
Neuroprotection and Disease Modification by Astrocytes and Microglia in Parkinson Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
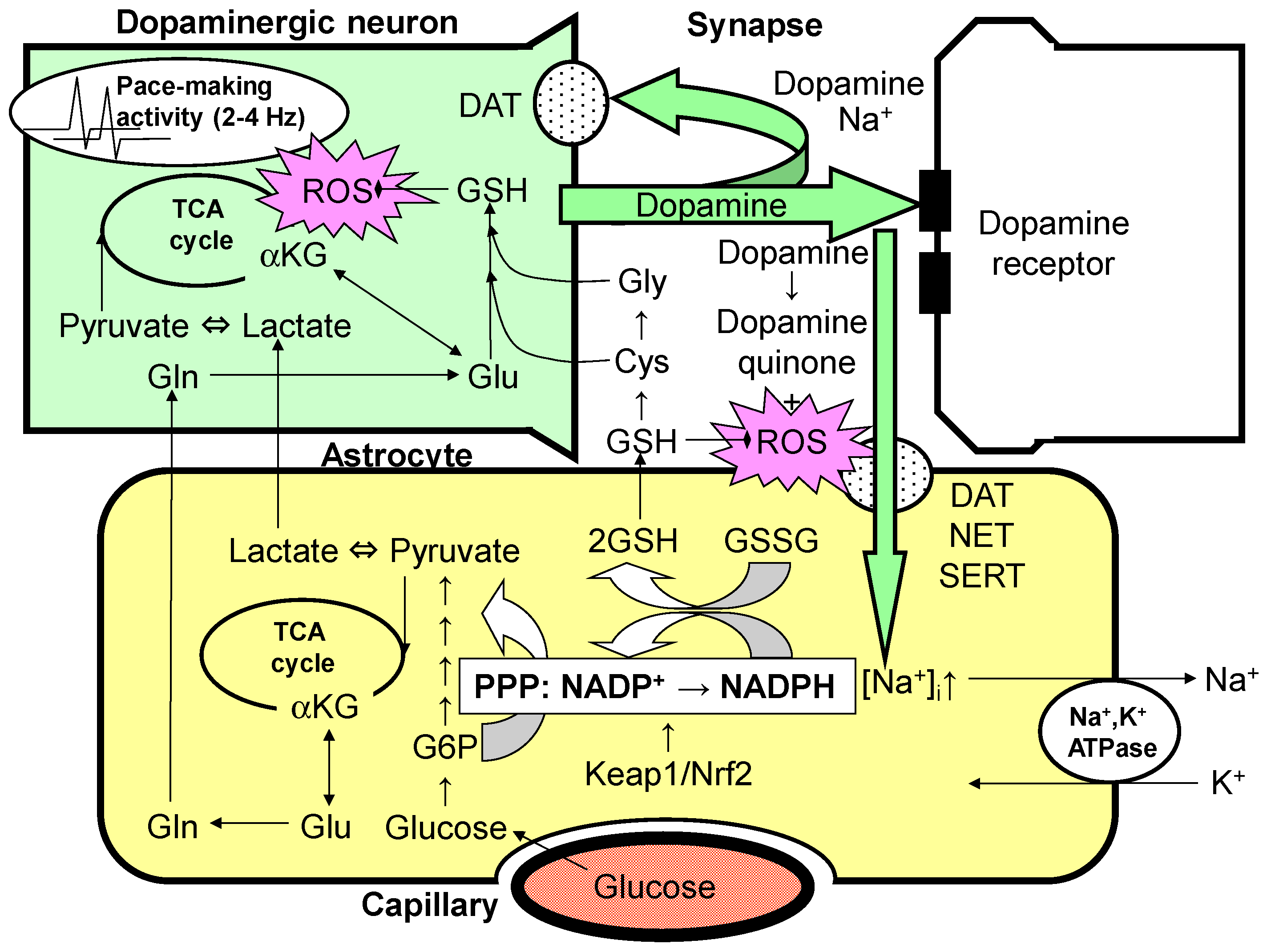
2. Dopaminergic Neurons and Astrocytes
3. Effects of Glutamate and Dopamine on Astrocytes
4. Dopamine and Astrocyte Activation of the Glycolytic System and PPP
5. Glutathione Synthesis System and Dopaminergic Neurons
6. PPP and Keap1/Nrf2 System
7. Parkinson Disease and the Keap1/Nrf2 System
8. Injurious Astrocytes in Parkinson Disease
9. Neuroinflammatory Microglia and Neuroprotective Astrocytes
10. Dopamine, Alpha-Synuclein, and TLR4
11. Issues to Be Resolved in the Future
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | amyotrophic lateral sclerosis |
| ANLS | astrocyte–neuron lactate shuttle |
| ATP | adenosine triphosphate |
| ARE | antioxidant response element |
| D2R | dopamine D2 receptor |
| DAMPs | damage-associated molecular patterns |
| DAT | dopamine transporter |
| G6PH | glucose 6-phosphate dehydrogenase |
| GSH | reduced form of glutathione |
| GSSG | oxidized form of glutathione |
| HCAR1 | hydroxycarboxylic acid receptor 1 |
| HO-1 | heme oxygenase 1 |
| iPSC | induced pluripotent stem cell |
| Keap1 | Kelch-like enoyl-CoA hydratase-associated protein 1 |
| l-DOPA | l-3,4-dihydroxyphenylalanine |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| MCT | monocarboxylate transporter |
| NET | norepinephrine transporter |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NMDA | N-methyl-D-aspartate |
| NF-κB | nuclear factor-κB |
| NO | nitric oxide |
| NOS | nitric oxide synthase |
| NRF-1 and NRF-2 | nuclear respiration factors 1 and 2 |
| Nrf2 | nuclear factor erythroid 2 p45 subunit-related factor 2 |
| GSSG | oxidized form of glutathione |
| PGC-1α | peroxisome-proliferator-activated γ co-activator-1α |
| PPAR-γ | peroxisome proliferator-activated receptor γ |
| PPP | pentose–phosphate pathway |
| PSD-95 | postsynaptic density protein 95 |
| ROS | reactive oxygen species |
| SERT | serotonin transporter |
| SIRT-1 | sirtuin 1 |
| TCA | tricarboxylic acid |
| TFAM | mitochondrial transcription factor A |
| TLR4 | Toll-like receptor 4 |
| VTA | ventral tegmental area |
| xCT | cystine/glutamate antiporter |
References
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Tolosa, E.; Garrido, A.; Scholz, S.W.; Poewe, W. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. 2021, 20, 385–397. [Google Scholar] [CrossRef]
- Vijiaratnam, N.; Simuni, T.; Bandmann, O.; Morris, H.R.; Foltynie, T. Progress towards therapies for disease modification in Parkinson’s disease. Lancet Neurol. 2021, 20, 559–572. [Google Scholar] [CrossRef]
- Latif, S.; Jahangeer, M.; Maknoon Razia, D.; Ashiq, M.; Ghaffar, A.; Akram, M.; El Allam, A.; Bouyahya, A.; Garipova, L.; Ali Shariati, M.; et al. Dopamine in Parkinson’s disease. Clin. Chim. Acta 2021, 522, 114–126. [Google Scholar] [CrossRef]
- Oosterveen, T.; Garção, P.; Moles-Garcia, E.; Soleilhavoup, C.; Travaglio, M.; Sheraz, S.; Peltrini, R.; Patrick, K.; Labas, V.; Combes-Soia, L.; et al. Pluripotent stem cell derived dopaminergic subpopulations model the selective neuron degeneration in Parkinson’s disease. Stem Cell Rep. 2021, 16, 2718–2735. [Google Scholar] [CrossRef] [PubMed]
- Alberico, S.L.; Cassell, M.D.; Narayanan, N.S. The Vulnerable Ventral Tegmental Area in Parkinson’s Disease. Basal Ganglia 2015, 5, 51–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brichta, L.; Greengard, P. Molecular determinants of selective dopaminergic vulnerability in Parkinson’s disease: An update. Front. Neuroanat. 2014, 8, 152. [Google Scholar] [CrossRef] [Green Version]
- Rey, F.; Ottolenghi, S.; Zuccotti, G.V.; Samaja, M.; Carelli, S. Mitochondrial dysfunctions in neurodegenerative diseases: Role in disease pathogenesis, strategies for analysis and therapeutic prospects. Neural Regen. Res. 2022, 17, 754–758. [Google Scholar]
- Prasuhn, J.; Brüggemann, N. Gene Therapeutic Approaches for the Treatment of Mitochondrial Dysfunction in Parkinson’s Disease. Genes 2021, 12, 1840. [Google Scholar] [CrossRef] [PubMed]
- Pena-DIaz, S.; Ventura, S. One ring is sufficient to inhibit α-synuclein aggregation. Neural Regen. Res. 2022, 17, 508–511. [Google Scholar]
