
Therapeutic Strategies Targeting Mitochondrial Calcium Signaling: A New Hope for Neurological Diseases?

 ,
,  , , , and
, , , and 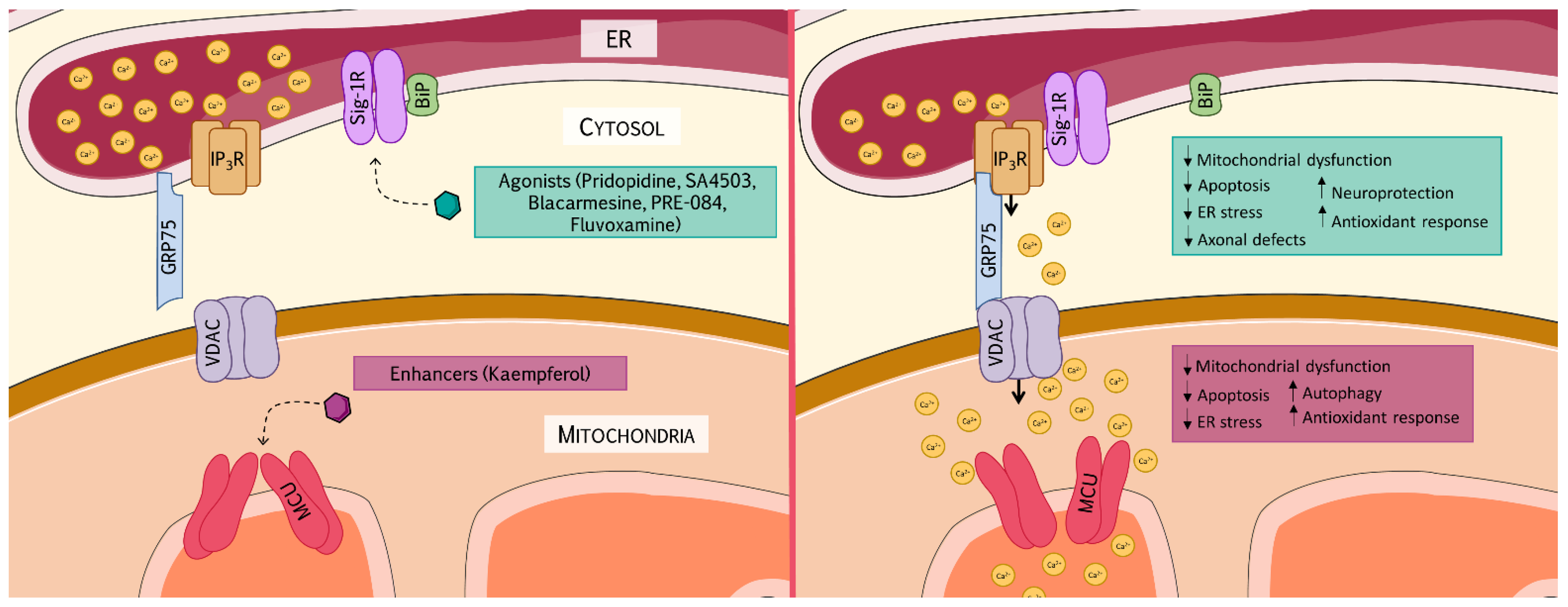{kind=link}
Abstract
:1. Introduction
2. MAMs’ Composition and Function
3. Ca2+ Regulates Mitochondrial Functions
- The first is distribution, because clustered mitochondria are able to buffer Ca2+ more efficiently than disperse mitochondria. Mitochondrial fusion/fission events require elevated amounts of cytosolic Ca2+ to be transferred to the mitochondria [33,55]. In this way, Ca2+ can activate the cytosolic GTPase dynamin-related protein (Drp-1), which is recruited to form a ring around mitochondria to promote mitochondrial fission [31]. On the other hand, mitofusin 2 (MFN2), the GTPase responsible for the OMM fusion, also participates as a key regulator of MAMs, contributing to intracellular Ca2+ homeostasis [33].
- Connectivity, because elongated mitochondria are better Ca2+ conductors, distributing the cation along the fused network. It has been described that, while fragmented mitochondria buffer Ca2+ from the ER in a heterogeneous manner, tubular mitochondria incorporate Ca2+ in an equilibrated and connected way [54,56].
- Vicinity is a dynamic property that also affects Ca2+ buffering, since elevated concentrations of Ca2+ near mitochondria are required to promote mitochondrial Ca2+ uptake. In this context, Csordás et al. demonstrated the importance of spacing distance in MAMs for an efficient Ca2+ transfer, outlining that it is essential that the tethered bridge is properly assembled to ensure Ca2+ influx into the mitochondria [44,57].
4. Ca2+ and Oxidative Stress
5. MAMs’ Communication and Neurological Diseases
6. Therapeutic Approaches Targeting MAMs
6.1. Sigma-1 Receptor as a Therapeutic Target
6.1.1. Pridopidine
6.1.2. SA4503
6.1.3. Blarcamesine
6.1.4. PRE-084
6.1.5. Fluvoxamine
6.2. Mitochondrial Calcium Uniporter as a Therapeutic Target
Kaempferol
6.3. Other Approaches
6.3.1. Taurine
6.3.2. Nerve Growth Factor
6.3.3. MiCUps
6.3.4. Antioxidants
7. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD—Alzheimer’s disease | ALS—Amyotrophic lateral sclerosis |
| Bcl-2—B-cell lymphoma 2 | BiP/GRP78—Binding immunoglobulin protein/Glucose response protein 78 |
| Ca2+—Calcium | CMT—Charcot–Marie–Tooth |
| Drp-1—Dynamin -related protein | EMRE—Essential MCU regulator |
| FRDA—Friedreich’s ataxia | GDAP1—Ganglioside-induced associated protein 1 |
| GRP75—Glucose-regulated protein 75 | GSH—Glutathione |
| H2O2—Hydrogen peroxide | HD—Huntington’s disease |
| IMM—Inner mitochondrial membrane | IP3R—Inositol 1,4,5-trisphosphate receptor |
| MAMs—Endoplasmic reticulum–mitochondria-associated membranes | MCU/Mcu—Mitochondrial calcium uniporter |
| MCUb—Mitochondrial calcium uniporter regulatory subunit | MCUR1—Mitochondrial calcium uniporter regulator 1 |
| MFN2—Mitofusin 2 | MICU1/2/3—Mitochondrial calcium uptake protein 1, 2, 3 |
| MiCUps—Mitochondrial Ca2+ uptake enhancers | NGF—Nerve Growth Factor |
| NRF2—Nuclear factor erythroid 2-related factor 2 | OMM—Outer mitochondrial membrane |
| PD—Parkinson’s disease | ROS—Reactive oxygen species |
| RyR—Ryanodine receptor | SCA2/3—Spinocerebellar ataxias type 2 and 3 |
| Sig-1R—Sigma non-opioid intracellular receptor 1 | SOD1—Superoxide dismutase 1 |
| SOD2—Superoxide dismutase 2 | SR/ER—Sarcoplasmic/endoplasmic reticulum |
| VDAC—Voltage-dependent anion channel |
References
- Skupin, A.; Thurley, K. Calcium signaling: From single channels to pathways. Adv. Exp. Med. Biol. 2012, 740, 531–551. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Taylor, C.W. Controlling calcium entry. Cell 2002, 111, 767–769. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.S. Calcium Signaling: From Basic to Bedside. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2020; Volume 1131, pp. 1–6. [Google Scholar]
- Farber, J.L. The role of calcium ions in toxic cell injury. In Proceedings of the Environmental Health Perspectives. Env. Health Perspect. 1990, 84, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Duchen, M.R.; Pozzan, T. Flirting in little space: The ER/mitochondria Ca2+ liaison. Sci. STKE 2004, 2004, re1. [Google Scholar] [CrossRef]
- Berridge, M.J. Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 933–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikoshiba, K. Role of IP3 receptor signaling in cell functions and diseases. Adv. Biol. Regul. 2015, 57, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J. The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev. 2016, 96, 1261–1296. [Google Scholar] [CrossRef] [Green Version]
- Bezprozvanny, I. Role of inositol 1,4,5-trishosphate receptors in pathogenesis of Huntington’s disease and spinocerebellar ataxias. Neurochem. Res. 2011, 36, 1186–1197. [Google Scholar] [CrossRef] [Green Version]
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H.T. Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 2017, 120, 1969–1993. [Google Scholar] [CrossRef] [PubMed]
- Smani, T.; Gallardo-Castillo, I.; Ávila-Médina, J.; Jimenez-Navarro, M.F.; Ordoñez, A.; Hmadcha, A. Impact of Diabetes on Cardiac and Vascular Disease: Role of Calcium Signaling. Curr. Med. Chem. 2017, 26, 4166–4177. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Torres, E.; Hernández-Oliveras, A.; Fuentes-García, G.; Zarain-Herzberg, Á. Calcium signaling and epigenetics: A key point to understand carcinogenesis. Cell Calcium 2020, 91, 102285. [Google Scholar] [CrossRef]
- Bezprozvanny, I. Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 2009, 15, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Connelly, J.; Levitan, E.S.; Sun, D.; Wang, J.Q. Calcium/Calmodulin–Dependent Protein Kinase II in Cerebrovascular Diseases. Transl. Stroke Res. 2021, 12, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Thakore, P.; Earley, S. Transient receptor potential channels and endothelial cell calcium signaling. Compr. Physiol. 2019, 9, 1249–1277. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.P. MAM: More than just a housekeeper. Trends Cell Biol. 2009, 19, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Perrone, M.; Caroccia, N.; Genovese, I.; Missiroli, S.; Modesti, L.; Pedriali, G.; Vezzani, B.; Vitto, V.A.M.; Antenori, M.; Lebiedzinska-Arciszewska, M.; et al. The role of mitochondria-associated membranes in cellular homeostasis and diseases. Int. Rev. Cell Mol. Biol. 2020, 350, 119–196. [Google Scholar] [CrossRef]
- Copeland, D.E.; Dalton, A.J. An association between mitochondria and the endoplasmic reticulum in cells of the pseudobranch gland of a teleost. J. Biophys. Biochem. Cytol. 1959, 5, 393–396. [Google Scholar] [CrossRef]
- Herrera-Cruz, M.S.; Simmen, T. Of yeast, mice and men: MAMs come in two flavors. Biol. Direct 2017, 12, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Zhang, A.; Williamson, C.D.; Wong, D.S.; Bullough, M.D.; Brown, K.J.; Hathout, Y.; Colberg-Poley, A.M. Quantitative proteomic analyses of human cytomegalovirus-induced restructuring of endoplasmic reticulum-mitochondrial contacts at late times of infection. Mol. Cell. Proteomics 2011, 10, M111.009936. [Google Scholar] [CrossRef] [Green Version]
- Poston, C.N.; Krishnan, S.C.; Bazemore-Walker, C.R. In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J. Proteomics 2013, 79, 219–230. [Google Scholar] [CrossRef]
