
The Interconnected Mechanisms of Oxidative Stress and Neuroinflammation in Epilepsy

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. OS and Epilepsy
3. Role of OS in Neuroinflammation and Epilepsy
4. Excitatory/Inhibitory Imbalance: Relevance to OS and Epilepsy
5. Pharmacological Evidence of the Interaction between OS, Inflammation and Epilepsy
5.1. AEDs and OS
5.2. Antioxidants with Anti-Epileptic Properties
5.3. Anti-Inflammatory Drugs with Anti-Epileptic Properties
6. Future Directions
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organisation. Epilepsy. Available online: www.who.int/news-room/fact-sheets/detail/epilepsy (accessed on 24 November 2021).
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Primers 2018, 4, 18024. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.S.; van Emde Boas, W.; Blume, W.; Elger, C.; Genton, P.; Lee, P.; Engel, J., Jr. Epileptic seizures and epilepsy: Definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005, 46, 470–472. [Google Scholar] [CrossRef]
- Stein, M.A.; Kanner, A.M. Management of newly diagnosed epilepsy: A practical guide to monotherapy. Drugs 2009, 69, 199–222. [Google Scholar] [CrossRef] [PubMed]
- Pennell, P.B. Unravelling the heterogeneity of epilepsy for optimal individualised treatment: Advances in 2019. Lancet Neurol. 2020, 1, 8–10. [Google Scholar] [CrossRef]
- Perucca, P.; Bahlo, M.; Berkovic, S.F. The Genetics of Epilepsy. Ann. Rev. Genom. Hum. Genet. 2020, 21, 205–230. [Google Scholar] [CrossRef]
- Hauser, W.A.; Annegers, J.F.; Kurland, L.T. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935–1984. Epilepsia 1993, 34, 453–468. [Google Scholar] [CrossRef]
- Carvill, G.L.; Matheny, T.; Hesselberth, J.; Demarest, S. Haploinsufficiency, dominant negative, and gain-of-function mechanisms in epilepsy: Matching therapeutic approach to the pathophysiology. Neurotherapeutics 2021, 3, 1500–1514. [Google Scholar] [CrossRef]
- Fukata, Y.; Fukata, M. Epilepsy and synaptic proteins. Curr. Opin. Neurobiol. 2017, 45, 1–8. [Google Scholar] [CrossRef]
- Yang, N.; Guan, Q.W.; Chen, F.H.; Xia, Q.X.; Yin, X.X.; Zhou, H.H.; Mao, X.Y. Antioxidants Targeting Mitochondrial Oxidative Stress: Promising Neuroprotectants for Epilepsy. Oxid. Med. Cell. Longev. 2020, 2020, 6687185. [Google Scholar] [CrossRef]
- Yiȿ, U.; Seςkin, E.; Kurul, S.H.; Kuralay, F.; Dirik, E. Effects of epilepsy and valproic acid on oxidant status in children with idiopathic epilepsy. Epilepsy Res. 2009, 84, 232–237. [Google Scholar] [CrossRef]
- Morimoto, M.; Satomura, S.; Hashimoto, T.; Ito, E.; Kyotani, S. Oxidative stress measurement and prediction of epileptic seizures in children and adults with severe motor and intellectual disabilities. J. Clin. Med. Res. 2016, 8, 437–444. [Google Scholar] [CrossRef] [Green Version]
- Magistretti, J.P.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 4, 883–901. [Google Scholar] [CrossRef] [Green Version]
- Hyder, F.; Fulbright, R.K.; Shulman, R.G.; Rothman, D.L. Glutamatergic function in the resting awake human brain is supported by uniformly high oxidative energy. J. Cereb. Blood Flow Metab. 2013, 3, 339–347. [Google Scholar] [CrossRef] [Green Version]
- Alyu, F.; Dikmen, M. Inflammatory aspects of epileptogenesis: Contribution of molecular inflammatory mechanisms. Acta Neuropsychiatr. 2017, 29, 1–16. [Google Scholar] [CrossRef]
- Mlodzikowska-Albrecht, J.; Steinborn, B.; Zarowski, M. Cytokines, epilepsy and epileptic drugs—Is there a mutual influence? Pharmacol. Rep. 2007, 59, 129–138. [Google Scholar]
- Vezzani, A.; Granata, T. Brain inflammation in epilepsy: Experimental and clinical evidence. Epilepsia 2005, 46, 1724–1743. [Google Scholar] [CrossRef]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Cobb, C.A.; Cole, M.P. Oxidative and nitrative stress in neurodegeneration. Neurobiol. Dis. 2015, 84, 4–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinós, C.; Galindo, M.I.; García-Gimeno, M.A.; Ibáñez-Cabellos, J.S.; Martínez-Rubio, D.; Millán, J.M.; Rodrigo, R.; Sanz, P.; Seco-Cervera, M.; Sevilla, T.; et al. Oxidative stress, a crossroad between rare diseases and neurodegeneration. Antioxidants 2020, 9, 313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marklund, S.L. Human copper-containing superoxide dismutase of high molecular weight. Proc. Natl. Acad. Sci. USA 1982, 79, 7634–7638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, L.P.; Waldbaum, S.; Rowley, S.; Huang, T.T.; Day, B.J.; Patel, M. Mitochondrial oxidative stress and epilepsy in SOD2 deficient mice: Attenuation by a lipophilic metalloporphyrin. Neurobiol. Dis. 2012, 3, 1068–1076. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.P.; Patel, M. Mitochondrial oxidative stress and increased seizure susceptibility in Sod2 (−/+) mice. Free Radic. Biol. Med. 2004, 36, 542–554. [Google Scholar] [CrossRef]
