
Effects of Erythrodiol on the Antioxidant Response and Proteome of HepG2 Cells

 , , , ,
, , , ,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. EO Solution
2.3. MTT Assay
2.4. Measurement of ROS Production
2.5. Enzyme Activity Assays
2.5.1. Protein Extracts
2.5.2. Superoxide Dismutase (SOD) Assay
2.5.3. Catalase Assay
2.5.4. Glutathione Peroxidase Assay
2.5.5. Glutathione Reductase Assay
2.5.6. Glutathione S-Transferase Assay
2.5.7. Glucose 6-Phosphate Dehydrogenase and 6-Phosphogluconate Dehydrogenase Enzymes
2.5.8. Determination of Protein Concentration
2.6. Measurement of Antioxidant Metabolite Concentrations
2.6.1. Measurement of the Concentrations of GSSG and GSH
2.6.2. Measurement of Concentrations of NADP+ and NADPH
2.7. nLC–MS Proteomic Method
2.7.1. Protein Extraction
2.7.2. Data Analysis
2.7.3. Label-Free Quantitative Data Analysis in PERSEUS Software
2.7.4. Functional Data Analysis
2.8. Statistical Analysis
3. Results
3.1. EO Has a Cytotoxic Effect on HepG2 Cells
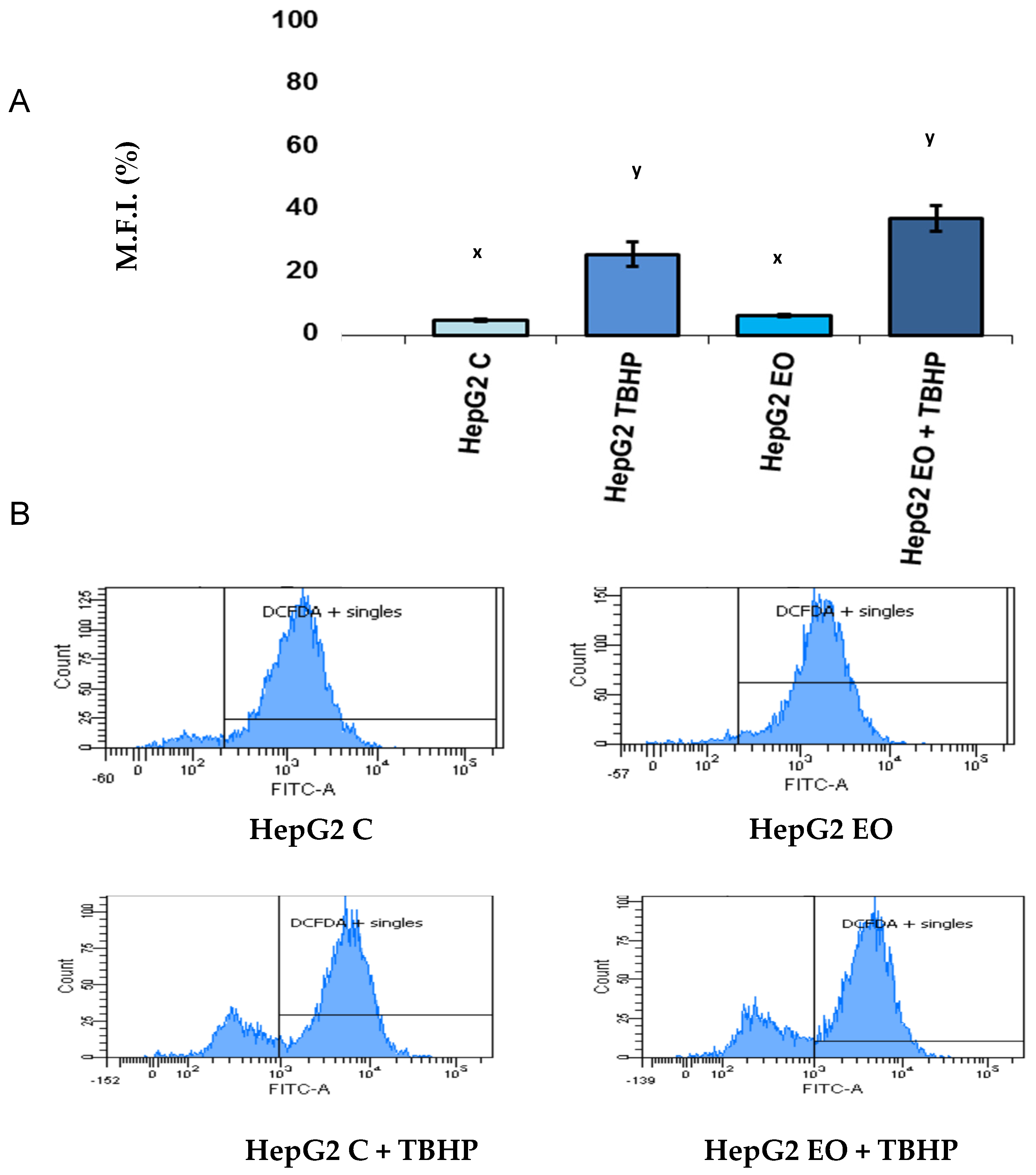
3.2. EO Does Not Change ROS Production in HepG2 Cells
3.3. EO Produces Selective Changes in the Specific Activity of Antioxidant Enzymes
3.4. EO Decreases the Levels of Antioxidant Metabolites
3.5. Changes in the Proteome of HepG2 Cells in the Presence of EO
4. Discussion
4.1. EO Has a Cytotoxic Effect on HepG2 Cells
4.2. EO Does Not Change Intracellular ROS Production, Decreases the Levels of Antioxidant Metabolites, and Produces Selective Changes in the Specific Activity of Antioxidant Enzymes in HepG2 Cells
4.3. Changes in the Proteome of HepG2 Cells in the Presence of EO
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peragón, J. Time course of pentacyclic triterpenoids from fruits and leaves of olive tree (Olea europaea L.) cv. Picual and cv. Cornezuelo during ripening. J. Agric. Food Chem. 2013, 61, 6671–6678. [Google Scholar] [CrossRef]
- Blanch, G.P.; Villen, J.; Herraiz, M. Rapid analysis of free erythrodiol and uvaol in olive oils by coupled reversed phase liquid chromatography gas chromatography. J. Agric. Food Chem. 1998, 46, 1027–1030. [Google Scholar] [CrossRef]
- Allouche, Y.; Jiménez, A.; Uceda, M.; Aguilera, M.P.; Gaforio, J.J.; Beltrán, G. Triterpenic content and chemometric analysis of virgin olive oils from forty olive cultivars. J. Agric. Food Chem. 2009, 57, 3604–3610. [Google Scholar] [CrossRef] [PubMed]
- Stiti, N.; Triki, S.; Hartmann, M.-A. Formation of triterpenoids throughout Olea europaea fruit ontogeny. Lipids 2007, 42, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Allouche, Y.; Warleta, F.; Campos, M.; Sánchez-Quesada, C.; Uceda, M.; Beltrán, G.; Gaforio, J.J. Antioxidant, antiproliferative, and pro-apoptotic capacities of pentayclic triterpenes found in the skin of olives on MCF-7 human breast cancer cells and their effects on DNA damage. J. Agric. Food Chem. 2011, 59, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Juan, M.E.; Wenzel, U.; Daniel, H.; Planas, J.M. Erythrodiol, a natural triterpenoid from olives, has antiproliferative and apoptotic activity in HT-29 human adenocarcinoma cells. Mol. Nutr. Food Res. 2008, 52, 595–599. [Google Scholar] [CrossRef]