- Cieri, D.; Vicario, M.; Giacomello, M.; Vallese, F.; Filadi, R.; Wagner, T.; Pozzan, T.; Pizzo, P.; Scorrano, L.; Brini, M.; et al. SPLICS: A split green fluorescent protein-based contact site sensor for narrow and wide heterotypic organelle juxtaposition. Cell Death Differ. 2018, 25, 1131–1145. [Google Scholar] [CrossRef] [Green Version]
- Tubbs, E.; Rieusset, J. Study of endoplasmic reticulum and mitochondria interactions by in situ proximity ligation assay in fixed cells. J. Vis. Exp. 2016, 2016, e54899. [Google Scholar] [CrossRef] [PubMed]
- Wieckowski, M.R.M.R.; Giorgi, C.; Lebiedzinska, M.; Duszynski, J.; Pinton, P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 2009, 4, 1582–1590. [Google Scholar] [CrossRef]
- Cho, K.F.; Branon, T.C.; Rajeev, S.; Svinkina, T.; Udeshi, N.D.; Thoudam, T.; Kwak, C.; Rhee, H.W.; Lee, I.K.; Carr, S.A.; et al. Split-TurboID enables contact-dependent proximity labeling in cells. Proc. Natl. Acad. Sci. USA 2020, 117, 12143–12154. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.J.; Vance, J.E. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J. Biol. Chem. 2000, 275, 34534–34540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voelker, D.R. Interorganelle transport of aminoglycerophospholipids. Biochim. Biophys. Acta 2000, 1486, 97–107. [Google Scholar] [CrossRef]
- Ingerman, E.; Perkins, E.M.; Marino, M.; Mears, J.A.; McCaffery, J.M.; Hinshaw, J.E.; Nunnari, J. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J. Cell Biol. 2005, 170, 1021–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [Green Version]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Pilling, A.D.; Horiuchi, D.; Lively, C.M.; Saxton, W.M. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol. Biol. Cell 2006, 17, 2057–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glater, E.E.; Megeath, L.J.; Stowers, R.S.; Schwarz, T.L. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J. Cell Biol. 2006, 173, 545–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–615. [Google Scholar] [CrossRef] [Green Version]
- Rutter, G.A. Moving Ca2+ from the endoplasmic reticulum to mitochondria: Is spatial intimacy enough? Biochem. Soc. Trans. 2006, 34, 351–355. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef]
- Su, T.P.; Hayashi, T.; Maurice, T.; Buch, S.; Ruoho, A.E. The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol. Sci. 2010, 31, 557–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [Green Version]
- Marchi, S.; Patergnani, S.; Pinton, P. The endoplasmic reticulum-mitochondria connection: One touch, multiple functions. Biochim. Biophys. Acta 2014, 1837, 461–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csordás, G.; Renken, C.; Várnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnóczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [Green Version]
- Drago, I.; Pizzo, P.; Pozzan, T. After half a century mitochondrial calcium in- and efflux machineries reveal themselves. EMBO J. 2011, 30, 4119–4125. [Google Scholar] [CrossRef] [Green Version]
- Boyman, L.; Karbowski, M.; Lederer, W.J. Regulation of Mitochondrial ATP Production: Ca2+ Signaling and Quality Control. Trends Mol. Med. 2020, 26, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Loshuertos, R.; Fern Andez-Silva, P. Tissue specificity of energy metabolism in mitochondria. Clin. Bioenerg. 2021, 3–60. [Google Scholar] [CrossRef]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic energy use and supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabadkai, G.; Duchen, M.R. Mitochondria: The hub of cellular Ca2+ signaling. Physiology 2008, 23, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Jouaville, L.S.; Pinton, P.; Bastianutto, C.; Rutter, G.A.; Rizzuto, R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 1999, 96, 13807–13812. [Google Scholar] [CrossRef] [Green Version]
- Denton, R.M.; Richards, D.A.; Chin, J.G. Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem. J. 1978, 176, 899–906. [Google Scholar] [CrossRef]
- McCormack, J.G.; Denton, R.M. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem. J. 1979, 180, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.-H.; Yang, J.; Parker, I.; et al. Essential Regulation of Cell Bioenergetics by Constitutive InsP3 Receptor Ca2+ Transfer to Mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Sagua, R.; Parra, V.; López-Crisosto, C.; Díaz, P.; Quest, A.F.G.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. Compr. Physiol. 2017, 7, 623–634. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frieden, M.; James, D.; Castelbou, C.; Danckaert, A.; Martinou, J.C.; Demaurex, N. Ca2+ homeostasis during mitochondrial fragmentation and perinuclear clustering induced by hFis1. J. Biol. Chem. 2004, 279, 22704–22714. [Google Scholar] [CrossRef] [Green Version]
- Csordás, G.; Várnai, P.; Golenár, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnóczky, G. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Eisner, V.; Lenaers, G.; Hajnóczky, G. Mitochondrial fusion is frequent in skeletal muscle and supports excitation-contraction coupling. J. Cell Biol. 2014, 205, 179–195. [Google Scholar] [CrossRef] [Green Version]
- Wozniak, A.L.; Wang, X.; Stieren, E.S.; Scarbrough, S.G.; Elferink, C.J.; Boehning, D. Requirement of biphasic calcium release from the endoplasmic reticulum for Fas-mediated apoptosis. J. Cell Biol. 2006, 175, 709–714. [Google Scholar] [CrossRef]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef]
- Baumgartner, H.K.; Gerasimenko, J.V.; Thorne, C.; Ferdek, P.; Pozzan, T.; Tepikin, A.V.; Petersen, O.H.; Sutton, R.; Watson, A.J.M.; Gerasimenko, O.V. Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J. Biol. Chem. 2009, 284, 20796–20803. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Nickel, A.; Kohlhaas, M.; Maack, C. Mitochondrial reactive oxygen species production and elimination. J. Mol. Cell. Cardiol. 2014, 73, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Aon, M.A.; Cortassa, S.; O’Rourke, B. Redox-optimized ROS balance: A unifying hypothesis. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 865–877. [Google Scholar] [CrossRef] [Green Version]
- Booth, D.M.; Enyedi, B.; Geiszt, M.; Várnai, P.; Hajnóczky, G. Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Mol. Cell 2016, 63, 240–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopper, R.K.; Carroll, S.; Aponte, A.M.; Johnson, D.T.; French, S.; Shen, R.-F.; Witzmann, F.A.; Harris, R.A.; Balaban, R.S. Mitochondrial matrix phosphoproteome: Effect of extra mitochondrial calcium. Biochemistry 2006, 45, 2524–2536. [Google Scholar] [CrossRef] [Green Version]
- Gerasimenko, J.V.; Gerasimenko, O.V.; Palejwala, A.; Tepikin, A.V.; Petersen, O.H.; Watson, A.J.M. Menadione-induced apoptosis: Roles of cytosolic Ca2+ elevations and the mitochondrial permeability transition pore. J. Cell Sci. 2002, 115, 485–497. [Google Scholar] [CrossRef]
- Zoccarato, F.; Cavallini, L.; Alexandre, A. Respiration-dependent Removal of Exogenous H2O2 in Brain Mitochondria. Inhibition by Ca2+. J. Biol. Chem. 2004, 279, 4166–4174. [Google Scholar] [CrossRef] [Green Version]
- Stepanova, A.; Magrané, J. Mitochondrial dysfunction in neurons in Friedreich’s ataxia. Mol. Cell. Neurosci. 2020, 102, 103419. [Google Scholar] [CrossRef] [PubMed]
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. The link between oxidative stress, redox status, bioenergetics and mitochondria in the pathophysiology of als. Int. J. Mol. Sci. 2021, 22, 6352. [Google Scholar] [CrossRef]