- Rotruck, J.T.; Pope, A.L.; Ganther, H.E.; Swanson, A.B.; Hafeman, D.G.; Hoekstra, W.G. Selenium: Biochemical role as a component of glutathione peroxidase. Science 1973, 4073, 588–590. [Google Scholar] [CrossRef]
- Keskin Guler, S.; Aytac, B.; Durak, Z.E.; Gokce Cokal, B.; Gunes, N.; Durak, I.; Yoldas, T. Antioxidative-oxidative balance in epilepsy patients on antiepileptic therapy: A prospective case-control study. Neurol. Sci. 2016, 5, 763–767. [Google Scholar] [CrossRef]
- Gluck, M.R.; Jayatilleke, E.; Shaw, S.; Rowan, A.J.; Haroutunian, V. CNS oxidative stress associated with the kainic acid rodent model of experimental epilepsy. Epilepsy Res. 2000, 39, 63–71. [Google Scholar] [CrossRef]
- Gutteridge, J.M.C. Antioxidant properties of the proteins caeruloplasmin, albumin and transferrin a study of their activity in serum and synovial fluid from patients with rheumatoid arthritis. Biochim. Biophys. Acta 1986, 2, 119–127. [Google Scholar] [CrossRef]
- Correale, J.; Rabinowicz, A.L.; Heck, C.N.; Smith, T.D.; Loskota, W.J.; DeGiorgio, C.M. Status epilepticus increases CSF levels of neuron-specific enolase and alters the blood-brain barrier. Neurology 1998, 50, 1388–1391. [Google Scholar] [CrossRef]
- Tumani, H.; Jobs, C.; Brettschneider, J.; Hoppner, A.C.; Kerling, F.; Fauser, S. Effect of epileptic seizures on the cerebrospinal fluid–A systematic retrospective analysis. Epilepsy Res. 2015, 114, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Favreau, L.V.; Pickett, C.B. Transcriptional regulation of the rat NAD(P)H: Quinone reductase gene. J. Biol. Chem. 1991, 7, 4556–4561. [Google Scholar] [CrossRef]
- Kwong, M.; Kan, Y.W.; Chan, J.Y. The CNC basic leucine zipper factor, Nrf1, is essential for cell survival in response to oxidative stress-inducing agents. J. Biol. Chem. 1999, 52, 37491–37498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, L.; Kwong, M.; Hou, S.; Lee, C.; Chan, J.Y. Deficiency of the Nrf1 and Nrf2 transcription factors results in early embryonic lethality and severe oxidative stress. J. Biol. Chem. 2003, 48, 48021–48029. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.C. The Nrf2-ARE pathway: An indicator and modulator of oxidative stress in neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Sherratt, P.J.; Nioi, P.; Yang, C.S.; Pickett, C.B. Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J. Biol. Chem. 2005, 37, 32485–32492. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, W.P.; Zhang, G.L.; Wu, Y.F.; Xie, T.; Kan, M.C.; Fang, H.B.; Wang, H.C. Activation of Nrf2-ARE signal pathway in hippocampus of amygdala kindling rats. Neurosci. Lett. 2013, 543, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Luo, F.; Zhang, Q.; Sang, Y.; Chen, X.; Zhang, L.; Liu, Y.; Li, X.; Li, J.; Ding, H.; et al. α-Lipoic acid alleviates pentetrazol-induced neurological deficits and behavioral dysfunction in rats with seizures via an Nrf2 pathway. RSC Adv. 2018, 8, 4084–4092. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.N.; Dong, H.T.; Yang, F.B.; Wang, Z.Q.; Ma, Z.H.; Ma, S.Z.; Ma, X.D.; Duan, L. Nrf2-ARE signaling pathway regulates the expressions of A1R and ENT1 in the brain of epileptic rats. Eur. Rev. Med. Pharmacol. Sci. 2018, 20, 6896–6904. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, C.; Meng, X.; Li, Z.; Lv, C.; Cao, P. Neuroprotection of edaravone on the hippocampus of kainate-induced epilepsy rats through Nrf2/HO-1 pathway. Neurochem. Int. 2018, 112, 159–165. [Google Scholar] [CrossRef]
- Oswald, M.C.W.; Garnham, N.; Sweeney, S.T.; Landgraf, M. Regulation of neuronal development and function by ROS. FEBS Letters 2018, 5, 679–691. [Google Scholar] [CrossRef]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-activated protein kinases and reactive oxygen species: How can ROS activate MAPK pathways? J. Signal Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef]
- Gloire, G.; Legrand-Poels, S.; Piette, J. NF-ΚB activation by reactive oxygen species: Fifteen years later. Biochem. Pharmacol. 2006, 11, 1493–1505. [Google Scholar] [CrossRef]
- Schreck, R.; Rieber, P.; Baeuerle, P.A. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-Kappa B transcription factor and HIV-1. EMBO J. 1991, 8, 2247–2258. [Google Scholar] [CrossRef]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef]
- Webster, K.M.; Sun, M.; Crack, P.; O’Brien, T.J.; Shultz, S.R.; Semple, B.D. Inflammation in epileptogenesis after traumatic brain injury. J. Neuroinflamm. 2017, 14, 10. [Google Scholar] [CrossRef] [Green Version]
- Eastman, C.L.; D’Ambrosio, R.; Ganesh, T. Modulating neuroinflammation and oxidative stress to prevent epilepsy and improve outcomes after traumatic brain injury. Neuropharmacology 2020, 172, 107907. [Google Scholar] [CrossRef] [PubMed]
