity Assay
2.12. ABTS Cation Radical Scavenging Activity Assay
2.13. ROS Scavenging Activity Assay
2.14. Peroxynitrite (ONOO−) Scavenging Activity Assay
2.15. Extraction of Cytosolic and Nuclear Proteins from Cells
2.16. Western Blot Analysis
2.17. RNA Extraction and Quantitative Real Time-Polymerase Chain Reaction (RT-qPCR)
2.18. Statistical Analysis
3. Results and Discussion
3.1. Chemistry
3.2. Mushroom Tyrosinase Inhibition
3.3. In Silico Docking Simulation between Tyrosinase and Analogues 2 and 3
3.3.1. Docking Simulations of Analogues 2 and 3 with Mushroom Tyrosinase
3.3.2. Docking Simulations of Analogs 2 and 3 with Human Tyrosinase
3.4. Kinetic Study of Analogs 2 and 3
3.5. Cytotoxicities of Analogs 2 and 3
3.6. Effects of Analog 3 on Melanin Production and Tyrosinase Activity in B16F10 Cells
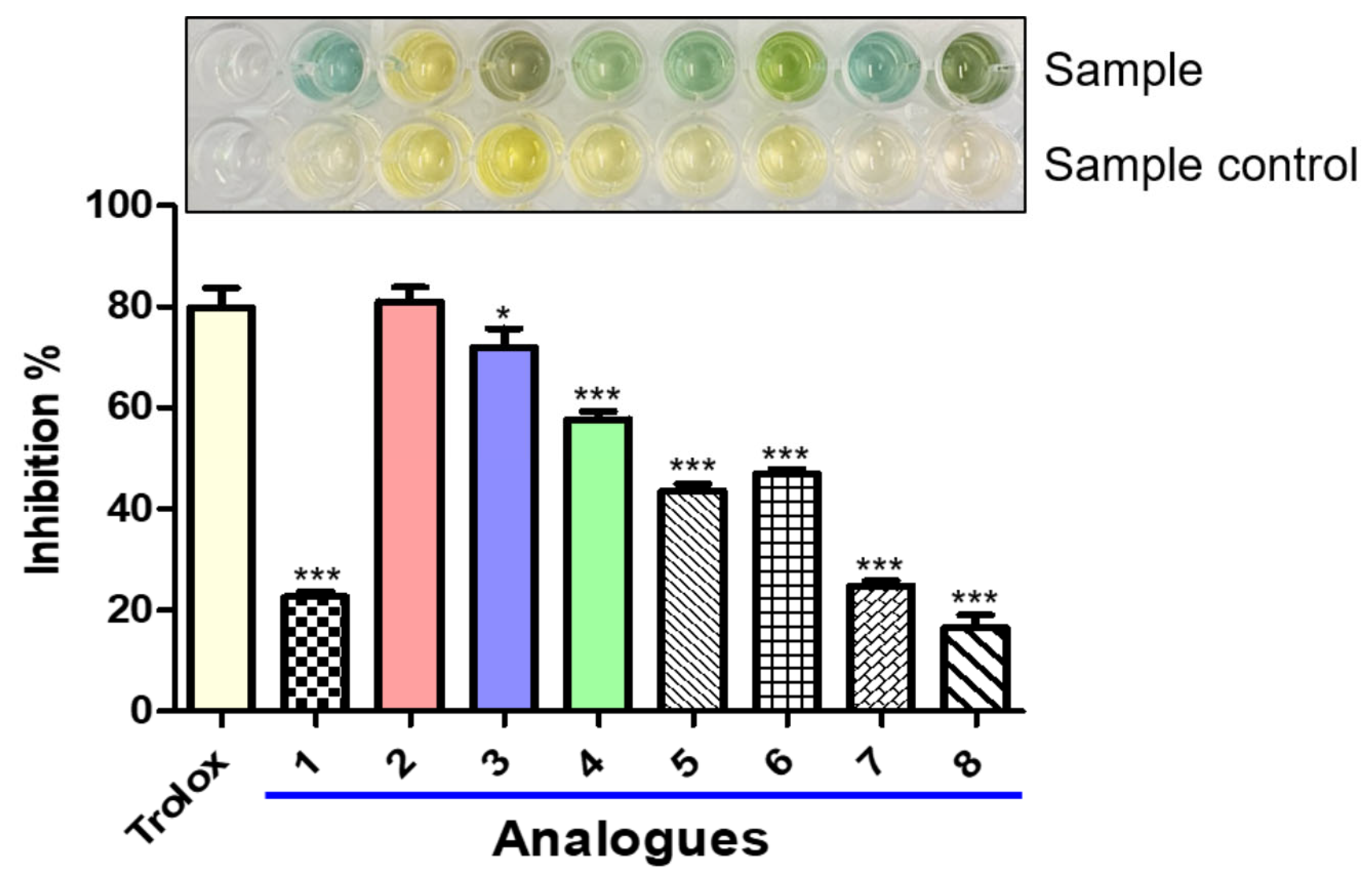
3.7. Effects of Analogs 1–8 on DPPH and ABTS Radical Scavenging Activities
3.8. ROS Scavenging Effects of Analogs 1–8
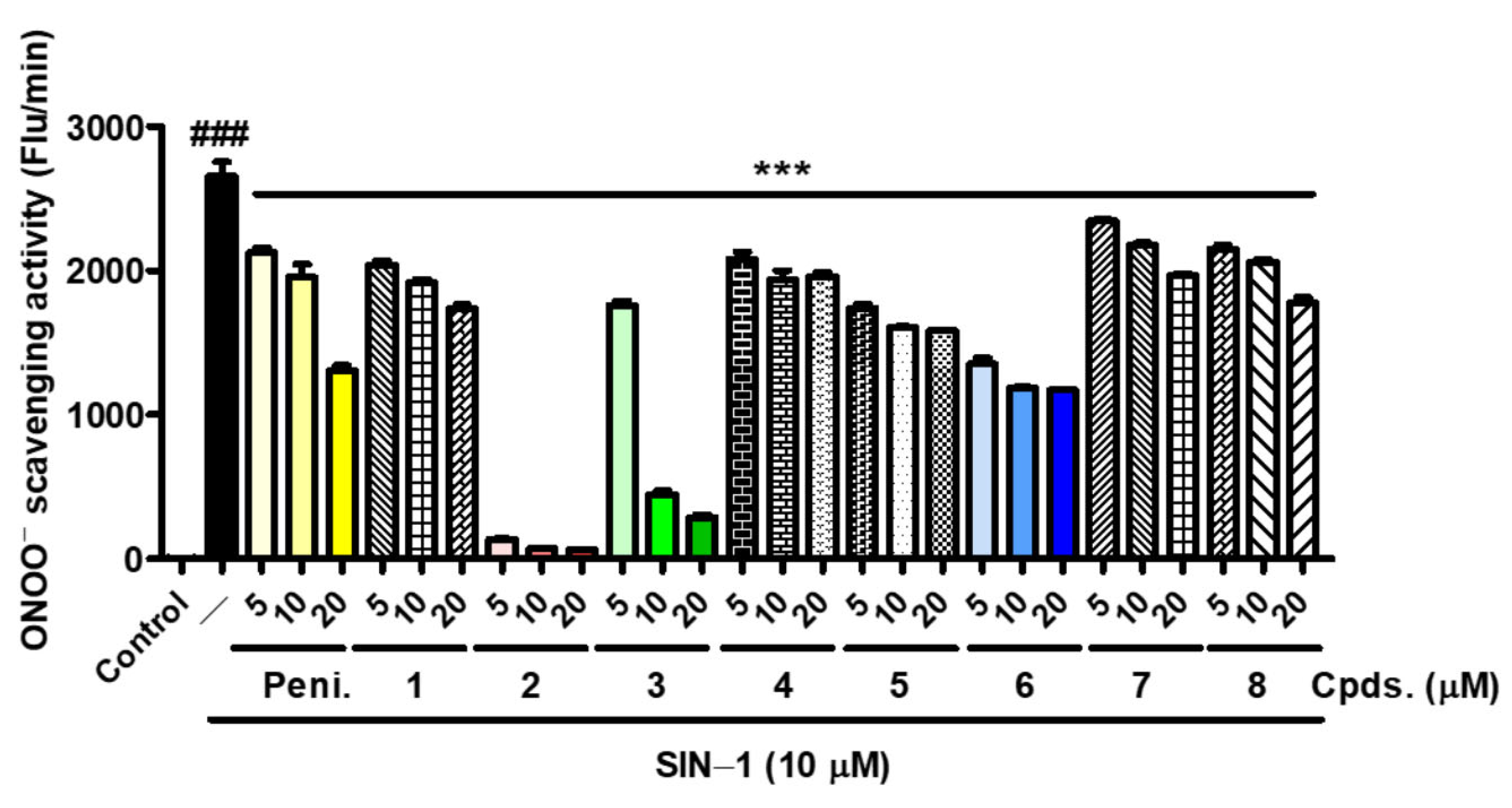
3.9. Effects of Analogs 1–8 on Peroxynitrite Scavenging Activity
3.10. Effect of Analog 3 on Melanogenesis-Related Proteins and Genes Expression in B16F10 Melanoma Cells Stimulated with α–MSH Plus IBMX
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brito, S.; Baek, J.M.; Cha, B.; Heo, H.; Lee, S.H.; Lei, L.; Jung, S.Y.; Lee, S.M.; Lee, S.H.; Kwak, B.M.; et al. Nicotinamide mononucleotide reduces melanin production in aged melanocytes by inhibiting cAMP/Wnt signaling. J. Dermatol. Sci. 2022, 106, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Weng, Q.Y.; Fisher, D.E. UV signaling pathways within the skin. J. Investig. Dermatol. 2014, 134, 2080–2085. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Young Ryu, I.; Choi, I.; Ullah, S.; Jin Jung, H.; Park, Y.; Hwang, Y.; Jeong, Y.; Hong, S.; Chun, P.; et al. Identification of (Z)-2-benzylidene-dihydroimidazothiazolone derivatives as tyrosinase inhibitors: Anti-melanogenic effects and in silico studies. Comput. Struct. Biotechnol. J. 2022, 20, 899–912. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Choi, D.C.; Noh, S.G.; Choi, H.; Choi, I.; Ryu, I.Y.; Chung, H.Y.; Moon, H.R. New Benzimidazothiazolone Derivatives as Tyrosinase Inhibitors with Potential Anti-Melanogenesis and Reactive Oxygen Species Scavenging Activities. Antioxidants 2021, 10, 1078. [Google Scholar] [CrossRef]