- Bernard-Marissal, N.; van Hameren, G.; Juneja, M.; Pellegrino, C.; Louhivuori, L.; Bartesaghi, L.; Rochat, C.; El Mansour, O.; Médard, J.J.; Croisier, M.; et al. Altered interplay between endoplasmic reticulum and mitochondria in Charcot-Marie-Tooth type 2A neuropathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2328–2337. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Zhu, P.P.; Parker, R.L.; Blackstone, C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J. Clin. Investig. 2010, 120, 1097–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villegas, R.; Martinez, N.W.; Lillo, J.; Pihan, P.; Hernandez, D.; Twiss, J.L.; Court, F.A. Calcium release from intra-axonal endoplasmic reticulum leads to axon degeneration through mitochondrial dysfunction. J. Neurosci. 2014, 34, 7179–7189. [Google Scholar] [CrossRef]
- Ellisman, M.H.; Porter, K.R. Microtrabecular structure of the axoplasmic matrix: Visualization of cross-linking structures and their distribution. J. Cell Biol. 1980, 87, 464. [Google Scholar] [CrossRef]
- Mironov, S.L.; Symonchuk, N. ER vesicles and mitochondria move and communicate at synapses. J. Cell Sci. 2006, 119, 4926–4934. [Google Scholar] [CrossRef] [Green Version]
- Manfredi, G.; Kawamata, H. Mitochondria and endoplasmic reticulum crosstalk in amyotrophic lateral sclerosis. Neurobiol. Dis. 2016, 90, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Arribas, M.; Yakhine-Diop, S.M.S.; Pedro, J.M.B.S.; Gómez-Suaga, P.; Gómez-Sánchez, R.; Martínez-Chacón, G.; Fuentes, J.M.; González-Polo, R.A.; Niso-Santano, M. Mitochondria-Associated Membranes (MAMs): Overview and Its Role in Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 6287–6303. [Google Scholar] [CrossRef] [Green Version]
- Eysert, F.; Kinoshita, P.F.; Mary, A.; Vaillant-Beuchot, L.; Checler, F.; Chami, M. Molecular dysfunctions of mitochondria-associated membranes (Mams) in alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 9521. [Google Scholar] [CrossRef]
- Veeresh, P.; Kaur, H.; Sarmah, D.; Mounica, L.; Verma, G.; Kotian, V.; Kesharwani, R.; Kalia, K.; Borah, A.; Wang, X.; et al. Endoplasmic reticulum–mitochondria crosstalk: From junction to function across neurological disorders. Ann. N. Y. Acad. Sci. 2019, 1457, 41–60. [Google Scholar] [CrossRef] [PubMed]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rüb, C.; Liu, Y.; Magrané, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. α-synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef]
- Calì, T.; Ottolini, D.; Negro, A.; Brini, M. α-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J. Biol. Chem. 2012, 287, 17914–17929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherubini, M.; Lopez-Molina, L.; Gines, S. Mitochondrial fission in Huntington’s disease mouse striatum disrupts ER-mitochondria contacts leading to disturbances in Ca2+ efflux and Reactive Oxygen Species (ROS) homeostasis. Neurobiol. Dis. 2020, 136, 104741. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, D.; Suski, J.M.; Oulès, B.; Debayle, D.; Gay, A.S.; Lacas-Gervais, S.; Bussiere, R.; Bauer, C.; Pinton, P.; Paterlini-Bréchot, P.; et al. Localization and Processing of the Amyloid-β Protein Precursor in Mitochondria-Associated Membranes. J. Alzheimer’s Dis. 2017, 55, 1549–1570. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Yi, J.; Fu, R.; Liu, E.; Siddique, T.; Rios, E.; Deng, H.X. Hyperactive intracellular calcium signaling associated with localized mitochondrial defects in skeletal muscle of an animal model of amyotrophic lateral sclerosis. J. Biol. Chem. 2010, 285, 705–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, M.K.; Keller, B.U. Cu/Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of mitochondria and perturbs Ca2+ homeostasis in SOD1G93A mice. Mol. Pharmacol. 2009, 75, 478–489. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.Y.C.; Hristova, E.; Ahlskog, N.; Tasse, L.A.; Ngsee, J.K.; Chudalayandi, P.; Bergeron, R. Aberrant subcellular dynamics of sigma-1 receptor mutants underlying neuromuscular diseases. Mol. Pharmacol. 2016, 90, 238–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, S.; Ilieva, H.; Tamada, H.; Nomura, H.; Komine, O.; Endo, F.; Jin, S.; Mancias, P.; Kiyama, H.; Yamanaka, K. Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR 1—And SOD 1 -linked ALS. EMBO Mol. Med. 2016, 8, 1421–1437. [Google Scholar] [CrossRef]
- Matilla-Dueñas, A.; Ashizawa, T.; Brice, A.; Magri, S.; McFarland, K.N.; Pandolfo, M.; Pulst, S.M.; Riess, O.; Rubinsztein, D.C.; Schmidt, J.; et al. Consensus Paper: Pathological Mechanisms Underlying Neurodegeneration in Spinocerebellar Ataxias. Cerebellum 2014, 13, 269. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Tang, T.S.; Tu, H.; Nelson, O.; Pook, M.; Hammer, R.; Nukina, N.; Bezprozvanny, I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J. Neurosci. 2008, 28, 12713–12724. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, L.R.; Calap-Quintana, P.; Lapeña-Luzón, T.; Pallardó, F.V.; Schneuwly, S.; Navarro, J.A.; Gonzalez-Cabo, P. Oxidative stress modulates rearrangement of endoplasmic reticulum-mitochondria contacts and calcium dysregulation in a Friedreich’s ataxia model. Redox Biol. 2020, 37, 101762. [Google Scholar] [CrossRef]
- Abeti, R.; Brown, A.F.; Maiolino, M.; Patel, S.; Giunti, P. Calcium Deregulation: Novel Insights to Understand Friedreich’s Ataxia Pathophysiology. Front. Cell. Neurosci. 2018, 12, 264. [Google Scholar] [CrossRef] [Green Version]
- Bolinches-Amorós, A.; Mollá, B.; Pla-Martín, D.; Palau, F.; González-Cabo, P. Mitochondrial dysfunction induced by frataxin deficiency is associated with cellular senescence and abnormal calcium metabolism. Front. Cell. Neurosci. 2014, 8, 124. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Carmona, S.; Muhammad, A.K.M.G.; Bell, S.; Landeros, J.; Vazquez, M.; Ho, R.; Franco, A.; Lu, B.; Dorn, G.W.; et al. Erratum: Restoring mitofusin balance prevents axonal degeneration in a Charcot-marie-tooth type 2A model. J. Clin. Investig. 2021, 131, e147307. [Google Scholar] [CrossRef] [PubMed]
- Civera-Tregón, A.; Domínguez, L.; Martínez-Valero, P.; Serrano, C.; Vallmitjana, A.; Benítez, R.; Hoenicka, J.; Satrústegui, J.; Palau, F. Mitochondria and calcium defects correlate with axonal dysfunction in GDAP1-related Charcot-Marie-Tooth mouse model. Neurobiol. Dis. 2021, 152, 105300. [Google Scholar] [CrossRef]
- Pla-Martín, D.; Rueda, C.B.; Estela, A.; Sánchez-Piris, M.; González-Sánchez, P.; Traba, J.; de la Fuente, S.; Scorrano, L.; Renau-Piqueras, J.; Alvarez, J.; et al. Silencing of the Charcot-Marie-Tooth disease-associated gene GDAP1 induces abnormal mitochondrial distribution and affects Ca2+ homeostasis by reducing store-operated Ca2+ entry. Neurobiol. Dis. 2013, 55, 140–151. [Google Scholar] [CrossRef]
- Wagner, M.; Osborn, D.P.S.; Gehweiler, I.; Nagel, M.; Ulmer, U.; Bakhtiari, S.; Amouri, R.; Boostani, R.; Hentati, F.; Hockley, M.M.; et al. Bi-allelic variants in RNF170 are associated with hereditary spastic paraplegia. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Zhu, P.P.; Denton, K.R.; Pierson, T.M.; Li, X.J.; Blackstone, C. Pharmacologic rescue of axon growth defects in a human iPSC model of hereditary spastic paraplegia SPG3A. Hum. Mol. Genet. 2014, 23, 5638–5648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.; Cho, I.T.; Schoel, L.J.; Cho, G.; Golden, J.A. Hereditary spastic paraplegia-linked REEP1 modulates endoplasmic reticulum/mitochondria contacts. Ann. Neurol. 2015, 78, 679–696. [Google Scholar] [CrossRef]
- SMART-Servier Medical ART. Available online: https://smart.servier.com/ (accessed on 13 December 2021).