- Bajwa, E.; Pointer, C.B.; Klegeris, A. The role of mitochondrial damage-associated molecular patterns in chronic neuroinflammation. Mediat. Inflamm. 2019, 2019, 4050796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugrin JRosenblatt-Velin, N.; Parapanov, R.; Liaudet, L. The role of oxidative stress during inflammatory processes. Biol. Chem. 2014, 395, 203–230. [Google Scholar] [CrossRef] [Green Version]
- Maroso, M.; Balosso, S.; Ravizza, T.; Liu, J.; Aronica, E.; Iyer, A.; Rossetti, C.; Molteni, M.; Casalgrandi, M.; Manfredi, A.A.; et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 2010, 16, 413–419. [Google Scholar] [CrossRef] [Green Version]
- Vezzani, B.; Carinci, M.; Patergnani, S.; Pasquin, M.P.; Guarino, A.; Aziz, N.; Pinton, P.; Simonato, M.; Giorgi, C. The dichotomous role of inflammation in the CNS: A mitochondrial point of view. Biomolecules. 2020, 10, 1437. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhang, L.; Teng, J.; Miao, W. HMGB1 mediates microglia activation via the TLR4/NF-ΚB pathway in coriaria lactone induced epilepsy. Mol. Med. Rep. 2018, 4, 5125–5131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, S.; Zheng, Y.; Wang, Y.; Chen, Z. HMGB1, neuronal excitability and epilepsy. Acta Epileptol. 2021, 3, 13. [Google Scholar] [CrossRef]
- Victor, T.R.; Tsirka, S.E. Microglial contributions to aberrant neurogenesis and pathophysiology of epilepsy. Neuroimmunol. Neuroinflamm. 2020, 7, 234–247. [Google Scholar] [CrossRef] [PubMed]
- McElroy, P.B.; Liang, L.P.; Day, B.J.; Patel, M. Scavenging reactive oxygen species inhibits status epilepticus-induced neuroinflammation. Exp. Neurol. 2017, 298, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Badawi, G.A.; Shokr, M.M.; Zaki, H.F.; Mohamed, A.F. Pentoxifylline prevents epileptic seizure via modulating HMGB1/RAGE/TLR4 signalling pathway and improves memory in pentylenetetrazol kindling rats. Clin. Exp. Pharmacol. Physiol. 2021, 8, 1111–1124. [Google Scholar] [CrossRef]
- De Deus, J.L.; Amorim, M.R.; de Barcellos Filho, P.C.G.; de Oliveira, J.A.C.; Batalhão, M.E.; Garcia-Cairasco, N.; Cárnio, E.C.; Leão, R.M.; Branco, L.G.S.; Cunha, A.O.S. Inflammatory markers in the hippocampus after audiogenic kindling. Neurosci. Lett. 2020, 721, 134830. [Google Scholar] [CrossRef] [PubMed]
- Terrone, G.; Balosso, S.; Pauletti, A.; Ravizza, T.; Vezzani, A. Inflammation and reactive oxygen species as disease modifiers in epilepsy. Neuropharmacology 2020, 167, 107742. [Google Scholar] [CrossRef]
- Terrone, G.; Frigerio, F.; Balosso, S.; Ravizza, T.; Vezzani, A. Inflammation and reactive oxygen species in status epilepticus: Biomarkers and implications for therapy. Epilepsy Behav. 2019, 101, 106275. [Google Scholar] [CrossRef]
- Xia, L.; Pan, S.Q.; Zhang, Q.M.; Zhou, Q.; Xia, L.; Lu, Z.N. Elevated IL-6 and IL-1β are associated with temporal lobe epilepsy: A study in chinese patients. Eur. J. Inflamm. 2018, 16, 205873921877893. [Google Scholar] [CrossRef] [Green Version]
- Ethemoglu, O.; Ay, H.; Koyuncu, I.; Gonel, A. Comparison of cytokines and prooxidants/antioxidants markers among adults with refractory versus well-controlled epilepsy: A cross-sectional study. Seizure 2018, 60, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Pecoraro-Bisogni, F.; Lignani, G.; Contestabile, A.; Castroflorio, E.; Pozzi, D.; Rocchi, A.; Prestigio, C.; Orlando, M.; Valente, P.; Massacesi, M.; et al. REST-Dependent presynaptic homeostasis induced by chronic neuronal hyperactivity. Mol. Neurobiol. 2018, 55, 4959–4972. [Google Scholar] [CrossRef]
- Turrigiano, G.G.; Nelson, S.B. Homeostatic plasticity in the developing nervous system. Nat. Rev. Neurosci. 2004, 5, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Guarnieri, F.C.; Pozzi, D.; Raimondi, A.; Fesce, R.; Valente, M.M.; Delvecchio, V.S.; Van Esch, H.; Matteoli, M.; Benfenati, F.; D’Adamo, P.; et al. A novel SYN1 missense mutation in non-syndromic X-linked intellectual disability affects synaptic vesicle life cycle, clustering and mobility. Hum. Mol. Genet. 2017, 26, 4699–4714. [Google Scholar] [CrossRef]
- Valente, P.; Castroflorio, E.; Rossi, P.; Fadda, M.; Sterlini, B.; Cervigni, R.I.; Prestigio, C.; Giovedì, S.; Onofri, F.; Mura, E.; et al. PRRT2 is a key component of the Ca(2+)-dependent neurotransmitter release machinery. Cell Rep. 2016, 15, 117–131. [Google Scholar] [CrossRef] [Green Version]
- Buckmaster, P.S.; Yamawaki, R.; Thind, K. More docked vesicles and larger active zones at basket cell-to-granule cell synapses in a rat model of temporal lobe epilepsy. J. Neurosci. 2016, 36, 3295–3308. [Google Scholar] [CrossRef] [Green Version]
- Colasante, G.; Qiu, Y.; Massimino, L.; Di Berardino, C.; Cornford, J.H.; Snowball, A.; Weston, M.; Jones, S.P.; Giannelli, S.; Lieb, A.; et al. In vivo CRISPRa decreases seizures and rescues cognitive deficits in a rodent model of epilepsy. Brain 2020, 143, 891–905. [Google Scholar] [CrossRef] [Green Version]