- Martín, R.; Ibeas, E.; Carvalho-Tavares, J.; Hernández, M.; Ruiz-Gutiérrez, V.; Nieto, M.L. Natural triterpenic diols promote apoptosis in astrocytoma cells through ROS-mediated mitochondrial depolarization and JNK activation. PLoS ONE 2009, 4, e5975. [Google Scholar] [CrossRef] [Green Version]
- Máñez, S.; Recio, M.C.; Giner, R.M.; Ríos, J.L. Effect of selected triterpenoids on chronic dermal inflammation. Eur. J. Pharmacol. 1997, 334, 103–105. [Google Scholar] [CrossRef]
- De la Puerta, R.; Martínez-Domínguez, E.; Ruiz-Gutiérrez, V. Effect of minor components of virgin olive oil on topical aintiinflammatory assays. Z. Naturforsch. 2000, 55c, 814–819. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Rodríguez, R.; Herrera, M.D.; Perona, J.S.; Ruíz-Gutiérrez, V. Potential vasorelaxant effects of oleanolic acid and erythrodiol, two triterpenoids contained in “orujo” olive oil, on rat aorta. Brit. J. Nutr. 2004, 92, 635–642. [Google Scholar] [CrossRef] [Green Version]
- Márquez-Martín, A.; De La Puerta, R.; Fernández-Arche, A.; Ruiz-Gutiérrez, V.; Yanqoob, P. Modulation of cytokine secretion by pentacyclic triterpenes from olive pomace oil in human mononuclear cells. Cytokine 2006, 36, 211–217. [Google Scholar] [CrossRef]
- Bonel-Pérez, G.C.; Pérez-Jiménez, A.; Gris-Cárdenas, I.; Parra-Pérez, A.M.; Lupiáñez, J.L.; Reyes-Zurita, F.J.; Siles, E.; Csuk, R.; Peragón, J.; Rufino-Palomares, E.E. Antiproliferative and pro-apoptotic effect of uvaol in human hepatocarcinoma HepG2 cells by affecting G0/G1 cell cycle arrest, ROS production and AKT/PI3K signalling pathway. Molecules 2020, 25, 4254. [Google Scholar] [CrossRef]
- Desagher, S.; Martinou, J.C. Mitochondria as the central control of apoptosis. Trends Cell Biol. 2000, 10, 369–377. [Google Scholar] [CrossRef]
- Wei, J.; Liu, M.; Liu, H.; Wang, H.; Wang, F.; Zhang, Y.; Han, L.; Lin, X. Oleanolic acid arrests cell cycle and induces apoptosis via ROS-mediated mitochondrial depolarization and lysosomal membrane permeabilization in human pancreatic cancer cells. J. Appl. Toxicol. 2013, 33, 756–765. [Google Scholar] [CrossRef]
- Pilz, R.B.; Willis, R.C.; Boss, G.R. The influence of ribose 5-phosphate availability on purine synthesis of cultured human lymphoblasts and mitogen-stimulated lymphocytes. J. Biol. Chem. 1984, 259, 2927–2935. [Google Scholar] [CrossRef]
- Rufino-Palomares, E.E.; Reyes-Zurita, F.J.; García-Salguero, L.; Peragón, J.; de la Higuera, M.; Lupiáñez, J.A. The role of maslinic acid in the pentose phosphate pathway during growth of Gilthead Sea bream (Sparus aurata). Aquac. Nutr. 2013, 19, 709–720. [Google Scholar] [CrossRef]
- Mokhtari, K.; Rufino-Palomares, E.E.; Pérez-Jiménez, A.; Reyes-Zurita, F.J.; Figuera, C.; García-Salguero, L.; Medina, P.P.; Peragón, J.; Lupiáñez, J.A. Maslinic acid, a triterpene from olive, affects the antioxidant and mitochondrial status of B16F10 melanoma cells grown under stressful conditions. Evid.-Based Complementary Altern. Med. 2015, 2015, 272457. [Google Scholar]