- Cardinale, A.; Calabrese, V.; de Iure, A.; Picconi, B. Alpha-Synuclein as a Prominent Actor in the Inflammatory Synaptopathy of Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 6517. [Google Scholar] [CrossRef]
- Guzman, J.N.; Sánchez-Padilla, J.; Chan, C.S.; Surmeier, D.J. Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci. 2009, 29, 11011–11019. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Izawa, Y.; Suzuki, N. Astrogliopathy as a loss of astroglial protective function against glycoxidative stress under hyperglycemia. Rinsho Shinkeigaku 2012, 52, 41–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, S.; Seki, M.; Suzuki, N. Roles of metabolic compartmentalization by astrocytes and neurons in the pathophysiology and treatment of Parkinson’s disease. Brain Nerves 2013, 65, 1497–1508. [Google Scholar]
- Vucic, S.; Pavey, N.; Haidar, M.; Turner, B.J.; Kiernan, M.C. Cortical hyperexcitability: Diagnostic and pathogenic biomarker of ALS. Neurosci. Lett. 2021, 759, 136039. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, F.; Monni, L.; Nistri, A. Functional up-regulation of the M-current by retigabine contrasts hyperexcitability and excitotoxicity on rat hypoglossal motoneurons. J. Physiol. 2018, 596, 2611–2629. [Google Scholar] [CrossRef] [PubMed]
- Grapperon, A.M.; Attarian, S. Disorders of motor neurons manifested by hyperactivity. Rev. Neurol. 2017, 173, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Wainger, B.J.; Macklin, E.A.; Vucic, S.; McIlduff, C.E.; Paganoni, S.; Maragakis, N.J.; Bedlack, R.; Goyal, N.A.; Rutkove, S.B.; Lange, D.J.; et al. Effect of Ezogabine on Cortical and Spinal Motor Neuron Excitability in Amyotrophic Lateral Sclerosis: A Randomized Clinical Trial. JAMA Neurol. 2021, 78, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Kovalchuk, M.O.; Heuberger, J.A.A.C.; Sleutjes, B.T.H.M.; Ziagkos, D.; van den Berg, L.H.; Ferguson, T.A.; Franssen, H.; Groeneveld, G.J. Acute Effects of Riluzole and Retigabine on Axonal Excitability in Patients with Amyotrophic Lateral Sclerosis: A Randomized, Double-Blind, Placebo-Controlled, Crossover Trial. Clin. Pharmacol. Ther. 2018, 104, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, B.; Mauricio, E.A.; Shah, J.S.; Li, Z.; Rogawski, M.A. Cortical excitability threshold can be increased by the AMPA blocker Perampanel in amyotrophic lateral sclerosis. Muscle Nerve 2021, 64, 215–219. [Google Scholar] [CrossRef]
- Aizawa, H.; Kato, H.; Oba, K.; Kawahara, T.; Okubo, Y.; Saito, T.; Naito, M.; Urushitani, M.; Tamaoka, A.; Nakamagoe, K.; et al. Randomized phase 2 study of perampanel for sporadic amyotrophic lateral sclerosis. J. Neurol. 2021. [Google Scholar] [CrossRef]
- Akamatsu, M.; Yamashita, T.; Hirose, N.; Teramoto, S.; Kwak, S. The AMPA receptor antagonist perampanel robustly rescues amyotrophic lateral sclerosis (ALS) pathology in sporadic ALS model mice. Sci. Rep. 2016, 6, 28649. [Google Scholar] [CrossRef] [Green Version]
- Ortner, N.J. Voltage-Gated Ca2+ Channels in Dopaminergic Substantia Nigra Neurons: Therapeutic Targets for Neuroprotection in Parkinson’s Disease? Front. Synaptic Neurosci. 2021, 13, 636103. [Google Scholar] [CrossRef]
- Benkert, J.; Hess, S.; Roy, S.; Beccano-Kelly, D.; Wiederspohn, N.; Duda, J.; Simons, C.; Patil, K.; Gaifullina, A.; Mannal, N.; et al. Cav2.3 channels contribute to dopaminergic neuron loss in a model of Parkinson’s disease. Nat. Commun. 2019, 10, 5094. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.S.; Gertler, T.S.; Surmeier, D.J. Calcium homeostasis, selective vulnerability and Parkinson’s disease. Trends Neurosci. 2009, 32, 249–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampton, O.L.; Buckley, R.F.; Manning, L.K.; Scott, M.R.; Properzi, M.J.; Peña-Gómez, C.; Jacobs, H.I.L.; Chhatwal, J.P.; Johnson, K.A.; Sperling, R.A.; et al. Resting-state functional connectivity and amyloid burden influence longitudinal cortical thinning in the default mode network in preclinical Alzheimer’s disease. Neurolmage Clin. 2020, 28, 102407. [Google Scholar] [CrossRef]