- Salpietro, V.; Dixon, C.L.; Guo, H.; Bello, O.D.; Vandrovcova, J.; Efthymiou, S.; Maroofian, R.; Heimer, G.; Burglen, L.; Valence, S.; et al. AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat. Commun. 2019, 10, 3094. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.L.; Bourne, J.N.; Cao, G.; Chirillo, M.A.; Ostroff, L.E.; Watson, D.J.; Harris, K.M. Mitochondrial support of persistent presynaptic vesicle mobilization with age-dependent synaptic growth after LTP. Elife 2016, 5, e15275. [Google Scholar] [CrossRef]
- Augustin, K.; Khabbush, A.; Williams, S.; Eaton, S.; Orford, M.; Cross, J.H.; Heales, S.J.R.; Walker, M.C.; Williams, R.S.B. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018, 17, 84–93. [Google Scholar] [CrossRef]
- Maru, E.; Kanda, M.; Ashida, H. Functional and morphological changes in the hippocampal neuronal circuits associated with epileptic seizures. Epilepsia 2002, 9, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Repetto, D.; Camera, P.; Melani, R.; Morello, N.; Russo, I.; Calcagno, E.; Tomasoni, R.; Bianchi, F.; Berto, G.; Giustetto, M.; et al. P140Cap regulates memory and synaptic plasticity through Src-mediated and citron-n-mediated actin reorganization. J. Neurosci. 2014, 34, 1542–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Südhof, T.C. The presynaptic active zone. Neuron 2012, 75, 11–25. [Google Scholar] [CrossRef] [Green Version]
- Sweatt, J.D. Dynamic DNA methylation controls glutamate receptor trafficking and synaptic scaling. J. Neurochem. 2016, 137, 312–330. [Google Scholar] [CrossRef] [Green Version]
- Clayton, D.F.; Anreiter, I.; Aristizabal, M.; Frankland, P.W.; Binder, E.B.; Citri, A. The role of the genome in experience-dependent plasticity: Extending the analogy of the genomic action potential. Proc. Natl. Acad. Sci. USA 2020, 117, 23252–23260. [Google Scholar] [CrossRef] [Green Version]
- Tien, N.W.; Kerschensteiner, D. Homeostatic plasticity in neural development. Neural Dev. 2018, 13, 9. [Google Scholar] [CrossRef]
- Méndez-Armenta, M.; Nava-Ruíz, C.; Juárez-Rebollar, D.; Rodríguez-Martínez, E.; Gómez, P.Y. Oxidative stress associated with neuronal apoptosis in experimental models of epilepsy. Oxid. Med. Cell. Longev. 2014, 2014, 293689. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; Orlando, M.; Andrzejak, E.; Bruns, C.; Trimbuch, T.; Rosenmund, C.; Garner, C.C.; Ackermann, F. Light-activated ROS production induces synaptic autophagy. J. Neurosci. 2019, 12, 2163–2183. [Google Scholar] [CrossRef] [Green Version]
- Sears, S.M.; Hewett, S.J. Influence of glutamate and GABA transport on brain excitatory/inhibitory balance. Exp. Biol. Med. 2021, 9, 1069–1083. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.R.; Binder, D.K. Regulation of synaptosomal GLT-1 and GLAST during epileptogenesis. Neuroscience 2019, 411, 185–201. [Google Scholar] [CrossRef] [PubMed]
- Schijns, O.E.; Bisschop, J.; Rijkers, K.; Dings, J.; Vanherle, S.; Lindsey, P.; Smeets, H.J.; Hoogland, G. GAT-1 (rs2697153) and GAT-3 (rs2272400) polymorphisms are associated with febrile seizures and temporal lobe epilepsy. Epileptic Disord. 2020, 2, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Petroff, O.A. GABA and glutamate in the human brain. Neuroscientist 2002, 8, 562–573. [Google Scholar] [CrossRef]
- During, M.J.; Spencer, D.D. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 1993, 341, 1607–1610. [Google Scholar] [CrossRef]
- Eid, T.; Williamson, A.; Lee, T.S.; Petroff, O.A.; de Lanerolle, N.C. Glutamate and astrocytes—key players in human mesial temporal lobe epilepsy? Epilepsia 2008, 49, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Rae, C.; Moussa, C.e.l.-H.; Griffin, J.L.; Parekh, S.B.; Bubb, W.A.; Hunt, N.H.; Balcar, V.J. A metabolomic approach to ionotropic glutamate receptor subtype function: A nuclear magnetic resonance in vitro investigation. J. Cereb. Blood Flow Metab. 2006, 8, 1005–1017. [Google Scholar] [CrossRef]
- Eid, T.; Gruenbaum, S.E.; Dhaher, R.; Lee, T.W.; Zhou, Y.; Danbolt, N.C. The glutamate-glutamine cycle in epilepsy. Adv. Neurobiol. 2016, 13, 351–400. [Google Scholar] [CrossRef] [PubMed]
- Hanada, T. Ionotropic glutamate receptors in epilepsy: A review focusing on AMPA and NMDA receptors. Biomolecules 2020, 10, 464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Vishnoi, S.; Raisuddin, S.; Parvez, S. Glutamate excitotoxicity and oxidative stress in epilepsy: Modulatory role of melatonin. J. Environ Pathol Toxicol. Oncol 2016, 35, 365–374. [Google Scholar] [CrossRef]
- Peng, W.F.; Ding, J.; Li, X.; Fan, F.; Zhang, Q.Q.; Wang, X. N-methyl-d-aspartate receptor NR2B subunit involved in depression-like behaviours in lithium chloride-pilocarpine chronic rat epilepsy model. Epilepsy Res. 2016, 119, 77–85. [Google Scholar] [CrossRef]
- Chen, K.; Baram, T.Z.; Soltesz, I. Febrile seizures in the developing brain result in persistent modification of neuronal excitability in limbic circuits. Nat. Med. 1999, 8, 888–894. [Google Scholar] [CrossRef]