- Mokhtari, K.; Pérez-Jiménez, A.; García-Salguero, L.; Lupiáñez, J.A.; Rufino-Palomares, E.E. Unveiling the differential antioxidant activity of maslinic acid in murine melanoma cells and in rat embryonic healthy cells following treatment with hydrogen peroxide. Molecules 2020, 25, 4020. [Google Scholar] [CrossRef]
- Šrejber, M.; Navrátilová, V.; Paloncýová, M.; Bazgier, V.; Berka, K.; Anzenbacher, P.; Otyepka, M. Membrane-attached mammalian cytochromes P450: An overview of the membrane’s effects on structure, drug binding, and interactions with redox partners. J. Inorg. Biochem. 2018, 183, 117–136. [Google Scholar] [CrossRef] [PubMed]
- Nóbrega-Pereira, S.; Fernández-Marcos, P.J.; Brioche, T.; Gómez-Cabrera, M.C.; Salvador-Pascual, A.; Flores, J.M.; Viña, J.; Serrano, M. G6PD protects from oxidative damage and improves health span in mice. Nature Comm. 2006, 7, 10894. [Google Scholar] [CrossRef] [Green Version]
- Peragón, J.; Barroso, J.B.; de la Higuera, M.; Lupiáñez, J.A. Relationship between growth and protein turnover and nucleic acids in the liver of rainbow trout (Oncorhynchus mykiss) during development. Can. J. Fish. Aquat. Sci. 1998, 55, 649–657. [Google Scholar] [CrossRef]
- Peragón, J.; Barroso, J.B.; García-Salguero, L.; Aranda, F.; de la Higuera, M.; Lupiáñez, J.A. Selective changes in the protein-turnover rates and nature of growth induced in trout liver by long-term starvation followed by re-feeding. Mol. Cell. Biochem. 1999, 201, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Peragón, J.; Barroso, J.B.; García-Salguero, L.; de la Higuera, M.; Lupiáñez, J.A. Dietary alterations in protein, carbohydrates and fat increases liver-protein turnover rate and decrease overall growth rate in the rainbow trout (Oncorhynchus mykiss). Mol. Cell. Biochem. 2000, 209, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Lupiáñez, J.A.; Adroher, F.-J.; Vargas, A.M.; Osuna, A. Differential behavior of glucose 6-phosphate dehydrogenase in two morphological forms of Trypanosoma cruzi. Int. J. Biochem. 1987, 19, 1085–1089. [Google Scholar] [CrossRef]
- Adroher, F.J.; Osuna, A.; Lupiáñez, J.A. Differential energetic metabolism during Trypanosoma cruzi differentiation. I. Citrate synthase, NADP-isocitrate and succinate dehydrogenases. Arch. Biochem. Biophys. 1988, 267, 252–261. [Google Scholar] [CrossRef]
- Adroher, F.J.; Osuna, A.; Lupiáñez, J.A. Differential energetic metabolism during Trypanosoma cruzi differentiation. II. Hexokinase, phosphofructokinase and pyruvate kinase. Mol. Cell. Biochem. 1990, 94, 71–82. [Google Scholar] [CrossRef]
- Peragón, J.; Barroso, J.B.; García-Salguero, L.; de la Higuera, M.; Lupiáñez, J.A. Dietary protein effects on growth and fractional protein synthesis and degradation rates in rainbow trout (Oncorhynchus mykiss) liver and white muscle. Aquaculture 1994, 124, 35–46. [Google Scholar] [CrossRef]
- Barroso, J.B.; Peragón, J.; García-Salguero, L.; Aranda, F.; de la Higuera, M.; Lupiáñez, J.A. Carbohydrate deprivation reduces NADPH-production in fish liver but not in adipose tissue. Int. J. Biochem. Cell Biol. 2001, 33, 785–796. [Google Scholar] [CrossRef]