- Ullah, S.; Son, S.; Yun, H.Y.; Kim, D.H.; Chun, P.; Moon, H.R. Tyrosinase inhibitors: A patent review (2011–2015). Expert. Opin. Ther. Pat. 2016, 26, 347–362. [Google Scholar] [CrossRef]
- Plant, N. Strategies for using in vitro screens in drug metabolism. Drug Discov. 2004, 9, 328–336. [Google Scholar] [CrossRef]
- Kim, S.J.; Yang, J.; Lee, S.; Park, C.; Kang, D.; Akter, J.; Ullah, S.; Kim, Y.J.; Chun, P.; Moon, H.R. The tyrosinase inhibitory effects of isoxazolone derivatives with a (Z)-β-phenyl-α, β-unsaturated carbonyl scaffold. Bioorg. Med. Chem. 2018, 26, 3882–3889. [Google Scholar] [CrossRef]
- Hearing, V.J.; Tsukamoto, K. Enzymatic control of pigmentation in mammals. FASEB J. 1991, 5, 2902–2909. [Google Scholar] [CrossRef]
- Land, E.; Ito, S.; Wakamatsu, K.; Riley, P. Rate constants for the first two chemical steps of eumelanogenesis. Pigment Cell Res. 2003, 16, 487–493. [Google Scholar] [CrossRef]
- Kim, H.R.; Lee, H.J.; Choi, Y.J.; Park, Y.J.; Woo, Y.; Kim, S.J.; Park, M.H.; Lee, H.W.; Chun, P.; Chung, H.Y.; et al. Benzylidene-linked thiohydantoin derivatives as inhibitors of tyrosinase and melanogenesis: Importance of the β-phenyl-α,β-unsaturated carbonyl functionality. MedChemComm 2014, 5, 1410–1417. [Google Scholar] [CrossRef]
- Ullah, S.; Kang, D.; Lee, S.; Ikram, M.; Park, C.; Park, Y.; Yoon, S.; Chun, P.; Moon, H.R. Synthesis of cinnamic amide derivatives and their anti-melanogenic effect in α-MSH-stimulated B16F10 melanoma cells. Eur. J. Med. Chem. 2019, 161, 78–92. [Google Scholar] [CrossRef]
- Ryu, I.Y.; Choi, I.; Jung, H.J.; Ullah, S.; Choi, H.; Al-Amin, M.; Chun, P.; Moon, H.R. In vitro anti-melanogenic effects of chimeric compounds, 2-(substituted benzylidene)-1,3-indanedione derivatives with a β-phenyl-α, β-unsaturated dicarbonyl scaffold. Bioorg. Chem. 2021, 109, 104688. [Google Scholar] [CrossRef]
- Jeong, Y.; Hong, S.; Jung, H.J.; Ullah, S.; Hwang, Y.; Choi, H.; Ko, J.; Lee, J.; Chun, P.; Chung, H.Y.; et al. Identification of a Novel Class of Anti-Melanogenic Compounds, (Z)-5-(Substituted benzylidene)-3-phenyl-2-thioxothiazolidin-4-one Derivatives, and Their Reactive Oxygen Species Scavenging Activities. Antioxidants 2022, 11, 948. [Google Scholar] [CrossRef]
- Denton, C.R.; Lerner, A.B.; Fitzpatrick, T.B. Inhibition of melanin formation by chemical agents. J. Investig. Dermatol. 1952, 18, 119–135. [Google Scholar] [CrossRef]