- Ouchi, Y.; Kikuchi, M. A review of the default mode network in aging and dementia based on molecular imaging. Rev. Neurosci. 2012, 23, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The science of stroke: Mechanisms in search of treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S. Neuroprotective Function of High Glycolytic Activity in Astrocytes: Common Roles in Stroke and Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 6568. [Google Scholar] [CrossRef]
- Takahashi, S. Lactate and ketone bodies act as energy substrates as well as signal molecules in the brain. In Psychology and Paho-Physiological Outcomes of Eating; Takada, A., Himmerich, H., Eds.; InTech Open: Rijeka, Croatia, 2021; pp. 1–20. [Google Scholar]
- Takahashi, S. Metabolic compartmentalization between astroglia and neurons in physiological and pathophysiological conditions of the neurovascular unit. Neuropathology 2020, 40, 121–137. [Google Scholar] [CrossRef] [Green Version]
- Asanuma, M.; Miyazaki, I. Glutathione and Related Molecules in Parkinsonism. Int. J. Mol. Sci. 2021, 22, 8689. [Google Scholar] [CrossRef] [PubMed]
- Mashima, K.; Takahashi, S.; Minami, K.; Izawa, Y.; Abe, T.; Tsukada, N.; Hishiki, T.; Suematsu, M.; Kajimura, M.; Suzuki, N. Neuroprotective Role of Astroglia in Parkinson Disease by Reducing Oxidative Stress Through Dopamine-Induced Activation of Pentose-Phosphate Pathway. ASN Neuro 2018, 10, 1759091418775562. [Google Scholar] [CrossRef] [PubMed]
- Trudler, D.; Sanz-Blasco, S.; Eisele, Y.S.; Ghatak, S.; Bodhinathan, K.; Akhtar, M.W.; Lynch, W.P.; Piña-Crespo, J.C.; Talantova, M.; Kelly, J.W.; et al. α-Synuclein Oligomers Induce Glutamate Release from Astrocytes and Excessive Extrasynaptic NMDAR Activity in Neurons, Thus Contributing to Synapse Loss. J. Neurosci. 2021, 41, 2264–2273. [Google Scholar] [CrossRef]
- Wang, J.; Wang, F.; Mai, D.; Qu, S. Molecular Mechanisms of Glutamate Toxicity in Parkinson’s Disease. Front. Neurosci. 2020, 14, 585584. [Google Scholar] [CrossRef]
- Misganaw, D. Heteromerization of dopaminergic receptors in the brain: Pharmacological implications. Pharmacol. Res. 2021, 170, 105600. [Google Scholar] [CrossRef]
- Bergonzoni, G.; Döring, J.; Biagioli, M. D1R- and D2R-Medium-Sized Spiny Neurons Diversity: Insights into Striatal Vulnerability to Huntington’s Disease Mutation. Front. Cell Neurosci. 2021, 15, 628010. [Google Scholar] [CrossRef]
- Hikima, T.; Lee, C.R.; Witkovsky, P.; Chesler, J.; Ichtchenko, K.; Rice, M.E. Activity-dependent somatodendritic dopamine release in the substantia nigra autoinhibits the releasing neuron. Cell Rep. 2021, 35, 108951. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bouabid, S.; Darvas, M.; Zhou, F.M. The antiparkinson drug ropinirole inhibits movement in a Parkinson’s disease mouse model with residual dopamine neurons. Exp. Neurol. 2020, 333, 113427. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.P. The role of D2-autoreceptors in regulating dopamine neuron activity and transmission. Neuroscience 2014, 282, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Bhat, S.; Niello, M.; Schicker, K.; Pifl, C.; Sitte, H.H.; Freissmuth, M.; Sandtner, W. Handling of intracellular K+ determines voltage dependence of plasmalemmal monoamine transporter function. elife 2021, 10, e67996. [Google Scholar] [CrossRef]
- Ryan, R.M.; Ingram, S.L.; Scimemi, A. Regulation of Glutamate, GABA and Dopamine Transporter Uptake, Surface Mobility and Expression. Front. Cell. Neurosci. 2021, 15, 670346. [Google Scholar] [CrossRef]
- Inazu, M.; Takeda, H.; Ikoshi, H.; Sugisawa, M.; Uchida, Y.; Matsumiya, T. Pharmacological characterization and visualization of the glial serotonin transporter. Neurochem. Int. 2001, 39, 39–49. [Google Scholar] [CrossRef]
- Inazu, M.; Takeda, H.; Matsumiya, T. Functional expression of the norepinephrine transporter in cultured rat astrocytes. J. Neurochem. 2003, 84, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Bega, D.; Kuo, P.H.; Chalkidou, A.; Grzeda, M.T.; Macmillan, T.; Brand, C.; Sheikh, Z.H.; Antonini, A. Clinical utility of DaTscan in patients with suspected Parkinsonian syndrome: A systematic review and meta-analysis. NPJ Parkinsons Dis. 2021, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, S.H.; Fisher, S.; Gupta, F.; Hermanowicz, N.; Kremens, D.E.; Lew, M.F.; Marek, K.; Pahwa, R.; Russell, D.S.; Seibyl, J. Clinical utility of DaTscan imaging in the evaluation of patients with parkinsonism: A US perspective. Expert Rev. Neurother. 2017, 17, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Calo, L.; Wegrzynowicz, M.; Santivañez-Perez, J.; Grazia Spillantini, M. Synaptic failure and alpha-synuclein. Mov. Disord. 2016, 31, 169–177. [Google Scholar] [CrossRef]