- Penke, B.; Fulop, L.; Szucs, M.; Frecska, E. The Role of Sigma-1 Receptor, an Intracellular Chaperone in Neurodegenerative Diseases. Curr. Neuropharmacol. 2018, 16, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.; Pasternak, G.W. Molecular cloning and pharmacological characterization of the rat sigma1 receptor. Biochem. Pharmacol. 2001, 62, 349–355. [Google Scholar] [CrossRef]
- Laurini, E.; Marson, D.; Fermeglia, M.; Pricl, S. 3D Homology Model of Sigma1 Receptor. Handb. Exp. Pharmacol. 2017, 244, 27–50. [Google Scholar] [CrossRef]
- Wu, N.-H.; Ye, Y.; Wan, B.-B.; Yu, Y.-D.; Liu, C.; Chen, Q.-J. Emerging Benefits: Pathophysiological Functions and Target Drugs of the Sigma-1 Receptor in Neurodegenerative Diseases. Mol. Neurobiol. 2021, 58, 5649–5666. [Google Scholar] [CrossRef]
- Maurice, T.; Grégoire, C.; Espallergues, J. Neuro(active)steroids actions at the neuromodulatory sigma1 (sigma1) receptor: Biochemical and physiological evidences, consequences in neuroprotection. Pharmacol. Biochem. Behav. 2006, 84, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Ryskamp, D.A.; Korban, S.; Zhemkov, V.; Kraskovskaya, N.; Bezprozvanny, I. Neuronal Sigma-1 Receptors: Signaling Functions and Protective Roles in Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 862. [Google Scholar] [CrossRef]
- Koshenov, Z.; Oflaz, F.E.; Hirtl, M.; Pilic, J.; Bachkoenig, O.A.; Gottschalk, B.; Madreiter-Sokolowski, C.T.; Rost, R.; Malli, R.; Graier, W.F. Sigma-1 Receptor Promotes Mitochondrial Bioenergetics by Orchestrating ER Ca2+ Leak during Early ER Stress. Metabolites 2021, 11, 442. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- Brimson, J.M.; Prasanth, M.I.; Malar, D.S.; Brimson, S.; Thitilertdecha, P.; Tencomnao, T. Drugs that offer the potential to reduce hospitalization and mortality from SARS-CoV-2 infection: The possible role of the sigma-1 receptor and autophagy. Expert Opin. Ther. Targets 2021, 25, 435–449. [Google Scholar] [CrossRef]
- Gromek, K.A.; Suchy, F.P.; Meddaugh, H.R.; Wrobel, R.L.; LaPointe, L.M.; Chu, U.B.; Primm, J.G.; Ruoho, A.E.; Senes, A.; Fox, B.G. The oligomeric states of the purified sigma-1 receptor are stabilized by ligands. J. Biol. Chem. 2014, 289, 20333–20344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, A.K.; Mavlyutov, T.; Singh, D.R.; Biener, G.; Yang, J.; Oliver, J.A.; Ruoho, A.; Raicu, V. The sigma-1 receptors are present in monomeric and oligomeric forms in living cells in the presence and absence of ligands. Biochem. J. 2015, 466, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Ye, N.; Qin, W.; Tian, S.; Xu, Q.; Wold, E.A.; Zhou, J.; Zhen, X.-C. Small Molecules Selectively Targeting Sigma-1 Receptor for the Treatment of Neurological Diseases. J. Med. Chem. 2020, 63, 15187–15217. [Google Scholar] [CrossRef] [PubMed]
- Meunier, J.; Hayashi, T. Sigma-1 receptors regulate Bcl-2 expression by reactive oxygen species-dependent transcriptional regulation of nuclear factor kappaB. J. Pharmacol. Exp. Ther. 2010, 332, 388–397. [Google Scholar] [CrossRef] [Green Version]
- Pal, A.; Fontanilla, D.; Gopalakrishnan, A.; Chae, Y.-K.; Markley, J.L.; Ruoho, A.E. The sigma-1 receptor protects against cellular oxidative stress and activates antioxidant response elements. Eur. J. Pharmacol. 2012, 682, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Couly, S.; Khalil, B.; Viguier, V.; Roussel, J.; Maurice, T.; Liévens, J.C. Sigma-1 receptor is a key genetic modulator in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2020, 29, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shanmugam, A.; Markand, S.; Zorrilla, E.; Ganapathy, V.; Smith, S.B. Sigma 1 receptor regulates the oxidative stress response in primary retinal Müller glial cells via NRF2 signaling and system xc(−), the Na(+)-independent glutamate-cystine exchanger. Free Radic. Biol. Med. 2015, 86, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Mavlyutov, T.A.; Nickells, R.W.; Guo, L.-W. Accelerated retinal ganglion cell death in mice deficient in the Sigma-1 receptor. Mol. Vis. 2011, 17, 1034–1043. [Google Scholar]
- Miki, Y.; Tanji, K.; Mori, F.; Wakabayashi, K. Sigma-1 receptor is involved in degradation of intranuclear inclusions in a cellular model of Huntington’s disease. Neurobiol. Dis. 2015, 74, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Maurice, T.; Strehaiano, M.; Duhr, F.; Chevallier, N. Amyloid toxicity is enhanced after pharmacological or genetic invalidation of the σ1 receptor. Behav. Brain Res. 2018, 339, 1–10. [Google Scholar] [CrossRef]
- Antonini, V.; Prezzavento, O.; Coradazzi, M.; Marrazzo, A.; Ronsisvalle, S.; Arena, E.; Leanza, G. Anti-amnesic properties of (±)-PPCC, a novel sigma receptor ligand, on cognitive dysfunction induced by selective cholinergic lesion in rats. J. Neurochem. 2009, 109, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Ruscher, K.; Shamloo, M.; Rickhag, M.; Ladunga, I.; Soriano, L.; Gisselsson, L.; Toresson, H.; Ruslim-Litrus, L.; Oksenberg, D.; Urfer, R.; et al. The sigma-1 receptor enhances brain plasticity and functional recovery after experimental stroke. Brain 2011, 134, 732–746. [Google Scholar] [CrossRef] [Green Version]
- Bernard-Marissal, N.; Médard, J.-J.; Azzedine, H.; Chrast, R. Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 2015, 138, 875–890. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Su, T.-P. Sigma-1 receptor ligands: Potential in the treatment of neuropsychiatric disorders. CNS Drugs 2004, 18, 269–284. [Google Scholar] [CrossRef]
- Nguyen, L.; Lucke-Wold, B.P.; Mookerjee, S.; Kaushal, N.; Matsumoto, R.R. Sigma-1 Receptors and Neurodegenerative Diseases: Towards a Hypothesis of Sigma-1 Receptors as Amplifiers of Neurodegeneration and Neuroprotection. Adv. Exp. Med. Biol. 2017, 964, 133–152. [Google Scholar] [CrossRef] [Green Version]
- Sahlholm, K.; Århem, P.; Fuxe, K.; Marcellino, D. The dopamine stabilizers ACR16 and (−)-OSU6162 display nanomolar affinities at the σ-1 receptor. Mol. Psychiatry 2013, 18, 12–14. [Google Scholar] [CrossRef]
- Sahlholm, K.; Sijbesma, J.W.A.; Maas, B.; Kwizera, C.; Marcellino, D.; Ramakrishnan, N.K.; Dierckx, R.A.J.O.; Elsinga, P.H.; van Waarde, A. Pridopidine selectively occupies sigma-1 rather than dopamine D2 receptors at behaviorally active doses. Psychopharmacology 2015, 232, 3443–3453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddings, C.R.; Arbez, N.; Akimov, S.; Geva, M.; Hayden, M.R.; Ross, C.A. Pridopidine protects neurons from mutant-huntingtin toxicity via the sigma-1 receptor. Neurobiol. Dis. 2019, 129, 118–129. [Google Scholar] [CrossRef] [PubMed]
- De Yebenes, J.G.; Landwehrmeyer, B.; Squitieri, F.; Reilmann, R.; Rosser, A.; Barker, R.A.; Saft, C.; Magnet, M.K.; Sword, A.; Rembratt, A.; et al. Pridopidine for the treatment of motor function in patients with Huntington’s disease (MermaiHD): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet. Neurol. 2011, 10, 1049–1057. [Google Scholar] [CrossRef]
- PRidopidine’s Outcome on Function in Huntington Disease, PROOF- HD-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04556656?term=Pridopidine&draw=2&rank=1 (accessed on 5 October 2021).