- Aghdam, M.S.; Palma, J.M.; Corpas, F.J. NADPH as a quality footprinting in horticultural crops marketability. Trends Food Sci. Technol. 2020, 103, 152–161. [Google Scholar] [CrossRef]
- Knasmuller, S.; Parzefall, W.; Sanyal, R.; Ecker, S.; Schwab, C.; Uhl, M.; Mersch-Sundermann, V.; Williamson, G.; Hietsch, G.; Langer, T.; et al. Use of metabolically competent human hepatoma cells for the detection of mutagens and antimutagens. Mutat. Res. 1998, 402, 185–202. [Google Scholar] [CrossRef]
- Knasmuller, S.; Mersch-Sundermann, V.; Kevekordes, S.; Darroudi, F.; Huber, W.W.; Hoelzl, C.; Bichler, J.; Majer, B.J. Use of human-derived liver cell lines for the detection of environmental and dietary genotoxicants: Current state of knowledge. Toxicology 2004, 198, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Mersch-Sundermann, V.; Knasmuller, S.; Wu, X.J.; Darroudi, F.; Kassie, F. Use of a human-derived liver cell line for the detection of cytoprotective, antigenotoxic and cogenotoxic agents. Toxicology 2004, 198, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Jiménez, A.; Rufino-Palomares, E.E.; Fernández-Gallego, N.; Ortuño-Costela, M.C.; Reyes-Zurita, F.J.; Peragón, J.; García-Salguero, L.; Mokhtari, K.; Medina, P.P.; Lupiáñez, J.A. Target Molecules in 3T3-L1adipocytes differentiation are regulated by maslinic acid, a natural triterpene from Olea europaea. Phytomedicine 2016, 23, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Lebel, C.P.; Bondy, S.C. Sensitive and rapid quantitation of oxygen reactive species formation in rat synaptosomes. Neurochem. Int. 1990, 17, 435–440. [Google Scholar] [CrossRef] [Green Version]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Aebi, H. Catalase in vitro. Methods Enzymol. 1984, 105, 121–126. [Google Scholar]
- Lawrence, R.A.; Burk, R.F. Glutahione peroxidase activity in selenium-deficient rat liver. Biochem. Biophys. Res. Comm. 1976, 71, 952–958. [Google Scholar] [CrossRef]
- Carlberg, I.; Mannervik, B. Purification and characterization of the flavoenzyme glutathione reductase from rat liver. J. Biol. Chem. 1975, 250, 5475–5480. [Google Scholar] [CrossRef]
- Habig, W.H.; Pabst, M.J.; Jakoby, W.B. Glutathione S transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 1974, 249, 7130–7139. [Google Scholar] [CrossRef]
- Peragón, J.; Aranda, F.; García-Salguero, L.; Corpas, F.J.; Lupiáñez, J.A. Stimulation of rat-kidney hexose monophosphate shunt dehydrogenase activity by chronic metabolic acidosis. Biochem. Int. 1989, 18, 1041–1059. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Rahman, I.; Kode, A.; Biswas, S.K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 2007, 1, 3159–3165. [Google Scholar] [CrossRef]
- Zhang, Z.; Yu, J.; Stanton, R.C. A method for determination of pyridine nucleotides using a single extract. Anal. Biochem. 2000, 285, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Fernández, B.; Fuentes-Almagro, C.; Peragón, J. Proteomics analysis reveals the implications of cytoskeleton and mitochondria in the response of the rat brain to starvation. Nutrients 2019, 11, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, J.; Mann, M. MaxQant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome, wide protein identification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Glutathione and apoptosis. Free Radic. Res. 2008, 42, 689–706. [Google Scholar] [CrossRef] [Green Version]