- Fu, B.; Li, H.; Wang, X.; Lee, F.S.; Cui, S. Isolation and identification of flavonoids in licorice and a study of their inhibitory effects on tyrosinase. J. Agric. Food Chem. 2005, 53, 7408–7414. [Google Scholar] [CrossRef]
- Criton, M.; Le Mellay-Hamon, V. Analogues of N-hydroxy-N′-phenylthiourea and N-hydroxy-N′-phenylurea as inhibitors of tyrosinase and melanin formation. Bioorg. Med. Chem. Lett. 2008, 18, 3607–3610. [Google Scholar] [CrossRef]
- Song, Y.M.; Ha, Y.M.; Kim, J.-A.; Chung, K.W.; Uehara, Y.; Lee, K.J.; Chun, P.; Byun, Y.; Chung, H.Y.; Moon, H.R. Synthesis of novel azo-resveratrol, azo-oxyresveratrol and their derivatives as potent tyrosinase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 7451–7455. [Google Scholar] [CrossRef]
- Moon, K.M.; Jeong, J.W.; Lee, B.; Kim, D.H.; Kim, H.R.; Woo, Y.W.; Lee, E.K.; An, H.J.; Kim, M.J.; Choi, Y.J.; et al. Antimelanogenic activity of MHY384 via inhibition of NO-induced cGMP signalling. Exp. Dermatol. 2016, 25, 652–654. [Google Scholar] [CrossRef]
- Ullah, S.; Park, C.; Ikram, M.; Kang, D.; Lee, S.; Yang, J.; Park, Y.; Yoon, S.; Chun, P.; Moon, H.R. Tyrosinase inhibition and anti-melanin generation effect of cinnamamide analogues. Bioorg. Chem. 2019, 87, 43–55. [Google Scholar] [CrossRef]
- Maeda, K.; Fukuda, M. Arbutin: Mechanism of its depigmenting action in human melanocyte culture. J. Pharmacol. Exp. Ther. 1996, 276, 765. [Google Scholar]
- Ogiwara, Y.; Sugiura, M.; Watanabe, K.; Tawara, J.; Endo, E.; Maruyama, H.; Tsuji, S.; Matsue, K.; Yamada, H.; Wako, Y.; et al. Evaluation of the repeated-dose liver, bone marrow and peripheral blood micronucleus and comet assays using kojic acid. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 780–781, 111–116. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Namasivayam, V.; Manickam, M.; Jung, S.-H. Inhibitors of Melanogenesis: An Updated Review. J. Med. Chem. 2018, 61, 7395–7418. [Google Scholar] [CrossRef]
- Chung, K.W.; Jeong, H.O.; Jang, E.J.; Choi, Y.J.; Kim, D.H.; Kim, S.R.; Lee, K.J.; Lee, H.J.; Chun, P.; Byun, Y.; et al. Characterization of a small molecule inhibitor of melanogenesis that inhibits tyrosinase activity and scavenges nitric oxide (NO). Biochim. Biophys. Acta 2013, 1830, 4752–4761. [Google Scholar] [CrossRef]