- Al-Saif, A.; Al-Mohanna, F.; Bohlega, S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 2011, 70, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Ionescu, A.; Gradus, T.; Altman, T.; Maimon, R.; Saraf Avraham, N.; Geva, M.; Hayden, M.; Perlson, E. Targeting the Sigma-1 Receptor via Pridopidine Ameliorates Central Features of ALS Pathology in a SOD1G93A Model. Cell Death Dis. 2019, 10, 210. [Google Scholar] [CrossRef]
- HEALEY ALS Platform Trial-Regimen D Pridopidine-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04615923?term=Pridopidine&cond=Amyotrophic+Lateral+Sclerosis&draw=2&rank=1 (accessed on 6 October 2021).
- Francardo, V.; Geva, M.; Bez, F.; Denis, Q.; Steiner, L.; Hayden, M.R.; Cenci, M.A. Pridopidine Induces Functional Neurorestoration Via the Sigma-1 Receptor in a Mouse Model of Parkinson’s Disease. Neurotherapeutics 2019, 16, 465–479. [Google Scholar] [CrossRef] [Green Version]
- A Study to Assess the Safety and Effectiveness of Pridopidine Compared to Placebo in the Treatment of Levodopa-Induced Dyskinesia in Patients With Parkinson’s Disease-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/study/NCT03922711?term=Pridopidine&draw=2&rank=7 (accessed on 7 October 2021).
- Maurice, T.; Su, T.P. The pharmacology of sigma-1 receptors. Pharmacol. Ther. 2009, 124, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Wang, P.; Li, Y.; Yao, L.; Liu, Y.; Yu, T.; Lin, J.; Fang, X.; Huang, Z. Activation of Sigma-1 Receptor by Cutamesine Attenuates Neuronal Apoptosis by Inhibiting Endoplasmic Reticulum Stress and Mitochondrial Dysfunction in a Rat Model of Asphyxia Cardiac Arrest. Shock 2019, 51, 105–113. [Google Scholar] [CrossRef]
- Tagashira, H.; Zhang, C.; Lu, Y.M.; Hasegawa, H.; Kanai, H.; Han, F.; Fukunaga, K. Stimulation of σ1-receptor restores abnormal mitochondrial Ca2+ mobilization and ATP production following cardiac hypertrophy. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3082–3094. [Google Scholar] [CrossRef]
- Tuerxun, T.; Numakawa, T.; Adachi, N.; Kumamaru, E.; Kitazawa, H.; Kudo, M.; Kunugi, H. SA4503, a sigma-1 receptor agonist, prevents cultured cortical neurons from oxidative stress-induced cell death via suppression of MAPK pathway activation and glutamate receptor expression. Neurosci. Lett. 2010, 469, 303–308. [Google Scholar] [CrossRef]
- Tadić, V.; Malci, A.; Goldhammer, N.; Stubendorff, B.; Sengupta, S.; Prell, T.; Keiner, S.; Liu, J.; Guenther, M.; Frahm, C.; et al. Sigma 1 receptor activation modifies intracellular calcium exchange in the G93AhSOD1 ALS model. Neuroscience 2017, 359, 105–118. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Shioda, N.; Yabuki, Y.; Zhang, C.; Han, F.; Fukunaga, K. SA4503, A Potent Sigma-1 Receptor Ligand, Ameliorates Synaptic Abnormalities and Cognitive Dysfunction in a Mouse Model of ATR-X Syndrome. Int. J. Mol. Sci. 2018, 19, 2811. [Google Scholar] [CrossRef] [Green Version]
- SA4503 8-Week Study in Major Depressive Disorder (MDD)-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00551109?term=cutamesine&draw=2&rank=1 (accessed on 8 October 2021).
- Goguadze, N.; Zhuravliova, E.; Morin, D.; Mikeladze, D.; Maurice, T. Sigma-1 Receptor Agonists Induce Oxidative Stress in Mitochondria and Enhance Complex I Activity in Physiological Condition but Protect Against Pathological Oxidative Stress. Neurotox. Res. 2019, 35, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Lahmy, V.; Meunier, J.; Malmström, S.; Naert, G.; Givalois, L.; Kim, S.H.; Villard, V.; Vamvakides, A.; Maurice, T. Blockade of Tau hyperphosphorylation and Aβ1–42 eneration by the aminotetrahydrofuran derivative ANAVEX2-73, a mixed muscarinic and σ1 receptor agonist, in a nontransgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 2013, 38, 1706–1723. [Google Scholar] [CrossRef] [PubMed]
- Lahmy, V.; Long, R.; Morin, D.; Villard, V.; Maurice, T. Mitochondrial protection by the mixed muscarinic/σ1 ligand ANAVEX2-73, a tetrahydrofuran derivative, in Aβ25-35 peptide-injected mice, a nontransgenic Alzheimer’s disease model. Front. Cell. Neurosci. 2014, 8, 463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villard, V.; Espallergues, J.; Keller, E.; Vamvakides, A.; Maurice, T. Anti-amnesic and neuroprotective potentials of the mixed muscarinic receptor/sigma 1 (σ1) ligand ANAVEX2-73, a novel aminotetrahydrofuran derivative. J. Psychopharmacol. 2011, 25, 1101–1117. [Google Scholar] [CrossRef] [PubMed]
- Phase 2a Dose Finding, PK/PD and 12 Month Exploratory Efficacy Study of ANAVEX2-73 in Patients with Alzheimer’s Disease -Full Text Vie-ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02244541 (accessed on 7 October 2021).
- Study of ANAVEX2-73 in Patients with Rett Syndrome-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03758924?term=anavex2&draw=2&rank=6 (accessed on 7 October 2021).
- OLE Study for Patients with Parkinson’s Disease With Dementia Enrolled in Study ANAVEX2-73-PDD-001-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04575259?term=anavex2&draw=2&rank=1 (accessed on 7 October 2021).
- OLE of Phase 2b/3 Study ANAVEX2-73-AD-004-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04314934?term=anavex2&draw=2&rank=2 (accessed on 7 October 2021).
- ANAVEX2-73 Study in Patients with Rett Syndrome-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03941444?term=anavex2&draw=2&rank=4 (accessed on 7 October 2021).
- ANAVEX2-73 Study in Pediatric Patients with Rett Syndrome-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04304482?term=anavex2&draw=2&rank=3 (accessed on 7 October 2021).