- Ballesteros-Vivas, D.; Álvarez-Rivera, G.; León, C.; Morantes, S.J.; Ibáñez, D.; Parada-Alfonso, F.; Cifuentes, A.; Valdés, A. Anti-proliferative bioactivity against HT-29 colon cancer cells of a withanolides-rich extract golden berry (Physalis peruviana L.) calyx investigated by Foodomics. J. Funct. Foods 2019, 63, 103567. [Google Scholar] [CrossRef]
- Dudgeon, C.; Qiu, W.; Sun, Q.; Zhang, L.; Yu, J. Transcriptional regulation of apoptosis. In Essentials of Apoptosis. A Guide for Basic and Clinical Research, 2nd ed.; Yin, X.M., Dong, Z., Eds.; Humana Press: Totowa, NJ, USA, 2009; Chapter 10; pp. 239–260. [Google Scholar]
- Rojas López, A.; Monzón, P.; Acerenza, L. A model for the regulation of apoptosis intrinsic pathway: The potential role of the transcriptional regulator E2F in the point of no return. J. Theo. Biol. 2021, 525, 110765. [Google Scholar] [CrossRef] [PubMed]
- Broeker, C.D.; Andrechek, E.R. E2F transcription factors in cancer, more than the cell cycle. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar] [CrossRef]
- Garneau, H.; Alvarez, L.; Paquin, M.C.; Lussier, C.; Rancourt, C.; Beaulieu, J.F.; Rivard, N. Nuclear expression of E2F4 induces cell death via multiple pathways in normal human intestinal epithelial crypt cells but not in colon cancer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G758–G772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, L.A.; Goluszko, E.; Chen, H.-Z.; Leone, G.; Post, S.; Lozano, G.; Chen, Z.; Chauchereau, A. E2F3 is a mediator of DNA damage-induced apoptosis. Mol. Cell. Biol. 2010, 30, 524–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeGregori, J.; Johnson, D.G. Distinct and overlapping roles for E2F family members in transcription, proliferation and apoptosis. Curr. Mol. Med. 2006, 6, 739–748. [Google Scholar] [PubMed]
- Johnson, D.G.; DeGregori, J. Putting the oncogenic and tumor suppressive activities of E2F into context. Curr. Mol. Med. 2006, 6, 731–738. [Google Scholar] [PubMed]
- Stark, G.R.; Taylor, W.R. Control of the G2/M transition. Mol. Biotechnol. 2006, 32, 227–248. [Google Scholar] [CrossRef]
- Frakhri, S.; Moradi, S.Z.; Farzaei, M.H.; Bishayee, A. Modulation of dysregulated cancer metabolism by plant secondary metabolites: A mechanistic review. Sem. Cancer Biol. 2020, 17, 1–30. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Control | EO | |
|---|---|---|
| SOD | 2.89 ± 0.05 a | 3.09 ± 0.25 a |
| CAT | 54.03 ± 1.91 a | 55.82 ± 2.60 a |
| GPX | 1.44 ± 0.17 a | 2.31 ± 0.14 b |
| GR | 54.58 ± 6.42 a | 40.48 ± 4.67 b |
| GST | 6.78 ± 0.25 a | 5.50 ± 0.59 a |
| G6PDH | 16.72 ± 1.74 a | 27.79 ± 1.05 b |
| 6PGDH | 75.44 ± 1.38 a | 82.89 ± 1.52 b |
| Control | EO | |
|---|---|---|
| [GSH] | 2571.00 ± 117.24 a | 503.10 ± 38.64 b |
| [GSSG] | 544.4 ± 27.84 a | 352.18 ± 14.86 b |
| [GSH] + [GSSG] | 3115.4 ± 150.68 a | 855.28 ± 50.91 b |
| [GSH]/[GSSG] | 4.72 ± 0.23 a | 1.43 ± 0.08 b |