- Kim, S.H.; Ha, Y.M.; Moon, K.M.; Choi, Y.J.; Park, Y.J.; Jeong, H.O.; Chung, K.W.; Lee, H.J.; Chun, P.; Moon, H.R.; et al. Anti-melanogenic effect of (Z)-5-(2,4-dihydroxybenzylidene) thiazolidine-2,4-dione, a novel tyrosinase inhibitor. Arch. Pharm. Res. 2013, 36, 1189–1197. [Google Scholar] [CrossRef]
- Choi, Y.J.; Uehara, Y.; Park, J.Y.; Kim, S.J.; Kim, S.R.; Lee, H.W.; Moon, H.R.; Chung, H.Y. MHY884, a newly synthesized tyrosinase inhibitor, suppresses UVB-induced activation of NF-κB signaling pathway through the downregulation of oxidative stress. Bioorg. Med. Chem. Lett. 2014, 24, 1344–1348. [Google Scholar] [CrossRef]
- Yun, H.Y.; Kim, D.H.; Son, S.; Ullah, S.; Kim, S.J.; Kim, Y.J.; Yoo, J.W.; Jung, Y.; Chun, P.; Moon, H.R. Design, synthesis, and anti-melanogenic effects of (E)-2-benzoyl-3-(substituted phenyl)acrylonitriles. Drug Des. Devel. Ther. 2015, 9, 4259–4268. [Google Scholar]
- Son, S.; Kim, H.; Yun, H.Y.; Kim, D.H.; Ullah, S.; Kim, S.J.; Kim, Y.J.; Kim, M.S.; Yoo, J.W.; Chun, P.; et al. (E)-2-Cyano-3-(substituted phenyl)acrylamide analogs as potent inhibitors of tyrosinase: A linear β-phenyl-α,β-unsaturated carbonyl scaffold. Bioorg. Med. Chem. 2015, 23, 7728–7734. [Google Scholar] [CrossRef]
- Choi, I.; Park, Y.; Ryu, I.Y.; Jung, H.J.; Ullah, S.; Choi, H.; Park, C.; Kang, D.; Lee, S.; Chun, P.; et al. In silico and in vitro insights into tyrosinase inhibitors with a 2-thioxooxazoline-4-one template. Comput. Struct. Biotechnol. J. 2021, 19, 37–50. [Google Scholar] [CrossRef]
- Hassan, M.; Ashraf, Z.; Abbas, Q.; Raza, H.; Seo, S.-Y. Exploration of Novel Human Tyrosinase Inhibitors by Molecular Modeling, Docking and Simulation Studies. Interdiscip. Sci. Comput. Life Sci. 2018, 10, 68–80. [Google Scholar] [CrossRef]
- Larik, F.A.; Saeed, A.; Channar, P.A.; Muqadar, U.; Abbas, Q.; Hassan, M.; Seo, S.-Y.; Bolte, M. Design, synthesis, kinetic mechanism and molecular docking studies of novel 1-pentanoyl-3-arylthioureas as inhibitors of mushroom tyrosinase and free radical scavengers. Eur. J. Med. Chem. 2017, 141, 273–281. [Google Scholar] [CrossRef]
- Saeed, A.; Mahesar, P.A.; Channar, P.A.; Abbas, Q.; Larik, F.A.; Hassan, M.; Raza, H.; Seo, S.-Y. Synthesis, molecular docking studies of coumarinyl-pyrazolinyl substituted thiazoles as non-competitive inhibitors of mushroom tyrosinase. Bioorg. Chem. 2017, 74, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farid, R.; Day, T.; Friesner, R.A.; Pearlstein, R.A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. [Google Scholar] [CrossRef] [PubMed]