- Barker-Haliski, M.; White, H.S. Glutamatergic mechanisms associated with seizures and epilepsy. Cold Spring Harb Perspect. Med. 2015, 5, 022863. [Google Scholar] [CrossRef] [Green Version]
- Levite, M.; Zelig, D.; Friedman, A.; Ilouz, N.; Eilam, R.; Bromberg, Z.; Lasu, A.A.R.; Arbel-Alon, S.; Edvardson, S.; Tarshish, M.; et al. Dual-targeted autoimmune sword in fatal epilepsy: Patient’s glutamate receptor AMPA GluR3B peptide autoimmune antibodies bind, induce reactive oxygen species (ROS) in, and kill both human neural cells and T cells. J. Autoimmun. 2020, 112, 102462. [Google Scholar] [CrossRef]
- Schousboe, A.; Scafidi, S.; Bak, L.K.; Waagepetersen, H.S.; McKenna, M.C. Glutamate metabolism in the brain focusing on astrocytes. Adv. Neurobiol. 2014, 11, 13–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, D.C.; Tewari, B.P.; Chaunsali, L.; Sontheimer, H. Neuron-glia interactions in the pathophysiology of epilepsy. Nat. Rev. Neurosci. 2019, 20, 282–297. [Google Scholar] [CrossRef] [PubMed]
- Twible, C.; Abdo, R.; Zhang, Q. Astrocyte role in temporal lobe epilepsy and development of mossy fiber sprouting. Front. Cell. Neurosci. 2021, 15, 725693. [Google Scholar] [CrossRef] [PubMed]
- Proper, E.A.; Hoogland, G.; Kappen, S.M.; Jansen, G.H.; Rensen, M.G.; Schrama, L.H.; van Veelen, C.W.; van Rijen, P.C.; van Nieuwenhuizen, O.; Gispen, W.H.; et al. Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain 2002, 125 Pt 1, 32–43. [Google Scholar] [CrossRef]
- Nikolic, L.; Nobili, P.; Shen, W.; Audinat, E. Role of astrocyte purinergic signaling in epilepsy. Glia 2020, 68, 1677–1691. [Google Scholar] [CrossRef]
- Nikolic, L.; Shen, W.; Nobili, P.; Virenque, A.; Ulmann, L.; Audinat, E. Blocking TNFα-driven astrocyte purinergic signaling restores normal synaptic activity during epileptogenesis. Glia 2018, 66, 2673–2683. [Google Scholar] [CrossRef]
- Greenfield Jr, L.J. Molecular mechanisms of antiseizure drug activity at GABAA receptors. Seizure 2013, 22, 589–600. [Google Scholar] [CrossRef] [Green Version]
- Ghit, A.; Assal, D.; Al-Shami, A.S.; Hussein, D.E.E. GABAA receptors: Structure, function, pharmacology, and related disorders. J. Genet. Eng. Biotechnol. 2021, 1, 123. [Google Scholar] [CrossRef]
- McDonald, J.W.; Garofalo, E.A.; Hood, T.; Sackellares, J.C.; Gilman, S.; McKeever, P.E.; Troncoso, J.C.; Johnston, M.V. Altered excitatory and inhibitory amino acid receptor binding in hippocampus of patients with temporal lobe epilepsy. Ann. Neurol. 1991, 29, 529–541. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.W.; de Lanerolle, N.C.; Kim, J.H.; Sundaresan, S.; Spencer, D.D.; Mattson, R.H.; Zoghbi, S.S.; Baldwin, R.M.; Hoffer, P.B.; Seibyl, J.P. Central and peripheral benzodiazepine receptors: Opposite changes in human epileptogenic tissue. Neurology 1992, 42, 811–815. [Google Scholar] [CrossRef]
- Savic, I.; Persson, A.; Roland, P.; Pauli, S.; Sedvall, G.; Widén, L. In-vivo demonstration of reduced benzodiazepine receptor binding in human epileptic foci. Lancet 1988, 2, 863–866. [Google Scholar] [CrossRef]
- Henry, T.R.; Frey, K.A.; Sackellares, J.C.; Gilman, S.; Koeppe, R.A.; Brunberg, J.A.; Ross, D.A.; Berent, S.; Young, A.B.; Kuhl, D.E. In vivo cerebral metabolism and central benzodiazepine-receptor binding in temporal lobe epilepsy. Neurology 1993, 43, 1998–2006. [Google Scholar] [CrossRef]
- Amato, A.; Connolly, C.N.; Moss, S.J.; Smart, T.G. Modulation of neuronal and recombinant GABAA receptors by redox reagents. J. Physiol. 1999, 517, 35–50. [Google Scholar] [CrossRef]
- Pan, Z.H.; Zhang, X.; Lipton, S.A. Redox modulation of recombinant human GABAA receptors. Neuroscience 2000, 98, 333–338. [Google Scholar] [CrossRef]
- Accardi, M.V.; Daniels, B.A.; Brown, P.M.G.E.; Fritschy, J.M.; Tyagarajan, S.K.; Bowie, D. Mitochondrial reactive oxygen species regulate the strength of inhibitory GABA-mediated synaptic transmission. Nat. Commun. 2014, 5, 3168. [Google Scholar] [CrossRef] [PubMed]
- Frantseva, M.V.; Perez, J.L.; Carlen, P.L. Changes in membrane and synaptic properties of thalamocortical circuitry caused by hydrogen peroxide. J. Neurophysiol. 1998, 80, 1317–1326. [Google Scholar] [CrossRef] [Green Version]
- Akyuz, E.; Polat, A.K.; Eroglu, E.; Kullu, I.; Angelopoulou, E.; Paudel, Y.N. Revisiting the role of neurotransmitters in epilepsy: An updated review. Life Sci. 2021, 15, 265–118826. [Google Scholar] [CrossRef] [PubMed]
- Kalilani, L.; Sun, X.; Pelgrims, B.; Noack-Rink, M.; Villanueva, V. The epidemiology of drug-resistant epilepsy: A systematic review and meta-analysis. Epilepsia 2018, 12, 2179–2193. [Google Scholar] [CrossRef] [Green Version]