| [NADPH] | 547.62 ± 21.79 a | 121.30 ± 8.14 b |
| [NADP+] | 609.26 ± 32.69 a | 423.11 ± 24.79 b |
| [NADPH]/[NADP+] | 0.90 ± 0.04 a | 0.29 ± 0.02 b |
| Protein IDs a | Gene Name | Protein Name | Score b | Coverage (%) c | Peptides d | kDa | Intensity | |
|---|---|---|---|---|---|---|---|---|
| 1 | A0A0G2JY07 | Mcm5 | DNA replication licensing factor MCM5 | 9.8013 | 12.1 | 7 | 82.467 | 90,380,000 |
| 2 | Q5HZY3 | Uchl5 | Ubiquitin carboxyl-terminal hydrolase | 13.129 | 22.5 | 5 | 37.097 | 97,660,000 |
| 3 | A0A0G2K3Q6 | Aldoc | Fructose-bisphosphate aldolase C | 3.8455 | 22.5 | 7 | 40.484 | 44,823,000 |
| 4 | F7EWC1 | Vasp | Vasodilator-stimulated phosphoprotein | 4.3293 | 3.5 | 1 | 39.485 | 152,990,000 |
| 5 | F7FG31 | Ctbp1 | C-terminal-binding protein 1 | 6.0241 | 11.7 | 4 | 43.847 | 67,520,000 |
| 6 | B4F786 | Cd2bp2 | CD2 antigen (Cytoplasmic tail) binding protein 2 (Predicted), isoform CRA_a | 3.1481 | 6.2 | 2 | 37.54 | 63,614,000 |
| 7 | B5DEX9 | Arid3a | AT-rich interaction domain 3A | 9.361 | 10.8 | 5 | 63.594 | 79,387,000 |
| 8 | D3ZAZ0 | Eif3m | Eukaryotic translation initiation factor 3 subunit M | 102.58 | 28.6 | 8 | 42.516 | 298,630,000 |
| 9 | D3ZD89 | Naa15 | N(alpha)-acetyltransferase 15, NatA auxiliary subunit | 22.247 | 5.2 | 3 | 101.01 | 51,300,000 |
| 10 | D3ZDK7 | Pgp | Glycerol-3-phosphate phosphatase | 25.181 | 17.1 | 4 | 34.6 | 141,480,000 |
| 11 | D3ZQ74 | Plod1 | Procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1 | 197.95 | 7.1 | 3 | 83.612 | 137,530,000 |
| 12 | D3ZU74 | Dync1i2 | Cytoplasmic dynein 1 intermediate chain 2 | 12.408 | 4.2 | 2 | 68.362 | 111,420,000 |
| 13 | D3ZVK3 | Trmt6 | tRNA (adenine(58)-N(1))-methyltransferase non-catalytic subunit TRM6 | 4.0587 | 4 | 2 | 55.278 | 57,655,000 |
| 14 | D3ZW08 | Adsl | Adenylosuccinate lyase | 14.433 | 6.6 | 3 | 54.852 | 56,954,000 |
| 15 | D4A355 | Mastl | Microtubule-associated serine/threonine-protein kinase-like | 2.5367 | 2.3 | 1 | 96.233 | 5,377,500,000 |
| 16 | D4ADZ9 | Pus7 | Pseudouridine synthase 7 | 5.5004 | 5.8 | 3 | 74.64 | 59,883,000 |
| 17 | D4AEP0 | Adss | Adenylosuccinate synthetase isozyme 2 | 12.613 | 17.8 | 5 | 50.085 | 206,830,000 |
| 18 | F1LPD6 | Acaa1b | 3-ketoacyl-CoA thiolase A, peroxisomal | 5.6309 | 7.2 | 3 | 44.524 | 123,980,000 |
| 19 | F1LVV4 | Rcc2 | Regulator of chromosome condensation 2 | 29.009 | 26.9 | 7 | 46.652 | 155,410,000 |
| 20 | F1LXV3 | Stk26 | Serine/threonine kinase 26 | 3.486 | 5.3 | 2 | 46.573 | 80,258,000 |
| 21 | F1M949 | Ckap5 | Cytoskeleton-associated protein 5 | 5.8607 | 2.1 | 3 | 187.92 | 53,088,000 |
| 22 | G3V8R0 | RGD1311703 | Small acidic protein | 28.294 | 28.2 | 4 | 19.961 | 65,264,000 |
| 23 | G3V9I9 | Srek1 | Splicing regulatory glutamine/lysine-rich protein 1 | 3.0994 | 5.2 | 2 | 69.235 | 38,385,000 |
| 24 | P09606 | Glul | Glutamine synthetase | 50.405 | 9.9 | 3 | 42.267 | 196,290,000 |
| 25 | P46413 | Gss | Glutathione synthetase | 12.553 | 15.2 | 5 | 52.344 | 170,440,000 |
| 26 | P62142 | Ppp1cb | Ser/thr-protein phosphatase PP1-beta catalytic subunit | 5.3181 | 40.4 | 10 | 37.186 | 266,690,000 |