- Bagherzadeh, K.; Shirgahi Talari, F.; Sharifi, A.; Ganjali, M.R.; Saboury, A.A.; Amanlou, M. A new insight into mushroom tyrosinase inhibitors: Docking, pharmacophore-based virtual screening, and molecular modeling studies. J. Biomol. Struct. Dyn. 2015, 33, 487–501. [Google Scholar] [CrossRef]
- Jung, H.J.; Noh, S.G.; Ryu, I.Y.; Park, C.; Lee, J.Y.; Chun, P.; Moon, H.R.; Chung, H.Y. (E)-1-(Furan-2-yl)-(substituted phenyl)prop-2-en-1-one Derivatives as Tyrosinase Inhibitors and Melanogenesis Inhibition: An In Vitro and In Silico Study. Molecules 2020, 25, 5460. [Google Scholar] [CrossRef]
- Jung, H.J.; Noh, S.G.; Park, Y.; Kang, D.; Chun, P.; Chung, H.Y.; Moon, H.R. In vitro and in silico insights into tyrosinase inhibitors with (E)-benzylidene-1-indanone derivatives. Comput. Struct. Biotechnol. J. 2019, 17, 1255–1264. [Google Scholar] [CrossRef]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Ali, S.F.; LeBel, C.P.; Bondy, S.C. Reactive oxygen species formation as a biomarker of methylmercury and trimethyltin neurotoxicity. Neurotoxicology 1992, 13, 637–648. [Google Scholar]
- LeBel, C.P.; Bondy, S.C. Sensitive and rapid quantitation of oxygen reactive species formation in rat synaptosomes. Neurochem. Int. 1990, 17, 435–440. [Google Scholar] [CrossRef]
- Kooy, N.W.; Royall, J.A.; Ischiropoulos, H.; Beckman, J.S. Peroxynitrite-mediated oxidation of dihydrorhodamine 123. Free Radic. Biol. Med. 1994, 16, 149–156. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Chung, K.W.; Park, Y.J.; Choi, Y.J.; Park, M.H.; Ha, Y.M.; Uehara, Y.; Yoon, J.H.; Chun, P.; Moon, H.R.; Chung, H.Y. Evaluation of in vitro and in vivo anti-melanogenic activity of a newly synthesized strong tyrosinase inhibitor (E)-3-(2,4 dihydroxybenzylidene)pyrrolidine-2,5-dione (3-DBP). Biochim. Biophys. Acta 2012, 1820, 962–969. [Google Scholar] [CrossRef]
- Park, J.W.; Ha, Y.M.; Moon, K.-m.; Kim, S.-r.; Jeong, H.O.; Park, Y.J.; Lee, H.J.; Park, J.Y.; Song, Y.M.; Chun, P.; et al. De novo tyrosinase inhibitor: 4-(6,7-Dihydro-5H-indeno [5,6-d]thiazol-2-yl)benzene-1,3-diol (MHY1556). Bioorg. Med. Chem. Lett. 2013, 23, 4172–4176. [Google Scholar] [CrossRef]