- Steriade, C.; French, J.; Devinsky, O. Epilepsy: Key experimental therapeutics in early clinical development. Exp. Opin. Investig. Drugs 2020, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Nazıroğlu, M.; Yürekli, V.A. Effects of antiepileptic drugs on antioxidant and oxidant molecular pathways: Focus on trace elements. Cell. Mol. Neurobiol. 2013, 33, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Mahle, C.; Dasgupta, A. Decreased total antioxidant capacity and elevated lipid hydroperoxide concentrations in sera of epileptic patients receiving phenytoin. Life Sci. 1997, 61, 437–443. [Google Scholar] [CrossRef]
- Liu, C.S.; Wu, H.M.; Kao, S.H.; Wei, Y.H. Phenytoin-mediated oxidative stress in serum of female epileptics: A possible pathogenesis in the fetal hydantoin syndrome. Hum. Exp. Toxicol. 1997, 16, 177–181. [Google Scholar] [CrossRef]
- Ficarra, S.; Misiti, F.; Russo, A.; Carelli-Alinovi, C.; Bellocco, E.; Barreca, D.; Laganà, G.; Leuzzi, U.; Toscano, G.; Giardina, B.; et al. Antiepileptic carbamazepine drug treatment indices alteration of membrane in red blood cells: Possible positive effects on metabolism and oxidative stress. Biochimie 2013, 95, 833–841. [Google Scholar] [CrossRef]
- Gathwala, G.; Marwah, A.; Gahlaut, V.; Marwah, P. Effect of high-dose phenobarbital on oxidative stress in perinatal asphyxia: An open label randomized controlled trial. Indian Pediatr. 2011, 48, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, K. Seizure-induced oxidative stress in rat brain regions: Blockade by nNOS inhibition. Pharmacol. Biochem. Behav. 2005, 80, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Krauss, G.L.; Klein, P.; Brandt, C.; Lee, S.K.; Milanov, I.; Milovanovic, M.; Steinhoff, B.J.; Kamin, M. Safety and efficacy of adjunctive cenobamate (YKP3089) in patients with uncontrolled focal seizures: A multicentre, double-blind, randomised, placebo-controlled, dose-response trial. Lancet Neurol. 2020, 19, 38–48. [Google Scholar] [CrossRef]
- Wiciński, M.; Puk, O.; Malinowski, B. Cenobamate: Neuroprotective potential of a new antiepileptic drug. Neurochem. Res. 2021, 46, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kalonia, H.; Kumar, A. Possible GABAergic mechanism in the neuroprotective effect of gabapentin and lamotrigine against 3-nitropropionic acid induced neurotoxicity. Eur. J. Pharmacol. 2012, 674, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Devinsky, O.; Marsh, E.; Friedman, D.; Thiele, E.; Laux, L.; Sullivan, J.; Miller, I.; Flamini, R.; Wilfong, A.; Filloux, F.; et al. Cannabidiol in patients with treatment-resistant epilepsy: An open-label interventional trial. Lancet Neurol. 2016, 270–278. [Google Scholar] [CrossRef]
- Gray, R.A.; Whalley, B.J. The proposed mechanisms of action of CBD in epilepsy. Epileptic Disord. 2020, S1, 10–15. [Google Scholar] [CrossRef]
- Atalay, S.; Jarocka-Karpowicz, I.; Skrzydlewska, E. Antioxidative and anti-inflammatory properties of cannabidiol. Antioxidants 2019, 9, 21. [Google Scholar] [CrossRef] [Green Version]
- Shakeel, S.; Rehman, M.U.; Tabassum, N.; Amin, U.; Mir, M.U.R. Effect of Naringenin (a naturally occurring flavanone) against pilocarpine-induced status epilepticus and oxidative stress in mice. Pharmacogn. Mag. 2017, 13 (Suppl. 1), S154–S160. [Google Scholar] [CrossRef]
- Tawfik, M.K. Coenzyme Q10 enhances the anticonvulsant effect of phenytoin in pilocarpine-induced seizures in rats and ameliorates phenytoin-induced cognitive impairment and oxidative stress. Epilepsy Behav. 2011, 22, 671–677. [Google Scholar] [CrossRef]
- Shin, E.J.; Suh, S.K.; Lim, Y.K.; Hjelle, O.P.; Ottersen, O.P.; Shin, C.Y.; Ko, K.H.; Kim, W.-K.; Kim, D.S.; Chun, W.; et al. Ascorbate attenuates trimethyltin-induced oxidative burden and neuronal degeneration in the rat hippocampus by maintaining glutathione homeostasis. Neuroscience 2005, 33, 715–727. [Google Scholar] [CrossRef] [Green Version]
- Dhir, A. Curcumin in epilepsy disorders: Curcumin and epilepsy. Phytother. Res. 2018, 10, 1865–1875. [Google Scholar] [CrossRef]
- Mehvari, J.; Motlagh, F.G.; Najafi, M.; Ghazvini, M.R.A.; Naeini, A.A.; Zare, M. Effects of vitamin E on seizure frequency, electroencephalogram findings, and oxidative stress status of refractory epileptic patients. Adv. Biomed. Res. 2016, 5, 36. [Google Scholar] [CrossRef]
- Wang, W.; Wu, Y.; Zhang, G.; Fang, H.; Wang, H.; Zang, H.; Xie, T.; Wang, W. Activation of Nrf2-ARE signal pathway protects the brain from damage induced by epileptic seizure. Brain Res. 2014, 1544, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Kenney-Jung, D.; Vezzani, A.; Kahoud, R.J.; LaFrance-Corey, R.G.; Ho Mai-Lan Muskardin, T.W.; Wirrell, E.C.; Howe, C.L.; Payne, E.T. Febrile infection- related epilepsy syndrome treated with anakinra. Ann. Neurol. 2016, 80, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Dilena, R.; Mauri, E.; Aronica, E.; Bernasconi, P.; Bana, C.; Cappelletti, C.; Carrabba, G.; Ferrero, S.; Giorda, R.; Guez, S.; et al. Therapeutic effect of anakinra in the relapsing chronic phase of febrile infection–related epilepsy syndrome. Epilepsia Open 2019, 4, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Jyonouchi, H.; Geng, L. Intractable epilepsy (IE) and responses to anakinra, a human recombinant IL-1 receptor antagonist (IL-1Ra): Case reports. J. Clin. Cell. Immunol. 2016, 7, 456–460. [Google Scholar] [CrossRef] [Green Version]