| 27 | Q64560 | Tpp2 | Tripeptidyl-peptidase 2 | 4.8866 | 2.5 | 3 | 138.29 | 40,240,000 |
| 28 | Q6PEC1 | Tbca | Tubulin-specific chaperone A | 7.2779 | 17.6 | 2 | 12.744 | 172,870,000 |
| 29 | Q9ES53 | Ufd1l | Ubiquitin fusion degradation protein 1 homolog | 6.6969 | 7.5 | 2 | 34.485 | 105,660,000 |
| Protein IDs a | Gene Names | Protein Names | Score b | Coverage (%) c | Peptides d | kDa | Intensity | |
|---|---|---|---|---|---|---|---|---|
| 1 | A0A096MJA9 | Asph | Aspartyl/asparaginyl beta-hydroxylase | 4.0672 | 4.3 | 2 | 116.29 | 138,230,000 |
| 2 | A0A096MK75 | Rhog | Rho-related GTP-binding protein RhoG | 7.9252 | 33.1 | 3 | 67.165 | 153,310,000 |
| 3 | A0A0G2JVW5 | Huwe1 | HECT-type E3 ubiquitin transferase | 16.925 | 1.2 | 3 | 71.614 | 322,680,000 |
| 4 | A0A0G2JZA2 | Grpel1 | GrpE protein homolog 1. mitochondrial | 4.6136 | 16.5 | 2 | 21.243 | 223,840,000 |
| 5 | A0A0G2K261 | Iars2 | Isoleucine--tRNA ligase, mitochondrial | 35.558 | 8 | 5 | 19.872 | 129,930,000 |
| 6 | A0A0G2K4Y1 | Dnaja3 | DnaJ homolog subfamily A member 3, mitochondrial | 5.2058 | 6.6 | 2 | 49.416 | 63,935,000 |
| 7 | B0BMT9 | Sqrdl | Sulfide:quinone oxidoreductase, mitochondrial | 14.818 | 9.8 | 3 | 45.347 | 93,481,000 |
| 8 | B0BNB5 | Nup43 | Nucleoporin Nup43 | 14.829 | 8.5 | 2 | 46.435 | 196,970,000 |
| 9 | B1WBQ7 | Msh2 | DNA mismatch repair protein Msh2 | 15.144 | 5.5 | 4 | 112.68 | 264,890,000 |
| 10 | B1WC25 | Tra2a | Transformer-2 protein homolog alpha | 11.134 | 11 | 4 | 58.286 | 122,620,000 |
| 11 | B2RYG5 | Taf15 | TATA-binding protein-associated factor 2N | 12.166 | 14.3 | 5 | 55.413 | 126,240,000 |
| 12 | B2RYQ5 | Erh | Enhancer of rudimentary homolog | 14.177 | 31.7 | 2 | 65.673 | 73,205,000 |
| 13 | D3ZIN7 | Mrps23 | 28S ribosomal protein S23, mitochondrial | 3.558 | 6.2 | 2 | 104.15 | 151,590,000 |
| 14 | D3ZIT4 | Anapc7 | Anaphase-promoting complex subunit 7 | 14.411 | 8.5 | 3 | 24.857 | 203,460,000 |
| 15 | D3ZM09 | Sars2 | Serine--tRNA ligase, mitochondrial | 5.5606 | 5.4 | 2 | 88.596 | 73,564,000 |
| 16 | D4A054 | Ranbp2 | E3 SUMO-protein ligase RanBP2 | 12.017 | 3 | 7 | 24.163 | 120,060,000 |
| 17 | F1LT09 | Wdr33 | pre-mRNA 3’ end processing protein WDR33 | 4.5551 | 2.3 | 2 | 43.931 | 87,020,000 |
| 18 | F1LTU4 | Mrto4 | mRNA turnover protein 4 homolog | 8.9808 | 26.4 | 3 | 23.397 | 550,130,000 |
| 19 | F1M7L9 | Uncharacterized protein | 7.6448 | 4.4 | 4 | 12.259 | 63,761,000 | |
| 20 | F1MAA3 | LOC100909464 | Serine/threonine-protein phosphatase 2A 56 kDa regulatory subunit | 6.3291 | 7.9 | 3 | 27.911 | 76,667,000 |
| 21 | F7EQ81 | Gns | N-acetylglucosamine-6-sulfatase | 42.435 | 5.5 | 2 | 74.889 | 91,214,000 |
| 22 | G3V7F6 | RGD1561590 | 18 kDa Sin3-associated polypeptide | 47.857 | 27.3 | 4 | 15.906 | 105,570,000 |
| 23 | G3V7Z1 | Rcl1 | RNA 3’-terminal phosphate cyclase-like protein | 108.33 | 8.3 | 2 | 69.183 | 64,529,000 |
| 24 | O70593 | Sgta | Small glutamine-rich tetratricopeptide repeat-containing protein alpha | 5.0856 | 12.1 | 3 | 69.152 | 154,570,000 |
| 25 | P00173 | Cyb5a | Cytochrome b5 | 18.796 | 25.4 | 2 | 341.4 | 112,790,000 |