- Ivanidze, J.; Skafida, M.; Pandya, S.; Patel, D.; Osborne, J.R.; Raj, A.; Gupta, A.; Henchcliffe, C.; Dyke, J.P. Molecular Imaging of Striatal Dopaminergic Neuronal Loss and the Neurovascular Unit in Parkinson Disease. Front. Neurosci. 2020, 14, 528809. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Driscoll, B.F.; Law, M.J.; Sokoloff, L. Role of sodium and potassium ions in regulation of glucose metabolism in cultured astroglia. Proc. Natl. Acad. Sci. USA 1995, 92, 4616–4620. [Google Scholar] [CrossRef] [Green Version]
- Sokoloff, L.; Takahashi, S.; Gotoh, J.; Driscoll, B.F.; Law, M.J. Contribution of astroglia to functionally activated energy metabolism. Dev. Neurosci. 1996, 18, 344–352. [Google Scholar] [CrossRef]
- Souza, D.G.; Almeida, R.F.; Souza, D.O.; Zimmer, E.R. The astrocyte biochemistry. Semin. Cell Dev. Biol. 2019, 95, 142–150. [Google Scholar] [CrossRef]
- Bordone, M.P.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; de Freitas, A.E.; et al. The energetic brain—A review from students to students. J. Neurochem. 2019, 151, 139–165. [Google Scholar] [CrossRef] [PubMed]
- Aubert, A.; Costalat, R.; Magistretti, P.J.; Pellerin, L. Brain lactate kinetics: Modeling evidence for neuronal lactate uptake upon activation. Proc. Natl. Acad. Sci. USA 2005, 102, 16448–16453. [Google Scholar] [CrossRef] [Green Version]
- Laughton, J.D.; Bittar, P.; Charnay, Y.; Pellerin, L.; Kovari, E.; Magistretti, P.J.; Bouras, C. Metabolic compartmentalization in the human cortex and hippocampus: Evidence for a cell- and region-specific localization of lactate dehydrogenase 5 and pyruvate dehydrogenase. BMC Neurosci. 2007, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, L.; Bouzier-Sore, A.K.; Aubert, A.; Serres, S.; Merle, M.; Costalat, R.; Magistretti, P. Activity-dependent regulation of energy metabolism by astrocytes: An update. Glia 2007, 55, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Jolivet, R.; Allaman, I.; Pellerin, L.; Magistretti, P.J.; Weber, B. Comment on recent modeling studies of astrocyte-neuron metabolic interactions. J. Cereb. Blood Flow Metab. 2010, 30, 1982–1986. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, L.; Magistretti, P.J. Sweet sixteen for ANLS. J. Cereb. Blood Flow Metab. 2012, 32, 1152–1166. [Google Scholar] [CrossRef]
- Mazuel, L.; Blanc, J.; Repond, C.; Bouchaud, V.; Raffard, G.; Déglon, N.; Bonvento, G.; Pellerin, L.; Bouzier-Sore, A.-K. A neuronal MCT2 knockdown in the rat somatosensory cortex reduces both the NMR lactate signal and the BOLD response during whisker stimulation. PLoS ONE 2017, 12, e0174990. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L. Neuroenergetics: Astrocytes have a sweet spot for glucose. Curr. Biol. 2018, 28, R1258–R1260. [Google Scholar] [CrossRef] [Green Version]
- Dienel, G.A.; Hertz, L. Glucose and lactate metabolism during brain activation. J. Neurosci. Res. 2001, 66, 824–838. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Dienel, G.A. Lactate transport and transporters: General principles and functional roles in brain cells. J. Neurosci. Res. 2005, 79, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Peng, L.; Dienel, G.A. Energy metabolism in astrocytes: High rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J. Cereb. Blood Flow Metab. 2007, 27, 219–249. [Google Scholar] [CrossRef]
- Dienel, G.A.; Ball, K.K.; Cruz, N.F. A glycogen phosphorylase inhibitor selectively enhances local rates of glucose utilization in brain during sensory stimulation of conscious rats: Implications for glycogen turnover. J. Neurochem. 2007, 102, 466–478. [Google Scholar] [CrossRef] [Green Version]