- Nitsche, C.; Schreier, V.N.; Behnam, M.A.M.; Kumar, A.; Bartenschlager, R.; Klein, C.D. Thiazolidinone–Peptide Hybrids as Dengue Virus Protease Inhibitors with Antiviral Activity in Cell Culture. J. Med. Chem. 2013, 56, 8389–8403. [Google Scholar] [CrossRef]
- Liang, X.; Fu, H.; Xiao, P.; Fang, H.; Hou, X. Design, synthesis and biological evaluation of imidazolidine-2, 4-dione and 2-thioxothiazolidin-4-one derivatives as lymphoid-specific tyrosine phosphatase inhibitors. Bioorg. Chem. 2020, 103, 104124. [Google Scholar] [CrossRef]
- Mandal, S.P.; Mithuna; Garg, A.; Sahetya, S.S.; Nagendra, S.R.; Sripad, H.S.; Manjunath, M.M.; Sitaram; Soni, M.; Baig, R.N.; et al. Novel rhodanines with anticancer activity: Design, synthesis and CoMSIA study. RSC Adv. 2016, 6, 58641–58653. [Google Scholar] [CrossRef]
- Vögeli, U.; von Philipsborn, W.; Nagarajan, K.; Nair, M.D. Structures of Addition Products of Acetylenedicarboxylic Acid Esters with Various Dinucleophiles. An application of C, H-spin-coupling constants. Helv. Chim. Acta 1978, 61, 607–617. [Google Scholar] [CrossRef]
- Choi, H.; Ryu, I.Y.; Choi, I.; Ullah, S.; Jung, H.J.; Park, Y.; Jeong, Y.; Hwang, Y.; Hong, S.; Yoon, I.-S.; et al. Novel Anti-Melanogenic Compounds, (Z)-5-(Substituted Benzylidene)-4-thioxothiazolidin-2-one Derivatives: In Vitro and In Silico Insights. Molecules 2021, 26, 4963. [Google Scholar] [CrossRef]
- Legoabe, L.J.; Petzer, A.; Petzer, J.P. Selected C7-substituted chromone derivatives as monoamine oxidase inhibitors. Bioorg. Chem. 2012, 45, 1–11. [Google Scholar] [CrossRef]
- Ha, Y.M.; Kim, J.-A.; Park, Y.J.; Park, D.; Choi, Y.J.; Kim, J.M.; Chung, K.W.; Han, Y.K.; Park, J.Y.; Lee, J.Y.; et al. Synthesis and biological activity of hydroxybenzylidenyl pyrrolidine-2,5-dione derivatives as new potent inhibitors of tyrosinase. MedChemComm 2011, 2, 542–549. [Google Scholar] [CrossRef]
- Ha, Y.M.; Kim, J.-A.; Park, Y.J.; Park, D.; Kim, J.M.; Chung, K.W.; Lee, E.K.; Park, J.Y.; Lee, J.Y.; Lee, H.J.; et al. Analogs of 5-(substituted benzylidene)hydantoin as inhibitors of tyrosinase and melanin formation. Biochim. Biophys. Acta BBA Gen. Subj. 2011, 1810, 612–619. [Google Scholar] [CrossRef]
- Ha, Y.M.; Park, Y.J.; Kim, J.-A.; Park, D.; Park, J.Y.; Lee, H.J.; Lee, J.Y.; Moon, H.R.; Chung, H.Y. Design and synthesis of 5-(substituted benzylidene)thiazolidine-2,4-dione derivatives as novel tyrosinase inhibitors. Eur. J. Med. Chem. 2012, 49, 245–252. [Google Scholar] [CrossRef]