- Bucolo, C.; Drago, F.; Lin, L.R.; Reddy, V.N. Sigma receptor ligands protect human retinal cells against oxidative stress. Neuroreport 2006, 17, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Hyrskyluoto, A.; Pulli, I.; Törnqvist, K.; Ho, T.H.; Korhonen, L.; Lindholm, D. Sigma-1 receptor agonist PRE084 is protective against mutant huntingtin-induced cell degeneration: Involvement of calpastatin and the NF-κB pathway. Cell Death Dis. 2013, 4, e646. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, B.; Zeng, H.; Cai, C.; Hu, Q.; Cai, S.; Xu, L.; Meng, X.; Zou, F. Role of the mitochondrial Ca2+ uniporter in Pb2+-induced oxidative stress in human neuroblastoma cells. Brain Res. 2014, 1575, 12–21. [Google Scholar] [CrossRef]
- Omi, T.; Tanimukai, H.; Kanayama, D.; Sakagami, Y.; Tagami, S.; Okochi, M.; Morihara, T.; Sato, M.; Yanagida, K.; Kitasyoji, A.; et al. Fluvoxamine alleviates ER stress via induction of Sigma-1 receptor. Cell Death Dis. 2014, 5, e1332. [Google Scholar] [CrossRef]
- Tagashira, H.; Bhuiyan, M.S.; Shioda, N.; Fukunaga, K. Fluvoxamine rescues mitochondrial Ca2+ transport and ATP production through σ1-receptor in hypertrophic cardiomyocytes. Life Sci. 2014, 95, 89–100. [Google Scholar] [CrossRef]
- Tagashira, H.; Bhuiyan, S.; Shioda, N.; Hasegawa, H.; Kanai, H.; Fukunaga, K. Σ1-Receptor Stimulation With Fluvoxamine Ameliorates Transverse Aortic Constriction-Induced Myocardial Hypertrophy and Dysfunction in Mice. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1535–H1545. [Google Scholar] [CrossRef] [Green Version]
- Elsaed, W.M.; Alahmadi, A.M.; Al-Ahmadi, B.T.; Taha, J.A.; Tarabishi, R.M. Gastroprotective and antioxidant effects of fluvoxamine on stress-induced peptic ulcer in rats. J. Taibah Univ. Med. Sci. 2018, 13, 422–431. [Google Scholar] [CrossRef]
- Abdel-Salam, O.M.E.; Youssef Morsy, S.M.; Sleem, A.A. The effect of different antidepressant drugs on oxidative stress after lipopolysaccharide administration in mice. EXCLI J. 2011, 10, 290–302. [Google Scholar] [CrossRef]
- Almási, N.; Török, S.; Valkusz, Z.; Tajti, M.; Csonka, Á.; Murlasits, Z.; Pósa, A.; Varga, C.; Kupai, K. Sigma-1 receptor engages an anti-inflammatory and antioxidant feedback loop mediated by peroxiredoxin in experimental colitis. Antioxidants 2020, 9, 1081. [Google Scholar] [CrossRef]
- Sukhatme, V.P.; Reiersen, A.M.; Vayttaden, S.J.; Sukhatme, V.V. Fluvoxamine: A Review of Its Mechanism of Action and Its Role in COVID-19. Front. Pharmacol. 2021, 12, 652688. [Google Scholar] [CrossRef]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabó, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Qi, H.; Li, L.; Shuai, J. Optimal microdomain crosstalk between endoplasmic reticulum and mitochondria for Ca2+ oscillations. Sci. Rep. 2015, 5, 7984. [Google Scholar] [CrossRef] [Green Version]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [Green Version]
- Oxenoid, K.; Dong, Y.; Cao, C.; Cui, T.; Sancak, Y.; Markhard, A.L.; Grabarek, Z.; Kong, L.; Liu, Z.; Ouyang, B.; et al. Architecture of the mitochondrial calcium uniporter. Nature 2016, 533, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Yang, X.; Li, S.; Wang, Z.; Liu, Y.; Feng, J.; Zhu, Y.; Shen, Y. Structural and mechanistic insights into MICU1 regulation of mitochondrial calcium uptake. EMBO J. 2014, 33, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.C.; Liu, J.; Holmström, K.M.; Menazza, S.; Parks, R.J.; Fergusson, M.M.; Yu, Z.X.; Springer, D.A.; Halsey, C.; Liu, C.; et al. MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cell Rep. 2016, 16, 1561–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logan, C.V.; Szabadkai, G.; Sharpe, J.A.; Parry, D.A.; Torelli, S.; Childs, A.M.; Kriek, M.; Phadke, R.; Johnson, C.A.; Roberts, N.Y.; et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet. 2014, 46, 188–193. [Google Scholar] [CrossRef]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; de Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2019, 26, 179–195. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovács-Bogdán, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 2013, 342, 1379–1382. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Yamagoshi, R.; Harada, K.; Kawano, M.; Minami, N.; Ido, Y.; Kuwahara, K.; Fujita, A.; Ozono, M.; Watanabe, A.; et al. Analysis of the structure and function of EMRE in a yeast expression system. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 831–839. [Google Scholar] [CrossRef]
- Mallilankaraman, K.; Cárdenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenár, T.; Csordás, G.; Madireddi, P.; Yang, J.; Müller, M.; et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 2012, 14, 1336–1343. [Google Scholar] [CrossRef] [Green Version]
- Tomar, D.; Dong, Z.; Shanmughapriya, S.; Koch, D.A.; Thomas, T.; Hoffman, N.E.; Timbalia, S.A.; Goldman, S.J.; Breves, S.L.; Corbally, D.P.; et al. MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Rep. 2016, 15, 1673–1685. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, N.E.; Chandramoorthy, H.C.; Shamugapriya, S.; Zhang, X.; Rajan, S.; Mallilankaraman, K.; Gandhirajan, R.K.; Vagnozzi, R.J.; Ferrer, L.M.; Sreekrishnanilayam, K.; et al. MICU1 motifs define mitochondrial calcium uniporter binding and activity. Cell Rep. 2013, 5, 1576–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustos, G.; Cruz, P.; Lovy, A.; Cárdenas, C. Endoplasmic reticulum-mitochondria calcium communication and the regulation of mitochondrial metabolism in cancer: A novel potential target. Front. Oncol. 2017, 7, 199. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Jin, M.; Wang, Y.; Zhu, J.; Tan, R.; Zhao, J.; Ji, X.; Jin, C.; Jia, Y.; Ren, T.; et al. MCU-induced mitochondrial calcium uptake promotes mitochondrial biogenesis and colorectal cancer growth. Signal Transduct. Target. Ther. 2020, 5, 59. [Google Scholar] [CrossRef]
- Liao, Y.; Dong, Y.; Cheng, J. The function of the mitochondrial calcium uniporter in neurodegenerative disorders. Int. J. Mol. Sci. 2017, 18, 248. [Google Scholar] [CrossRef]
- Venugopal, A.; Iyer, M.; Balasubramanian, V.; Vellingiri, B. Mitochondrial calcium uniporter as a potential therapeutic strategy for Alzheimer’s disease. Acta Neuropsychiatr. 2020, 32, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Soman, S.; Keatinge, M.; Moein, M.; da Costa, M.; Mortiboys, H.; Skupin, A.; Sugunan, S.; Bazala, M.; Kuznicki, J.; Bandmann, O. Inhibition of the mitochondrial calcium uniporter rescues dopaminergic neurons in pink1−/− zebrafish. Eur. J. Neurosci. 2017, 45, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.H.; Wei, Y.H. Role of mitochondrial dysfunction and dysregulation of Ca2+ homeostasis in the pathophysiology of insulin resistance and type 2 diabetes. J. Biomed. Sci. 2017, 24, 70. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Cheng, R.; Liang, C.; Yao, Y.; Zhang, W.; Zhang, J.; Zhang, M.; Li, B.; Xu, C.; Zhang, R. MicroRNA-20b Promotes Cardiac Hypertrophy by the Inhibition of Mitofusin 2-Mediated Inter-organelle Ca2+ Cross-Talk. Mol. Ther. Nucleic Acids 2020, 19, 1343–1356. [Google Scholar] [CrossRef]
- Drago, I.; de Stefani, D.; Rizzuto, R.; Pozzan, T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 12986–12991. [Google Scholar] [CrossRef] [Green Version]
- Hussein, R.M.; Mohamed, W.R.; Omar, H.A. A neuroprotective role of kaempferol against chlorpyrifos-induced oxidative stress and memory deficits in rats via GSK3β-Nrf2 signaling pathway. Pestic. Biochem. Physiol. 2018, 152, 29–37. [Google Scholar] [CrossRef]
- Feng, H.; Cao, J.; Zhang, G.; Wang, Y. Kaempferol Attenuates Cardiac Hypertrophy via Regulation of ASK1/MAPK Signaling Pathway and Oxidative Stress. Planta Med. 2017, 83, 837–845. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Liao, Z.; Huang, L.; Liu, D.; Yin, D.; He, M. Kaempferol protects cardiomyocytes against anoxia/reoxygenation injury via mitochondrial pathway mediated by SIRT1. Eur. J. Pharmacol. 2015, 761, 245–253. [Google Scholar] [CrossRef]
- Schweitzer, M.K.; Wilting, F.; Sedej, S.; Dreizehnter, L.; Dupper, N.J.; Tian, Q.; Moretti, A.; My, I.; Kwon, O.; Priori, S.G.; et al. Suppression of Arrhythmia by Enhancing Mitochondrial Ca2+ Uptake in Catecholaminergic Ventricular Tachycardia Models. JACC Basic to Transl. Sci. 2017, 2, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Sanganahalli, B.G.; Herman, P.; Hyder, F.; Kannurpatti, S.S. Mitochondrial calcium uptake capacity modulates neocortical excitability. J. Cereb. Blood Flow Metab. 2013, 33, 1115–1126. [Google Scholar] [CrossRef] [Green Version]