| 26 | P08461 | Dlat | Dihydrolipoyllysine-residue acetyltransferase component of pyruvate dehydrogenase complex. mitochondrial | 10.385 | 9.7 | 4 | 14.441 | 341,840,000 |
| 27 | P12007 | Ivd | Isovaleryl-CoA dehydrogenase. mitochondrial | 3.6522 | 4.2 | 2 | 20.321 | 62,345,000 |
| 28 | P97546 | Nptn | Neuroplastin | 5.8398 | 7.9 | 2 | 67.603 | 27,068,000 |
| 29 | Q04970 | Nras | GTPase NRas | 10.771 | 39.2 | 4 | 22.162 | 265,030,000 |
| 30 | Q06647 | Atp5o | ATP synthase subunit O. mitochondrial | 12.515 | 11.7 | 2 | 40.81 | 108,260,000 |
| 31 | Q3B8N8 | Pes1 | Pescadillo homolog | 15.961 | 3.2 | 3 | 145.41 | 40,168,000 |
| 32 | Q5U2N0 | Ctps2 | CTP synthase 2 | 3.4722 | 3.8 | 2 | 32.578 | 93,367,000 |
| 33 | Q5XI78 | Ogdh | 2-oxoglutarate dehydrogenase. mitochondrial | 8.0893 | 8 | 5 | 62.993 | 44,703,000 |
| 34 | Q63396 | Sub1 | Activated RNA polymerase II transcriptional coactivator p15 | 11.65 | 20.5 | 2 | 25.811 | 139,220,000 |
| 35 | Q63584 | Tmed10 | Transmembrane emp24 domain-containing protein 10 | 8.5838 | 19.2 | 4 | 15.355 | 85,833,000 |
| 36 | Q6AXT5 | Rab21 | Ras-related protein Rab-21 | 60.667 | 16.6 | 4 | 447.65 | 64,474,000 |
| 37 | Q6AY02 | Rbm17 | Splicing factor 45 | 10.643 | 14.6 | 4 | 50.201 | 190,840,000 |
| 38 | Q6AY58 | Bcap31 | B-cell receptor-associated protein | 25.526 | 8.6 | 3 | 41.806 | 124,930,000 |
| 39 | Q7TSA0 | Rhot2 | Mitochondrial Rho GTPase 2 | 8.3952 | 7.9 | 4 | 60.068 | 80,485,000 |
| 40 | Q920L2 | Sdha | Succinate dehydrogenase [ubiquinone] flavoprotein subunit. mitochondrial | 102.6 | 7.2 | 3 | 19.586 | 96,290,000 |
| 41 | Q9WU49 | Carhsp1 | Calcium-regulated heat-stable protein 1 | 4.3794 | 26.5 | 3 | 34.157 | 81,131,000 |
| Protein IDs a | Gene Names | Protein Names | Score b | Coverage (%) c | Peptides d | kDa | Intensity (×106) | Fold Change e | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | B0BMT9 | Sqrdl | Sqrdl protein | 14.818 | 9.8 | 3 | 50.201 | 190.84 | −2 |
| 2 | Q06647 | Atp5o | ATP synthase subunit O. mitochondrial | 12.515 | 11.7 | 2 | 23.397 | 550.13 | −2.8 |
| 3 | Q920L2 | Sdha | Succinate dehydrogenase [ubiquinone] flavoprotein subunit. mitochondrial | 102.6 | 7.2 | 3 | 71.614 | 322.68 | −2.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peñas-Fuentes, J.L.; Siles, E.; Rufino-Palomares, E.E.; Pérez-Jiménez, A.; Reyes-Zurita, F.J.; Lupiáñez, J.A.; Fuentes-Almagro, C.; Peragón-Sánchez, J. Effects of Erythrodiol on the Antioxidant Response and Proteome of HepG2 Cells. Antioxidants 2022, 11, 73. https://doi.org/10.3390/antiox11010073
Peñas-Fuentes JL, Siles E, Rufino-Palomares EE, Pérez-Jiménez A, Reyes-Zurita FJ, Lupiáñez JA, Fuentes-Almagro C, Peragón-Sánchez J. Effects of Erythrodiol on the Antioxidant Response and Proteome of HepG2 Cells. Antioxidants. 2022; 11(1):73. https://doi.org/10.3390/antiox11010073
Chicago/Turabian StylePeñas-Fuentes, Juan Luis, Eva Siles, Eva E. Rufino-Palomares, Amalia Pérez-Jiménez, Fernando J. Reyes-Zurita, José A. Lupiáñez, Carlos Fuentes-Almagro, and Juan Peragón-Sánchez. 2022. "Effects of Erythrodiol on the Antioxidant Response and Proteome of HepG2 Cells" Antioxidants 11, no. 1: 73. https://doi.org/10.3390/antiox11010073