- Jung, H.J.; Lee, A.K.; Park, Y.J.; Lee, S.; Kang, D.; Jung, Y.S.; Chung, H.Y.; Moon, H.R. (2E,5E)-2,5-Bis(3-hydroxy-4-methoxybenzylidene) cyclopentanone Exerts Anti-Melanogenesis and Anti-Wrinkle Activities in B16F10 Melanoma and Hs27 Fibroblast Cells. Molecules 2018, 23, 1415. [Google Scholar] [CrossRef] [Green Version]
- Yin, S.J.; Si, Y.X.; Qian, G.Y. Inhibitory effect of phthalic Acid on tyrosinase: The mixed-type inhibition and docking simulations. Enzyme Res. 2011, 2011, 294724. [Google Scholar] [CrossRef]
- Logan, D.W.; Burn, S.F.; Jackson, I.J. Regulation of pigmentation in zebrafish melanophores. Pigment. Cell Res. 2006, 19, 206–213. [Google Scholar] [CrossRef]
- Chang, T.-S. Natural Melanogenesis Inhibitors Acting Through the Down-Regulation of Tyrosinase Activity. Materials 2012, 5, 1661–1685. [Google Scholar] [CrossRef]
- Ashooriha, M.; Khoshneviszadeh, M.; Khoshneviszadeh, M.; Moradi, S.E.; Rafiei, A.; Kardan, M.; Emami, S. 1, 2, 3-Triazole-based kojic acid analogs as potent tyrosinase inhibitors: Design, synthesis and biological evaluation. Bioorg. Chem. 2019, 82, 414–422. [Google Scholar] [CrossRef]
- Li, Q.; Mo, J.; Xiong, B.; Liao, Q.; Chen, Y.; Wang, Y.; Xing, S.; He, S.; Lyu, W.; Zhang, N.; et al. Discovery of Resorcinol-Based Polycyclic Structures as Tyrosinase Inhibitors for Treatment of Parkinson’s Disease. ACS Chem. Neurosci. 2022, 13, 81–96. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, S.J.; Ullah, S.; Yun, H.Y.; Chun, P.; Moon, H.R. Design, synthesis, and antimelanogenic effects of (2-substituted phenyl-1,3-dithiolan-4-yl)methanol derivatives. Drug Des. Devel. Ther. 2017, 11, 827–836. [Google Scholar] [CrossRef]
- Venza, I.; Venza, M.; Visalli, M.; Lentini, G.; Teti, D.; d’Alcontres, F.S. ROS as Regulators of Cellular Processes in Melanoma. Oxid. Med. Cell. Longev. 2021, 2021, 1208690. [Google Scholar] [CrossRef]
- Padmaja, S.; Madison, S. Reaction of peroxynitrite with the melanin precursor, 5, 6-dihydroxyindole-2-carboxylic acid. Res. Chem. Intermed. 1999, 25, 441–458. [Google Scholar] [CrossRef]