- Montero, M.; Lobatón, C.D.; Hernández-Sanmiguel, E.; Santodomingo, J.; Vay, L.; Moreno, A.; Alvarez, J. Direct activation of the mitochondrial calcium uniporter by natural plant flavonoids. Biochem. J. 2004, 384, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Ashrafizadeh, M.; Tavakol, S.; Ahmadi, Z.; Roomiani, S.; Mohammadinejad, R.; Samarghandian, S. Therapeutic effects of kaempferol affecting autophagy and endoplasmic reticulum stress. Phyther. Res. 2020, 34, 911–923. [Google Scholar] [CrossRef]
- Wang, H.; Chen, L.; Zhang, X.; Xu, L.; Xie, B.; Shi, H.; Duan, Z.; Zhang, H.; Ren, F. Kaempferol protects mice from D-GalN/LPS-induced acute liver failure by regulating the ER stress-Grp78-CHOP signaling pathway. Biomed. Pharmacother. 2019, 111, 468–475. [Google Scholar] [CrossRef]
- Rajendran, P.; Ammar, R.B.; Al-Saeedi, F.J.; Mohamed, M.E.; Elnaggar, M.A.; Al-Ramadan, S.Y.; Bekhet, G.M.; Soliman, A.M. Kaempferol Inhibits Zearalenone-Induced Oxidative Stress and Apoptosis via the PI3K/Akt-Mediated Nrf2 Signaling Pathway: In Vitro and In Vivo Studies. Int. J. Mol. Sci. 2020, 22, 217. [Google Scholar] [CrossRef]
- Samhan-Arias, A.K.; Martín-Romero, F.J.; Gutiérrez-Merino, C. Kaempferol blocks oxidative stress in cerebellar granule cells and reveals a key role for reactive oxygen species production at the plasma membrane in the commitment to apoptosis. Free Radic. Biol. Med. 2004, 37, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Yang, W.; He, Z.; He, H.; Yang, X.; Lu, Y.; Li, H. Kaempferol improves lung ischemia-reperfusion injury via antiinflammation and antioxidative stress regulated by SIRT1/HMGB1/NF-κB axis. Front. Pharmacol. 2020, 10, 1635. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Yang, W.; He, Z.; Guo, J.; Yang, X.; Wang, R.; Li, H. Kaempferol Alleviates Oxidative Stress and Apoptosis Through Mitochondria-dependent Pathway During Lung Ischemia-Reperfusion Injury. Front. Pharmacol. 2021, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Mao, J.; Wang, R.; Li, S.; Wu, B.; Yuan, Y. Kaempferol Protects Against Cerebral Ischemia Reperfusion Injury Through Intervening Oxidative and Inflammatory Stress Induced Apoptosis. Front. Pharmacol. 2020, 11, 424. [Google Scholar] [CrossRef] [Green Version]
- Naz, F.; Jyoti, S.; Siddique, Y.H. Effect of kaempferol on the transgenic Drosophila model of Parkinson’s disease. Sci. Rep. 2020, 10, 13793. [Google Scholar] [CrossRef]
- Beg, T.; Jyoti, S.; Naz, F.; Rahul; Ali, F.; Ali, S.K.; Reyad, A.M.; Siddique, Y.H. Protective Effect of Kaempferol on the Transgenic Drosophila Model of Alzheimer’s Disease. CNS Neurol. Disord. Drug Targets 2018, 17, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Holland, T.M.; Agarwal, P.; Wang, Y.; Leurgans, S.E.; Bennett, D.A.; Booth, S.L.; Morris, M.C. Dietary flavonols and risk of Alzheimer dementia. Neurology 2020, 94, E1749–E1756. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, E.; Rajasekaran, R. Comparative binding of kaempferol and kaempferide on inhibiting the aggregate formation of mutant (G85R) SOD1 protein in familial amyotrophic lateral sclerosis: A quantum chemical and molecular mechanics study. BioFactors 2018, 44, 431–442. [Google Scholar] [CrossRef]
- Foos, T.M.; Wu, J.Y. The role of taurine in the central nervous system and the modulation of intracellular calcium homeostasis. Neurochem. Res. 2002, 27, 21–26. [Google Scholar] [CrossRef] [PubMed]
- El Idrissi, A.; Trenkner, E. Taurine regulates mitochondrial calcium homeostasis. Adv. Exp. Med. Biol. 2003, 526, 527–536. [Google Scholar] [CrossRef] [PubMed]
- El Idrissi, A. Taurine increases mitochondrial buffering of calcium: Role in neuroprotection. Amino Acids 2008, 34, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Watson, J.J.; Shoemark, D.K.; Barua, N.U.; Patel, N.K. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 2013, 138, 155–175. [Google Scholar] [CrossRef]
- Martinou, I.; Desagher, S.; Eskes, R.; Antonsson, B.; André, E.; Fakan, S.; Martinou, J.-C. The Release of Cytochrome c from Mitochondria during Apoptosis of NGF-deprived Sympathetic Neurons Is a Reversible Event. J. Cell Biol. 1999, 144, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Carito, V.; Pingitore, A.; Cione, E.; Perrotta, I.; Mancuso, D.; Russo, A.; Genchi, G.; Caroleo, M.C. Localization of nerve growth factor (NGF) receptors in the mitochondrial compartment: Characterization and putative role. Biochim. Biophys. Acta 2012, 1820, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Martorana, F.; Gaglio, D.; Bianco, M.R.; Aprea, F.; Virtuoso, A.; Bonanomi, M.; Alberghina, L.; Papa, M.; Colangelo, A.M. Differentiation by nerve growth factor (NGF) involves mechanisms of crosstalk between energy homeostasis and mitochondrial remodeling. Cell Death Dis. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Jiang, H.; Takeda, K.; Lazarovici, P.; Katagiri, Y.; Yu, Z.X.; Dickens, G.; Chabuk, A.; Liu, X.W.; Ferrans, V.; Guroff, G. Nerve Growth Factor (NGF)-induced Calcium Influx and Intracellular Calcium Mobilization in 3T3 Cells Expressing NGF Receptors. J. Biol. Chem. 1999, 274, 26209–26216. [Google Scholar] [CrossRef] [Green Version]
- De Bernardi, M.A.; Rabin, S.J.; Colangelo, A.M.; Brooker, G.; Mocchetti, I. TrkA mediates the nerve growth factor-induced intracellular calcium accumulation. J. Biol. Chem. 1996, 271, 6092–6098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacHado, A.; Ferreira, D.; Grothe, M.J.; Eyjolfsdottir, H.; Almqvist, P.M.; Cavallin, L.; Lind, G.; Linderoth, B.; Seiger, Å.; Teipel, S.; et al. The cholinergic system in subtypes of Alzheimer’s disease: An in vivo longitudinal MRI study. Alzheimers. Res. Ther. 2020, 12, 51. [Google Scholar] [CrossRef] [PubMed]
- Wilting, F.; Kopp, R.; Gurnev, P.A.; Schedel, A.; Dupper, N.J.; Kwon, O.; Nicke, A.; Gudermann, T.; Schredelseker, J. The antiarrhythmic compound efsevin directly modulates voltage-dependent anion channel 2 by binding to its inner wall and enhancing mitochondrial Ca2+ uptake. Br. J. Pharmacol. 2020, 177, 2947–2958. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, H.; Schredelseker, J.; Huang, J.; Lu, K.; Naghdi, S.; Lu, F.; Franklin, S.; Fiji, H.D.G.; Wang, K.; Zhu, H.; et al. Mitochondrial Ca2+ uptake by the voltage-dependent anion channel 2 regulates cardiac rhythmicity. eLife 2015, 4, e04801. [Google Scholar] [CrossRef]
- Sander, P.; Feng, M.; Schweitzer, M.K.; Wilting, F.; Gutenthaler, S.M.; Arduino, D.M.; Fischbach, S.; Dreizehnter, L.; Moretti, A.; Gudermann, T.; et al. Approved drugs ezetimibe and disulfiram enhance mitochondrial Ca2+ uptake and suppress cardiac arrhythmogenesis. Br. J. Pharmacol. 2021, 178, 4518–4532. [Google Scholar] [CrossRef]
- Dietl, A.; Maack, C. Targeting Mitochondrial Calcium Handling and Reactive Oxygen Species in Heart Failure. Curr. Heart Fail. Rep. 2017, 14, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Tarnopolsky, M.A. The mitochondrial cocktail: Rationale for combined nutraceutical therapy in mitochondrial cytopathies. Adv. Drug Deliv. Rev. 2008, 60, 1561–1567. [Google Scholar] [CrossRef]
- Pagano, G.; Talamanca, A.A.; Castello, G.; Cordero, M.D.; d’Ischia, M.; Gadaleta, M.N.; Pallardó, F.V.; Petrović, S.; Tiano, L.; Zatterale, A. Current experience in testing mitochondrial nutrients in disorders featuring oxidative stress and mitochondrial dysfunction: Rational design of chemoprevention trials. Int. J. Mol. Sci. 2014, 15, 20169–20208. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Bioenergetic approaches for neuroprotection in Parkinson’s disease. Ann. Neurol. 2003, 53, S39–S48. [Google Scholar] [CrossRef]
- Pallardó, F.V.; Pagano, G.; Rodríguez, L.R.; Gonzalez-Cabo, P.; Lyakhovich, A.; Trifuoggi, M. Friedreich Ataxia: Current state-of-the-art, and future prospects for mitochondrial-focused therapies. Transl. Res. 2021, 229, 135–141. [Google Scholar] [CrossRef]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- Zhao, H.; Li, H.; Hao, S.; Chen, J.; Wu, J.; Song, C.; Zhang, M.; Qiao, T.; Li, K. Peptide SS-31 upregulates frataxin expression and improves the quality of mitochondria: Implications in the treatment of Friedreich ataxia. Sci. Rep. 2017, 7, 9840. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Xu, Z.; Cao, J.; Fu, Q.; Wu, Y.; Zhang, X.; Long, Y.; Zhang, X.; Yang, Y.; Li, Y.; et al. Elamipretide (SS-31) improves mitochondrial dysfunction, synaptic and memory impairment induced by lipopolysaccharide in mice. J. Neuroinflammation 2019, 16, 1–19. [Google Scholar] [CrossRef]
- Karaa, A.; Haas, R.; Goldstein, A.; Vockley, J.; Cohen, B.H. A randomized crossover trial of elamipretide in adults with primary mitochondrial myopathy. J. Cachexia. Sarcopenia Muscle 2020, 11, 909–918. [Google Scholar] [CrossRef] [Green Version]
- An Intermediate Size Expanded Access Protocol of Elamipretide-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04689360?term=elamipretide&draw=2&rank=8 (accessed on 26 October 2021).