- Dienel, G.A. Astrocytes are ’good scouts’: Being prepared also helps neighboring neurons. J. Cereb. Blood Flow Metab. 2010, 30, 1893–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienel, G.A. Brain lactate metabolism: The discoveries and the controversies. J. Cereb. Blood Flow Metab. 2012, 32, 1107–1138. [Google Scholar] [CrossRef] [Green Version]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiologicaland pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Dienel, G.A.; Cruz, N.F. Contributions of glycogen to astrocytic energetics during brain activation. Metab. Brain Dis. 2015, 30, 281–298. [Google Scholar] [CrossRef] [Green Version]
- Dienel, G.A. The metabolic trinity, glucose-glycogenlactate, links astrocytes and neurons in brain energetics, signaling, memory, and gene expression. Neurosci. Lett. 2017, 637, 18–25. [Google Scholar] [CrossRef]
- Dienel, G.A. Lack of appropriate stoichiometry: Strong evidence against an energetically important astrocyte-neuron lactate shuttle in brain. J. Neurosci. Res. 2017, 95, 2103–2125. [Google Scholar] [CrossRef] [Green Version]
- Dienel, G.A. Brain glucose metabolism: Integration ofenergetics with function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef] [PubMed]
- De Castro Abrantes, H.; Briquet, M.; Schmuziger, C.; Restivo, L.; Puyal, J.; Rosenberg, N.; Rocher, A.B.; Offermanns, S.; Chatton, J.Y. The Lactate Receptor HCAR1 Modulates Neuronal Network Activity through the Activation of Gα and Gβγ Subunits. J. Neurosci. 2019, 39, 4422–4433. [Google Scholar] [CrossRef] [Green Version]
- Morland, C.; Lauritzen, K.H.; Puchades, M.; Holm-Hansen, S.; Andersson, K.; Gjedde, A.; Attramadal, H.; Storm-Mathisen, J.; Bergersen, L.H. The lactate receptor, G-protein-coupled receptor 81/hydroxycarboxylic acid receptor 1: Expression and action in brain. J. Neurosci. Res. 2015, 93, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, K.H.; Morland, C.; Puchades, M.; Holm-Hansen, S.; Hagelin, E.M.; Lauritzen, F.; Attramadal, H.; Storm-Mathisen, J.; Gjedde, A.; Bergersen, L.H. Lactate receptor sites link neurotransmission, neurovascular coupling, and brain energy metabolism. Cereb. Cortex 2014, 24, 2784–2795. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Nakamura, Y.; Li, W.; Hamanaka, G.; Arai, K.; Lo, E.H.; Hayakawa, K. Effects of O-GlcNAcylation on functional mitochondrial transfer from astrocytes. J. Cereb Blood Flow Metab. 2021, 41, 1523–1535. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Lo, E.H.; Hayakawa, K. Placental Mitochondria Therapy for Cerebral Ischemia-Reperfusion Injury in Mice. Stroke 2020, 51, 3142–3146. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Chan, S.J.; Mandeville, E.T.; Park, J.H.; Bruzzese, M.; Montaner, J.; Arai, K.; Rosell, A.; Lo, E.H. Protective Effects of Endothelial Progenitor Cell-Derived Extracellular Mitochondria in Brain Endothelium. Stem Cells 2018, 36, 1404–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, K.; Bruzzese, M.; Chou, S.H.; Ning, M.; Ji, X.; Lo, E.H. Extracellular Mitochondria for Therapy and Diagnosis in Acute Central Nervous System Injury. JAMA Neurol. 2018, 75, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.H.; Lan, J.; Esposito, E.; Ning, M.; Balaj, L.; Ji, X.; Lo, E.H.; Hayakawa, K. Extracellular Mitochondria in Cerebrospinal Fluid and Neurological Recovery after Subarachnoid Hemorrhage. Stroke 2017, 48, 2231–2237. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Lo, E.H.; Hayakawa, K. Endoplasmic Reticulum Interaction Supports Energy Production and Redox Homeostasis in Mitochondria Released from Astrocytes. Transl. Stroke Res. 2021, 12, 1045–1054. [Google Scholar] [CrossRef]
- Bonsack, B.; Borlongan, M.C.; Lo, E.H.; Arai, K. Brief overview: Protective roles of astrocyte-derived pentraxin-3 in blood-brain barrier integrity. Brain Circ. 2019, 5, 145–149. [Google Scholar]
- Cardanho-Ramos, C.; Morais, V.A. Mitochondrial Biogenesis in Neurons: How and Where. Int. J. Mol. Sci. 2021, 22, 13059. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Rueda, D.; Guerra-Ojeda, S.; Aldasoro, M.; Iradi, A.; Obrador, E.; Ortega, A.; Mauricio, M.D.; Vila, J.M.; Valles, S.L. Astrocytes protect neurons from Aβ1-42 peptide-induced neurotoxicity increasing TFAM and PGC-1 and decreasing PPAR-γ and SIRT-1. Int. J. Med. Sci. 2015, 12, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, D.D.; Sokoloff, L. Circulation and energy metabolism of the brain. In Basic Neurochemistry: Molecular, Cellular, and Medical Aspects, 6th ed.; Siegel, G., Agranoff, B., Albers, R.W., Fisher, S., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 1999; pp. 637–669. [Google Scholar]