- DeSena, A.D.; Do, T.; Schulert, G.S. Systemic autoinflammation with intractable epilepsy managed with interleukin-1 blockade. J. Neuroinflamm. 2018, 15, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, J.S.; Lee, S.T.; Kim, R.; Chu, K.; Lee, S.K. Tocilizumab treatment for new onset refractory status epilepticus. Ann. Neurol. 2018, 84, 940–945. [Google Scholar] [CrossRef]
- Cantarín-Extremera, V.; Jiménez-Legido, M.; Duat-Rodríguez, A.; García-Fernández, M.; Ortiz-Cabrera, N.V.; Ruiz-Falcó-Rojas, M.L.; González-Gutiérrez-Solana, L. Tocilizumab in pediatric refractory status epilepticus and acute epilepsy: Experience in two patients. J. Neuroimmunol. 2020, 340, 577142. [Google Scholar] [CrossRef]
- Nowak, M.; Strzelczyk, A.; Reif, P.S.; Schorlemmer, K.; Bauer, S.; Norwood, B.A.; Oertel, W.H.; Rosenow, F.; Strik, H.; Hamer, H.M. Minocycline as potent anticonvulsivant in a patient with astrocytoma and drug resistant epilepsy. Seizure 2012, 21, 227–228. [Google Scholar] [CrossRef] [Green Version]
- Lagarde, S.; Villeneuve, N.; Trébuchon, A.; Kaphan, E.; Lepine, A.; McGonigal, A.; Roubertie, A.; Barthez, M.J.; Trommsdorff, V.; Lefranc, J.; et al. Anti-tumor necrosis factor alpha therapy (adalimumab) in Rasmussen’s encephalitis: An open pilot study. Epilepsia 2016, 57, 956–966. [Google Scholar] [CrossRef]
- Lance, E.I.; Sreenivasan, A.K.; Zabel, T.A.; Kossoff, E.H.; Comi, A.M. Aspirin use in Sturge- Weber syndrome: Side effects and clinical outcomes. J. Child Neurol. 2013, 28, 213–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godfred, R.M.; Parikh, M.S.; Haltiner, A.M.; Caylor, L.M.; Sepkuty, J.P.; Doherty, M.J. Does aspirin use make it harder to collect seizures during elective video- EEG telemetry? Epilepsy Behav. 2013, 27, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Bialer, M.; Johannessen, S.I.; Levy, R.H.; Perucca, E.; Tomson, T.; HWhite, S. Progress report on new antiepileptic drugs: A summary of the Eleventh Eilat Conference (EILAT XI). Epilepsy Res. 2013, 103, 2–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, E.G.; Chaffe, H.; Schwartz, R.H.; Lawson, M.S.; Edwards, N.; Fitzsimmons, G.; Whitney, A.; Cross, J.H. The ketogenic diet for the treatment of childhood epilepsy: A randomised controlled trial. Lancet Neurol. 2008, 6, 500–506. [Google Scholar] [CrossRef]
- Simeone, T.A.; Matthews, S.A.; Samson, K.K.; Simeone, K.A. Regulation of brain PPARgamma2 contributes to ketogenic diet anti-seizure efficacy. Exp. Neurol. 2017, 287, 54–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, S.; Budney, S.; Deodhar, M.; Matthews, S.A.; Simeone, K.A.; Simeone, T.A. Ketogenic diet regulates the antioxidant catalase via the transcription factor PPARγ2. Epilepsy Res. 2018, 147, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by -hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Dalle-Donne, I.; Scaloni, A.; Giustarini, D.; Cavarra, E.; Tell, G.; Lungarella, G.; Colombo, R.; Rossi, R.; Milzani, A. Proteins as biomarkers of oxidative/nitrosative stress in diseases: The contribution of redox proteomics. Mass Spectrom. Rev. 2005, 24, 55–99. [Google Scholar] [CrossRef]
- Farah, M.E.; Sirotkin, V.; Haarer, B.; Kakhniashvili, D.; Amberg, D.C. Diverse protective roles of the actin cytoskeleton during oxidative stress. Cytoskeleton 2011, 68, 340–354. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, C.; Carrasco, M.A.; Muñoz, P.; Núñez, M.T. A role for reactive oxygen/nitrogen species and iron on neuronal synaptic plasticity. Antioxid. Redox Signal. 2007, 9, 245–255. [Google Scholar] [CrossRef]
- Ulasov, A.V.; Rosenkranz, A.A.; Georgiev, G.P.; Sobolev, A.S. Nrf2/Keap1/ARE signaling: Towards specific regulation. Life Sci. 2021, 120111. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.D.; Hsu, A.; Williams, D.E.; Dashwood, R.H.; Stevens, J.F.; Yamamoto, M.; Ho, E. Metabolism and tissue distribution of sulforaphane in Nrf2 knockout and wild-type mice. Pharm. Res. 2011, 28, 3171–3179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shekh-Ahmad, T.; Eckel, R.; Naidu, S.D.; Higgins, M.; Yamamoto, M.; Dinkova-Kostova, A.T.; Kovac, S.; Abramov, A.Y.; Walker, M.C. KEAP1 inhibition is neuroprotective and suppresses the development of epilepsy. Brain 2018, 5, 1390–1403. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.R.; Chin, M.P.; Delatycki, M.B.; Subramony, S.H.; Corti, M.; Hoyle, J.C.; Boesch, S.; Nachbauer, W.; Mariotti, C.; Mathews, K.D.; et al. Safety and efficacy of Omaveloxolone in Friedreich ataxia (MOXIe Study). Ann. Neurol. 2021, 2, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Shekh-Ahmad, T.; Lieb, A.; Kovac, S.; Gola, L.; Wigley, C.; Abramov, A.Y.; Walker, M.C. Combination antioxidant therapy prevents epileptogenesis and modifies chronic epilepsy. Redox. Biol. 2019, 26, 101278. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.K.; Chen, S.D.; Lin, K.J.; Yao-Chung, C. Seizure-Induced Oxidative Stress in Status Epilepticus: Is Antioxidant Beneficial? Antioxidants 2020, 9, 1029. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.J.; Yu, B.C. Mitochondrial matters of the brain: Mitochondrial dysfunction and oxidative status in epilepsy. J. Bioenerg. Biomembr. 2010, 42, 457–459. [Google Scholar] [CrossRef] [PubMed]