- Tauskela, J.S. MitoQ--a mitochondria-targeted antioxidant. IDrugs 2007, 10, 399–412. [Google Scholar]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, Q.; Li, Y.; Zhang, Y.; Wu, Y. MitoQ Modulates Lipopolysaccharide-Induced Intestinal Barrier Dysfunction via Regulating Nrf2 Signaling. Mediators Inflamm. 2020, 2020, 3276148. [Google Scholar] [CrossRef] [Green Version]
- Escribano-Lopez, I.; Bañuls, C.; Diaz-Morales, N.; Iannantuoni, F.; Rovira-Llopis, S.; Gomis, R.; Rocha, M.; Hernandez-Mijares, A.; Murphy, M.P.; Victor, V.M. The mitochondria-targeted antioxidant MitoQ modulates mitochondrial function and endoplasmic reticulum stress in pancreatic β cells exposed to hyperglycaemia. Cell. Physiol. Biochem. 2019, 52, 186–197. [Google Scholar] [CrossRef]
- Ribeiro, R.F., Jr.; Dabkowski, E.R.; Shekar, K.C.; O’Connell, K.A.; Hecker, P.A.; Murphy, M.P. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic. Biol. Med. 2018, 117, 18–29. [Google Scholar] [CrossRef]
- Kang, L.; Liu, S.; Li, J.; Tian, Y.; Xue, Y.; Liu, X. The mitochondria-targeted anti-oxidant MitoQ protects against intervertebral disc degeneration by ameliorating mitochondrial dysfunction and redox imbalance. Cell Prolif. 2020, 53, e12779. [Google Scholar] [CrossRef]
- Effects of Mitochondrial-targeted Antioxidant on Mild Cognitive Impairment (MCI) Patients-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03514875?term=mitoQ&draw=3&rank=14 (accessed on 27 October 2021).
- Vascular Function in Health and Disease-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02966665?term=mitoQ&draw=3&rank=19 (accessed on 27 October 2021).
- MitoQ for Fatigue in Multiple Sclerosis (MS)-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04267926?term=mitoQ&draw=2&rank=3 (accessed on 27 October 2021).
- Goodfellow, M.J.; Borcar, A.; Proctor, J.L.; Greco, T.; Rosenthal, R.E.; Fiskum, G. Transcriptional activation of antioxidant gene expression by Nrf2 protects against mitochondrial dysfunction and neuronal death associated with acute and chronic neurodegeneration. Exp. Neurol. 2020, 328, 113247. [Google Scholar] [CrossRef]
- Rodríguez, L.R.; Lapeña, T.; Calap-Quintana, P.; Moltó, M.D.; Gonzalez-Cabo, P.; Langa, J.A.N. Antioxidant therapies and oxidative stress in friedreich’s ataxia: The right path or just a diversion? Antioxidants 2020, 9, 664. [Google Scholar] [CrossRef]
- Huang, M.L.H.; Chiang, S.; Kalinowski, D.S.; Bae, D.H.; Sahni, S.; Richardson, D.R. The role of the antioxidant response in mitochondrial dysfunction in degenerative diseases: Cross-talk between antioxidant defense, autophagy, and apoptosis. Oxid. Med. Cell. Longev. 2019, 2019, 6392763. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.D.; Dziedzic, A.; Saluk-Bijak, J.; Bijak, M. A review of various antioxidant compounds and their potential utility as complementary therapy in multiple sclerosis. Nutrients 2019, 11, 1528. [Google Scholar] [CrossRef] [Green Version]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 2020, 12, 12. [Google Scholar] [CrossRef]
- Silvestro, S.; Sindona, C.; Bramanti, P.; Mazzon, E. A state of the art of antioxidant properties of curcuminoids in neurodegenerative diseases. Int. J. Mol. Sci. 2021, 22, 3168. [Google Scholar] [CrossRef] [PubMed]
- Larrea, D.; Pera, M.; Gonnelli, A.; Quintana-Cabrera, R.; Akman, H.O.; Guardia-Laguarta, C.; Velasco, K.R.; Area-Gomez, E.; Dal Bello, F.; de Stefani, D.; et al. MFN2 mutations in charcot-marie-Tooth disease alter mitochondria-Associated er membrane function but do not impair bioenergetics. Hum. Mol. Genet. 2019, 28, 1782–1800. [Google Scholar] [CrossRef] [Green Version]
- Gdynia, H.J.; Kurt, A.; Endruhn, S.; Ludolph, A.C.; Sperfeld, A.D. Cardiomyopathy in motor neuron diseases. J. Neurol. Neurosurg. Psychiatry 2006, 77, 671–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.J.; Goodsell, K.; Grogan, M.; Ackerman, M.J. LMNA-Mediated Arrhythmogenic Right Ventricular Cardiomyopathy and Charcot-Marie-Tooth Type 2B1: A Patient-Discovered Unifying Diagnosis. J. Cardiovasc. Electrophysiol. 2016, 27, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Störk, S.; Liu, D.; Hu, K.; Herrmann, S.; Ertl, G.; Niemann, M. Cardiomyopathy of Friedreich ataxia. J. Neurochem. 2013, 126, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Espinós, C.; Galindo, M.I.; García-Gimeno, M.A.; Ibáñez-Cabellos, J.S.; Martínez-Rubio, D.; Millán, J.M.; Rodrigo, R.; Sanz, P.; Seco-Cervera, M.; Sevilla, T.; et al. Oxidative stress, a crossroad between rare diseases and neurodegeneration. Antioxidants 2020, 9, 313. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Sun, Q.; Chen, S. Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog. Neurobiol. 2016, 147, 1–19. [Google Scholar] [CrossRef]
- Lu, X.; Yang, H.; Chen, Y.; Li, Q.; He, S.; Jiang, X.; Feng, F.; Qu, W.; Sun, H. The Development of Pharmacophore Modeling: Generation and Recent Applications in Drug Discovery. Curr. Pharm. Des. 2018, 24, 3424–3439. [Google Scholar] [CrossRef]
- Pinzi, L.; Rastelli, G. Molecular docking: Shifting paradigms in drug discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [Green Version]
- Kutlushina, A.; Khakimova, A.; Madzhidov, T.; Polishchuk, P. Ligand-Based Pharmacophore Modeling Using Novel 3D Pharmacophore Signatures. Molecules 2018, 23, 3094. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez, L.R.; Lapeña-Luzón, T.; Benetó, N.; Beltran-Beltran, V.; Pallardó, F.V.; Gonzalez-Cabo, P.; Navarro, J.A. Therapeutic Strategies Targeting Mitochondrial Calcium Signaling: A New Hope for Neurological Diseases? Antioxidants 2022, 11, 165. https://doi.org/10.3390/antiox11010165
Rodríguez LR, Lapeña-Luzón T, Benetó N, Beltran-Beltran V, Pallardó FV, Gonzalez-Cabo P, Navarro JA. Therapeutic Strategies Targeting Mitochondrial Calcium Signaling: A New Hope for Neurological Diseases? Antioxidants. 2022; 11(1):165. https://doi.org/10.3390/antiox11010165
Chicago/Turabian StyleRodríguez, Laura R., Tamara Lapeña-Luzón, Noelia Benetó, Vicent Beltran-Beltran, Federico V. Pallardó, Pilar Gonzalez-Cabo, and Juan Antonio Navarro. 2022. "Therapeutic Strategies Targeting Mitochondrial Calcium Signaling: A New Hope for Neurological Diseases?" Antioxidants 11, no. 1: 165. https://doi.org/10.3390/antiox11010165