- Dienel, G.A. Energy metabolism in the brain. In From Molecules to Networks: An Introduction to Cellular and Molecular Neuroscience, 2nd ed.; Byrne, J.H., Roberts, J.L., Eds.; Academic Press: London, UK, 2009; pp. 49–110. [Google Scholar]
- Takahashi, S.; Izawa, Y.; Suzuki, N. Astroglial pentose phosphate pathway rates in response to high-glucose environments. ASN Neuro 2012, 4, e00078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zgorzynska, E.; Dziedzic, B.; Walczewska, A. An Overview of the Nrf2/ARE Pathway and Its Role in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 9592. [Google Scholar] [CrossRef]
- Kim, S.; Indu Viswanath, A.N.; Park, J.H.; Lee, H.E.; Park, A.Y.; Choi, J.W.; Kim, H.J.; Londhe, A.M.; Jang, B.K.; Lee, J.; et al. Nrf2 activator via interference of Nrf2-Keap1 interaction has antioxidant and anti-inflammatory properties in Parkinson’s disease animal model. Neuropharmacology 2020, 167, 107989. [Google Scholar] [CrossRef] [PubMed]
- Anandhan, A.; Nguyen, N.; Syal, A.; Dreher, L.A.; Dodson, M.; Zhang, D.D.; Madhavan, L. NRF2 Loss Accentuates Parkinsonian Pathology and Behavioral Dysfunction in Human alpha-Synuclein Overexpressing Mice. Aging Dis. 2021, 12, 964–982. [Google Scholar] [CrossRef]
- De Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. BEACON Trial Investigators: Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef] [Green Version]
- Kanda, H.; Yamawaki, K. Bardoxolone methyl: Drug development for diabetic kidney disease. Clin. Exp. Nephrol. 2020, 24, 857–864. [Google Scholar] [CrossRef]
- Wang, P.; Ye, Y. Astrocytes in Neurodegenerative Diseases: A Perspective from Tauopathy and alpha-Synucleinopathy. Life 2021, 11, 938. [Google Scholar] [CrossRef]
- Iizumi, T.; Takahashi, S.; Mashima, K.; Minami, K.; Izawa, Y.; Abe, T.; Hishiki, T.; Suematsu, M.; Kajimura, M.; Suzuki, N. A possible role of microglia-derived nitric oxide by lipopolysaccharide in activation of astroglial pentose-phosphate pathway via the Keap1/Nrf2 system. J. Neuroinflamm. 2016, 13, 99. [Google Scholar] [CrossRef] [Green Version]
- Kam, T.I.; Hinkle, J.T.; Dawson, T.M.; Dawson, V.L. Microglia and astrocyte dysfunction in parkinson’s disease. Neurobiol. Dis. 2020, 144, 105028. [Google Scholar] [CrossRef]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballarin, B.; Tymianski, M. Discovery and development of NA-1 for the treatment of acute ischemic stroke. Acta Pharmacol. Sin. 2018, 39, 661–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.J.; Tymianski, M. Targeting NMDA receptors in stroke: New hope in neuroprotection. Mol. Brain. 2018, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, A.; Ospel, J.M.; Menon, B.K.; Demchuk, A.M.; McTaggart, R.A.; Nogueira, R.G.; Poppe, A.Y.; Almekhlafi, M.A.; Hanel, R.A.; Thomalla, G.; et al. ESCAPE-NA1 Trial Investigators: Assessment of Discrepancies Between Follow-up Infarct Volume and 90-Day Outcomes Among Patients With Ischemic Stroke Who Received Endovascular Therapy. JAMA Netw. Open. 2021, 4, e2132376. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.F. ESCAPE-NA1 Trial Brings Hope of Neuroprotective Drugs for Acute Ischemic Stroke: Highlights of the Phase 3 Clinical Trial on Nerinetide. Neurosci. Bull. 2021, 37, 579–581. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.D.; Goyal, M.; Menon, B.K.; Nogueira, R.G.; McTaggart, R.A.; Demchuk, A.M.; Poppe, A.Y.; Buck, B.H.; Field, T.S.; Dowlatshahi, D.; et al. ESCAPE-NA1 Investigators: Efficacy and safety of nerinetide for the treatment of acute ischaemic stroke (ESCAPE-NA1): A multicentre, double-blind, randomised controlled trial. Lancet 2020, 395, 878–887. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Marsh, S.E.; Stevens, B. Microglia and Astrocytes in Disease: Dynamic Duo or Partners in Crime? Trends Immunol. 2020, 41, 820–835. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Shinozaki, Y.; Nomura, M.; Iwatsuki, K.; Moriyama, Y.; Gachet, C.; Koizumi, S. Microglia trigger astrocyte-mediated neuroprotection via purinergic gliotransmission. Sci. Rep. 2014, 4, 4329. [Google Scholar] [CrossRef] [Green Version]