- Witt, J.A.; Helmstaedter, C. Cognition in epilepsy: Current clinical issues of interest. Curr. Opin. Neurol. 2017, 30, 174–179. [Google Scholar] [CrossRef]


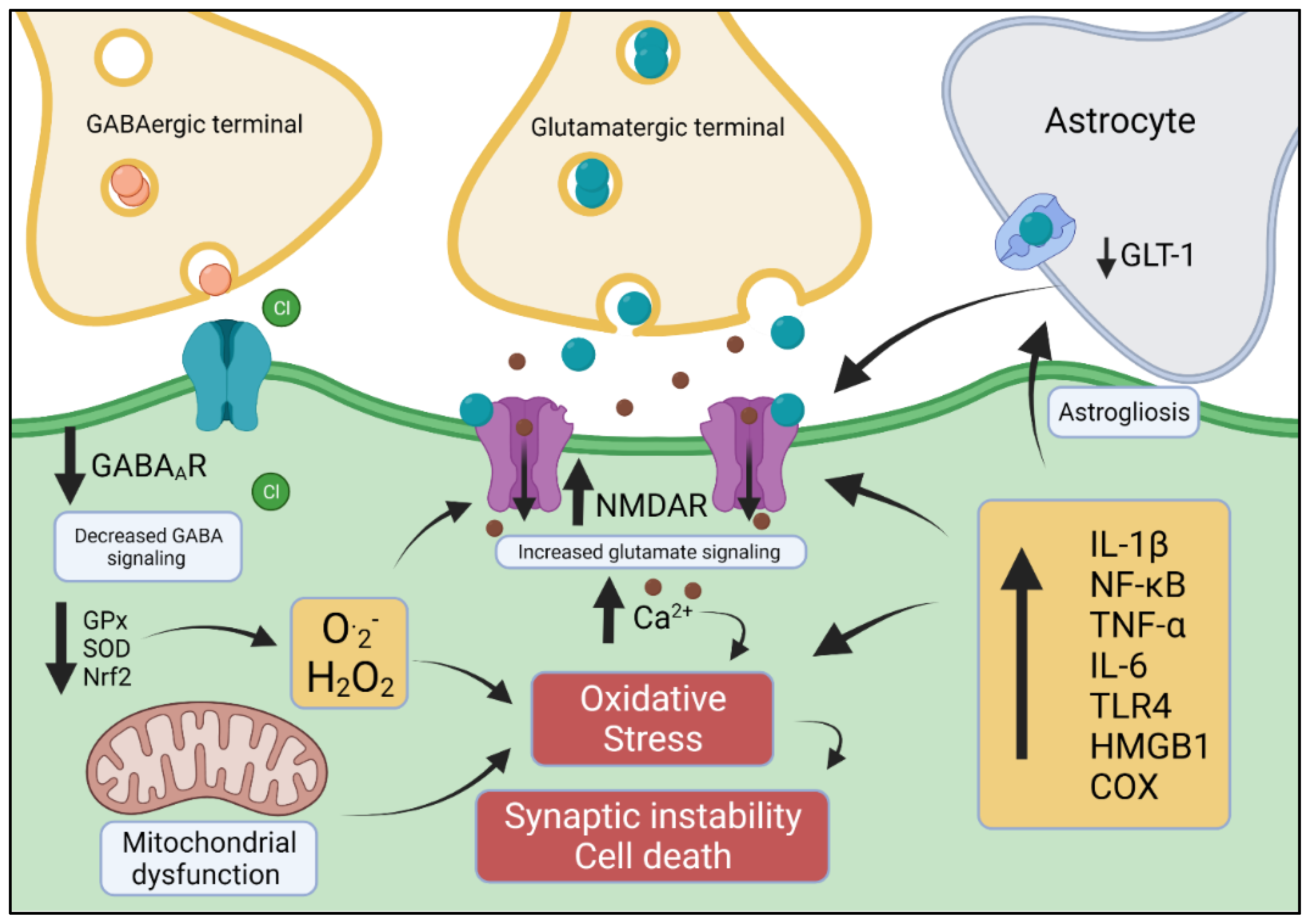{kind=link}
| Class | Drug | Mechanism of Action | Type of Seizure Targeted | Effects on OS and Inflammation Markers in Epilepsy (Pre- and Clinical Data) | References |
|---|---|---|---|---|---|
| Classical anti-epileptic | Valproic acid | Blocks voltage-gated ion channels | Focal and generalized | Increased lipid peroxidation | [111] |
| Phenytoin | Blocks voltage-gated sodium channels | Tonic-clonic | Reduced antioxidant capacity and glutathione concentration; Increased lipid peroxidation | [112,113] | |
| Carbamazepine | Blocks sodium channels | Focal and generalized | Decreased lipid peroxidation; Increased NO release | [114] | |
| Barbitures | Potentiates GABA signalling | Generalized | Decreased lipid peroxidation; Reduced levels of antioxidant enzymes | [115] | |
| Benzodiazepines | Facilitates GABA binding to GABAA receptors | Status epilepticus | Decreased lipid peroxidation | [116] | |
| Cenobamate | Blocks voltage-gated sodium channels. Allosteric agonist of GABA receptors | Uncontrolled focal | Activation of the PI3K/Akt-CREB-BDNF pathway | [117,118] | |
| Lamotrigine | Binds to the inactive sodium channel | Focal and generalized | Increased antioxidant defence; Reduced mitochondrial redox activity | [119] | |
| Antioxidant | Cannabidiol | Inhibits GRP55; Desensitizes receptor potential vanilloid type-1; Inhibits adenosine uptake, | Drug-resistant | Decreased ROS production; Increased antioxidant defences | [120,121,122] |
| Naringenin | Free radical scavenger | Pilocarpine-induced | Increased glutathione and antioxidant enzymes levels | [123] | |
| Coenzyme Q10 | Increases the levels of TCA and antioxidant enzymes | Pilocarpine-induced | Increased SOD and GSH levels, reduced lipid peroxidation | [124] | |
| N-acetylcysteine | Reduces glutathione precursor | Pentylenetetrazole-induced | Attenuated the impairment in glutathione homeostasis | [125] | |
| Curcumin | Free radical scavenger and metal chelator | Pentylenetetrazole-induced | Increased superoxide dismutase levels Reduced the expression of inflammatory cytokines and chemokines Reduced GFAP and IBA-1 markers | [126] | |
| Vitamin E | Peroxyl radical scavenger | Refractory | Increased antioxidant capacity. Increased catalase and glutathione levels | [127] | |
| Sulforaphane | Activates NRF2/ARE pathway | Status epilepticus | Decreased malondialdehyde levels and increased glutathione levels | [128] | |
| Anti-inflammatory | Anakinra | Antagonist of IL-1 receptor | Febrile infection- related epilepsy syndrome and Intractable epilepsy | Reduced IL-1 driven systemic autoinflammation | [129,130,131,132] |
| Anakinra + Canakinumab | Antagonist of IL-1 receptor; Monoclonal antibody against the IL-1 receptor | Generalized | |||
| Tocilizumab | Anti-IL-6 monoclonal antibody | Status epilepticus, acute epilepsy | Reduced IL-6 levels | [133,134] | |
| Minocycline | Inhibitor of microglia activation | Drug-resistant | Supressed IL-1β release from microglia | [135] | |
| Adalimumab | Anti-TNF monoclonal antibody | Partial and focal motor seizures | Reduced TNF-α levels | [136] | |
| Aspirin | Cyclooxygenase inhibitor | Focal-onset | Not reported | [137,138] | |
| VX09-765-401 | IL-1β inhibitor | Partial seizures | Not reported | [139] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parsons, A.L.M.; Bucknor, E.M.V.; Castroflorio, E.; Soares, T.R.; Oliver, P.L.; Rial, D. The Interconnected Mechanisms of Oxidative Stress and Neuroinflammation in Epilepsy. Antioxidants 2022, 11, 157. https://doi.org/10.3390/antiox11010157
Parsons ALM, Bucknor EMV, Castroflorio E, Soares TR, Oliver PL, Rial D. The Interconnected Mechanisms of Oxidative Stress and Neuroinflammation in Epilepsy. Antioxidants. 2022; 11(1):157. https://doi.org/10.3390/antiox11010157
Chicago/Turabian StyleParsons, Anna L. M., Eboni M. V. Bucknor, Enrico Castroflorio, Tânia R. Soares, Peter L. Oliver, and Daniel Rial. 2022. "The Interconnected Mechanisms of Oxidative Stress and Neuroinflammation in Epilepsy" Antioxidants 11, no. 1: 157. https://doi.org/10.3390/antiox11010157