- Kim, Y.J.; Yokozawa, T. Modulation of oxidative stress and melanogenesis by proanthocyanidins. Biol. Pharm. Bull. 2009, 32, 1155–1159. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, Y.; Yuan, L.; Fu, L.; Mei, C. Rosiglitazone inhibits insulin-like growth factor-1-induced polycystic kidney disease cell growth and p70S6 kinase activation. Mol. Med. Rep. 2013, 8, 861–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Gene Accession Number a | Ensembl Version a | Primers Sequence (Forward: 5’→3’, Reverse: 3’→5’) |
|---|---|---|---|
| Tyrosinase | NC_000073.7 | ENSMUSG00000004651.7 | Forward = GGAACAGCAACGAGCTAAGG Reverse = TGATGATCCGATTCACCAGA |
| TRP-1 | NC_000070.7 | ENSMUSG00000005994.15 | Forward = GGAACAGCAACGAGCTAAGG Reverse = TGATGATCCGATTCACCAGA |
| GAPDH | NC_000072.7 | ENSMUSG00000057666 | Forward = ACCACAGTCCATGCCATCAC Reverse = CCACCACCCTGTTGCTGTAG |
 | |||||||
| Compounds | R1 | R2 | R3 | R4 | IC50 Values (µM) a | Synthetic Yield (%) | Log p b |
| 1 | H | H | OH | H | 65.39 ± 7.82 | 91 | 3.43 |
| 2 | H | OH | OH | H | 5.21 ± 0.86 | 89 | 3.04 |
| 3 | OH | H | OH | H | 1.03 ± 0.14 | 65 | 3.04 |
| 4 | H | OMe | OH | H | >200 | 84 | 3.30 |
| 5 | H | OH | OMe | H | 54.98 ± 11.91 | 87 | 3.30 |
| 6 | H | OMe | OH | OMe | >200 | 65 | 3.18 |
| 7 | H | Br | OH | H | 66.23 ± 9.44 | 79 | 4.26 |
| 8 | H | Br | OH | Br | >200 | 89 | 5.09 |
| KA c | 25.26 ± 1.10 | −2.45 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ko, J.; Lee, J.; Jung, H.J.; Ullah, S.; Jeong, Y.; Hong, S.; Kang, M.K.; Park, Y.J.; Hwang, Y.; Kang, D.; et al. Design and Synthesis of (Z)-5-(Substituted benzylidene)-3-cyclohexyl-2-thioxothiazolidin-4-one Analogues as Anti-Tyrosinase and Antioxidant Compounds: In Vitro and In Silico Insights. Antioxidants 2022, 11, 1918. https://doi.org/10.3390/antiox11101918
Ko J, Lee J, Jung HJ, Ullah S, Jeong Y, Hong S, Kang MK, Park YJ, Hwang Y, Kang D, et al. Design and Synthesis of (Z)-5-(Substituted benzylidene)-3-cyclohexyl-2-thioxothiazolidin-4-one Analogues as Anti-Tyrosinase and Antioxidant Compounds: In Vitro and In Silico Insights. Antioxidants. 2022; 11(10):1918. https://doi.org/10.3390/antiox11101918
Chicago/Turabian StyleKo, Jeongin, Jieun Lee, Hee Jin Jung, Sultan Ullah, Yeongmu Jeong, Sojeong Hong, Min Kyung Kang, Yu Jung Park, YeJi Hwang, Dongwan Kang, and et al. 2022. "Design and Synthesis of (Z)-5-(Substituted benzylidene)-3-cyclohexyl-2-thioxothiazolidin-4-one Analogues as Anti-Tyrosinase and Antioxidant Compounds: In Vitro and In Silico Insights" Antioxidants 11, no. 10: 1918. https://doi.org/10.3390/antiox11101918