- Shinozaki, Y.; Shibata, K.; Yoshida, K.; Shigetomi, E.; Gachet, C.; Ikenaka, K.; Tanaka, K.F.; Koizumi, S. Transformation of Astrocytes to a Neuroprotective Phenotype by Microglia via P2Y(1) Receptor Downregulation. Cell Rep. 2017, 19, 1151–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Huck, J.H.; Freyer, D.; Böttcher, C.; Mladinov, M.; Muselmann-Genschow, C.; Thielke, M.; Gladow, N.; Bloomquist, D.; Mergenthaler, P.; Priller, J. De novo expression of dopamine D2 receptors on microglia after stroke. J. Cereb. Blood Flow Metab. 2015, 35, 1804–1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Färber, K.; Pannasch, U.; Kettenmann, H. Dopamine and noradrenaline control distinct functions in rodent microglial cells. Mol. Cell. Neurosci. 2005, 29, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Ruscher, K.; Kuric, E.; Wieloch, T. Levodopa treatment improves functional recovery after experimental stroke. Stroke 2012, 43, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Thomas Broome, S.; Louangaphay, K.; Keay, K.A.; Leggio, G.M.; Musumeci, G.; Castorina, A. Dopamine: An immune transmitter. Neural Regen. Res. 2020, 15, 2173–2185. [Google Scholar]
- Okano, H.; Yasuda, D.; Fujimori, K.; Morimoto, S.; Takahashi, S. Ropinirole, a New ALS Drug Candidate Developed Using iPSCs. Trends Pharmacol. Sci. 2020, 41, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, D.K.; Seo, B.A.; Ghosh, A.; Ma, S.X.; Hernandez-Quijada, K.; Andersen, J.K.; Ko, H.S.; Kim, Y.H. Alpha-Synuclein Preformed Fibrils Induce Cellular Senescence in Parkinson’s Disease Models. Cells 2021, 10, 1694. [Google Scholar] [CrossRef]
- Rostami, J.; Mothes, T.; Kolahdouzan, M.; Eriksson, O.; Moslem, M.; Bergström, J.; Ingelsson, M.; O’Callaghan, P.; Healy, L.M.; Falk, A.; et al. Crosstalk between astrocytes and microglia results in increased degradation of alpha-synuclein and amyloid-beta aggregates. J. Neuroinflamm. 2021, 18, 124. [Google Scholar] [CrossRef] [PubMed]
- Izco, M.; Blesa, J.; Verona, G.; Cooper, J.M.; Alvarez-Erviti, L. Glial activation precedes alpha-synuclein pathology in a mouse model of Parkinson’s disease. Neurosci. Res. 2021, 170, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.W.; Chang, N.P.; Krishnagiri, M.; Patel, A.P.; Lindman, M.; Angel, J.P.; Kung, P.L.; Atkins, C.; Daniels, B.P. Fibrillar alpha-synuclein induces neurotoxic astrocyte activation via RIP kinase signaling and NF-kappaB. Cell Death Dis. 2021, 12, 756. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released alpha-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.R.; Park, S.J.; Park, S.M. Molecular events underlying the cell-to-cell transmission of α-synuclein. FEBS J. 2021, 288, 6593–6602. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.T.; Kim, Y.J.; Nguyen, P.T.; Koh, Y.H.; Nguyen, T.T.; Ma, H.I.; Kim, Y.E. Temporal Evolution of Inflammation and Neurodegeneration with Alpha-Synuclein Propagation in Parkinson’s Disease Mouse Model. Front. Integr. Neurosci. 2021, 15, 715190. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Lai, T.T.; Kim, Y.J.; Jeon, B. Preferential microglial activation associated with pathological alpha synuclein transmission. J. Clin. Neurosci. 2020, 81, 469–476. [Google Scholar] [CrossRef]
- Tsunemi, T.; Ishiguro, Y.; Yoroisaka, A.; Valdez, C.; Miyamoto, K.; Ishikawa, K.; Saiki, S.; Akamatsu, W.; Hattori, N.; Krainc, D. Astrocytes Protect Human Dopaminergic Neurons from α-Synuclein Accumulation and Propagation. J. Neurosci. 2020, 40, 8618–8628. [Google Scholar] [CrossRef]
- Valdinocci, D.; Radford, R.A.; Siow, S.M.; Chung, R.S.; Pountney, D.L. Potential Modes of Intercellular α-Synuclein Transmission. Int. J. Mol. Sci. 2017, 18, 469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, E.; Mocanu, E.; Uruk, G.; Swanson, R.A. Glucose availability limits microglial nitric oxide production. J. Neurochem. 2021, 159, 1008–1015. [Google Scholar] [CrossRef]
- Xiang, X.; Wind, K.; Wiedemann, T.; Blume, T.; Shi, Y.; Briel, N.; Beyer, L.; Biechele, G.; Eckenweber, F.; Zatcepin, A.; et al. Microglial activation states drive glucose uptake and FDG-PET alterations in neurodegenerative diseases. Sci. Transl. Med. 2021, 13, eabe5640. [Google Scholar] [CrossRef]
- Zhang, S.; Lachance, B.B.; Mattson, M.P.; Jia, X. Glucose metabolic crosstalk and regulation in brain function and diseases. Prog. Neurobiol. 2021, 204, 102089. [Google Scholar] [CrossRef]
- Choi, H.; Choi, Y.; Lee, E.J.; Kim, H.; Lee, Y.; Kwon, S.; Hwang, D.W.; Lee, D.S. Alzheimer’s Disease Neuroimaging Initiative: Hippocampal glucose uptake as a surrogate of metabolic change of microglia in Alzheimer’s disease. J. Neuroinflamm. 2021, 18, 190. [Google Scholar] [CrossRef]
- Aldana, B.I. Microglia-Specific Metabolic Changes in Neurodegeneration. J. Mol. Biol. 2019, 431, 1830–1842. [Google Scholar] [CrossRef] [Green Version]
- Di Stadio, A.; Angelini, C. Microglia polarization by mitochondrial metabolism modulation: A therapeutic opportunity in neurodegenerative diseases. Mitochondrion 2019, 46, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.I.; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507.e6. [Google Scholar] [CrossRef] [PubMed]
- Johmura, Y.; Yamanaka, T.; Omori, S.; Wang, T.W.; Sugiura, Y.; Matsumoto, M.; Suzuki, N.; Kumamoto, S.; Yamaguchi, K.; Hatakeyama, S.; et al. Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science 2021, 371, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Li, C.; Zhu, J.; Wang, Y.; Huang, Y.; Zhao, S.; Sheng, S.; Song, Y.; Ji, C.; Li, C.; et al. Glutaminase 1 Regulates Neuroinflammation After Cerebral Ischemia Through Enhancing Microglial Activation and Pro-Inflammatory Exosome Release. Front. Immunol. 2020, 11, 161. [Google Scholar] [CrossRef] [Green Version]
- Fedotova, E.I.; Dolgacheva, L.P.; Abramov, A.Y.; Berezhnov, A.V. Lactate and Pyruvate Activate Autophagy and Mitophagy that Protect Cells in Toxic Model of Parkinson’s Disease. Mol. Neurobiol. 2021. [Google Scholar] [CrossRef]
- Komilova, N.R.; Angelova, P.R.; Berezhnov, A.V.; Stelmashchuk, O.A.; Mirkhodjaev, U.Z.; Houlden, H.; Gourine, A.V.; Esteras, N.; Abramov, A.Y. Metabolically induced intracellular pH changes activate mitophagy, autophagy, and cell protection in familial forms of Parkinson’s disease. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Pan, L.; Pembroke, W.G.; Rexach, J.E.; Godoy, M.I.; Condro, M.C.; Alvarado, A.G.; Harteni, M.; Chen, Y.W.; Stiles, L.; et al. Conservation and divergence of vulnerability and responses to stressors between human and mouse astrocytes. Nat. Commun. 2021, 12, 3958. [Google Scholar] [CrossRef] [PubMed]
- Leventoux, N.; Morimoto, S.; Imaizumi, K.; Sato, Y.; Takahashi, S.; Mashima, K.; Ishikawa, M.; Sonn, I.; Kondo, T.; Watanabe, H.; et al. Human Astrocytes Model Derived from Induced Pluripotent Stem Cells. Cells 2020, 9, 2680. [Google Scholar] [CrossRef] [PubMed]
- Sonnay, S.; Chakrabarti, A.; Thevenet, J.; Wiederkehr, A.; Christinat, N.; Masoodi, M. Differential Metabolism of Medium-Chain Fatty Acids in Differentiated Human-Induced Pluripotent Stem Cell-Derived Astrocytes. Front. Physiol. 2019, 10, 657. [Google Scholar] [CrossRef] [PubMed]
- Thevenet, J.; De Marchi, U.; Domingo, J.S.; Christinat, N.; Bultot, L.; Lefebvre, G.; Sakamoto, K.; Descombes, P.; Masoodi, M.; Wiederkehr, A. Medium-chain fatty acids inhibit mitochondrial metabolism in astrocytes promoting astrocyte-neuron lactate and ketone body shuttle systems. FASEB J. 2016, 30, 1913–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takahashi, S.; Mashima, K. Neuroprotection and Disease Modification by Astrocytes and Microglia in Parkinson Disease. Antioxidants 2022, 11, 170. https://doi.org/10.3390/antiox11010170
Takahashi S, Mashima K. Neuroprotection and Disease Modification by Astrocytes and Microglia in Parkinson Disease. Antioxidants. 2022; 11(1):170. https://doi.org/10.3390/antiox11010170
Chicago/Turabian StyleTakahashi, Shinichi, and Kyoko Mashima. 2022. "Neuroprotection and Disease Modification by Astrocytes and Microglia in Parkinson Disease" Antioxidants 11, no. 1: 170. https://doi.org/10.3390/antiox11010170