
Induction of Paraptotic Cell Death in Breast Cancer Cells by a Novel Pyrazolo[3,4-h]quinoline Derivative through ROS Production and Endoplasmic Reticulum Stress


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
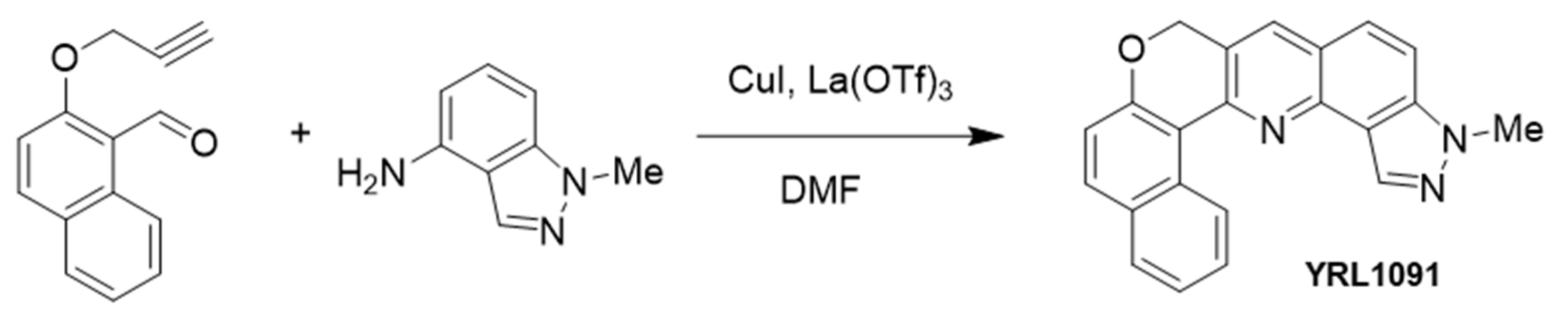
2.1. Synthesis of YRL1091
2.2. Chemicals and Reagents
2.3. Cell Culture
2.4. Cell Viability
2.5. Cell Morphology
2.6. Measurement of ROS
2.7. Transfection of Cells with siRNA for LC3B
2.8. Western Blotting
2.9. Immunofluorescence and Lysosomal Staining
2.10. Measurement of Apoptosis by Annexin V-FITC/PI Staining
2.11. Statistical Analyses
3. Results
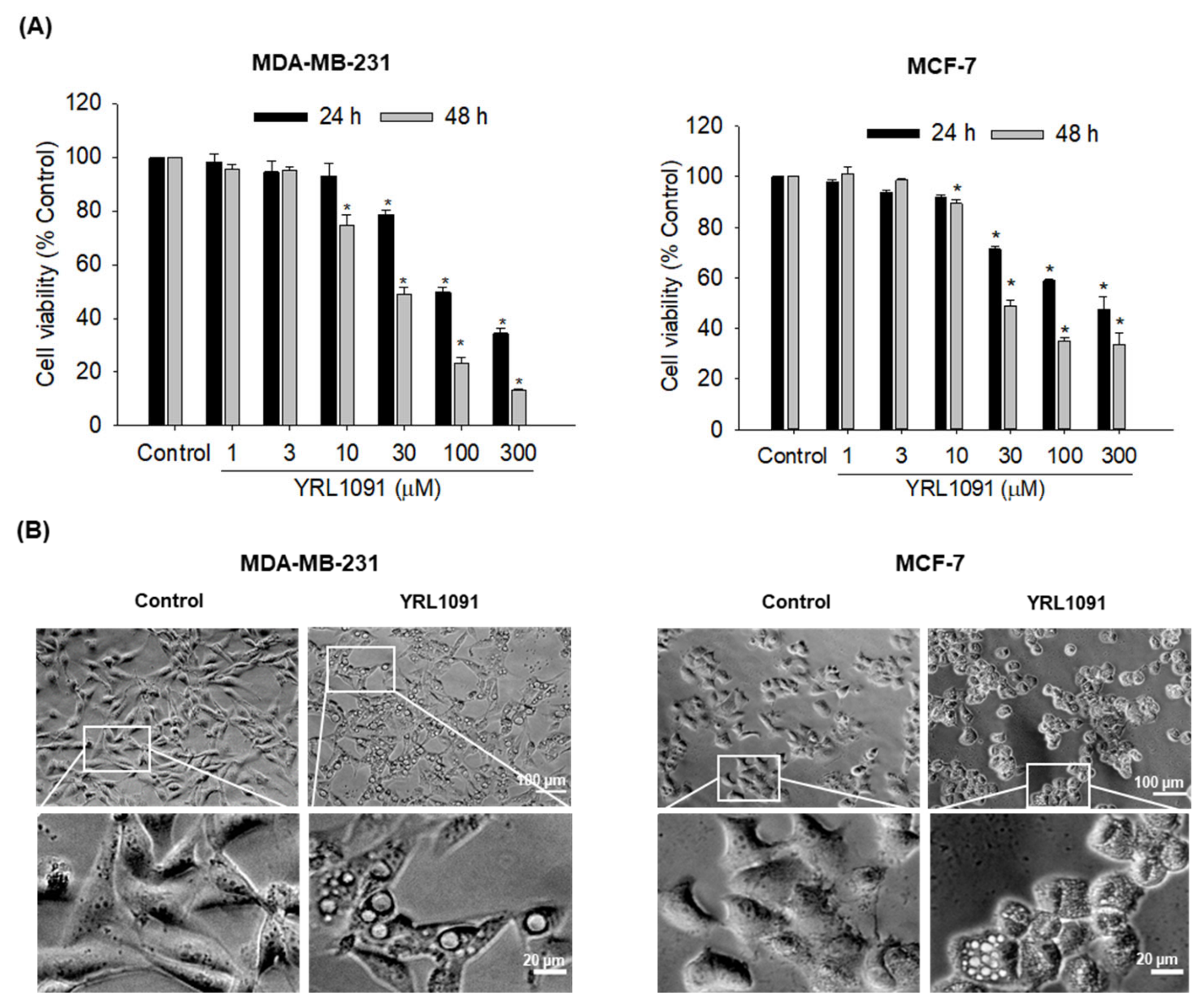
3.1. YRL1091 Induces Cytotoxicity and Cytoplasmic Vacuolization in BC Cells
3.2. YRL1091-Induced Cell Vacuolization Shows the Features of Paraptosis
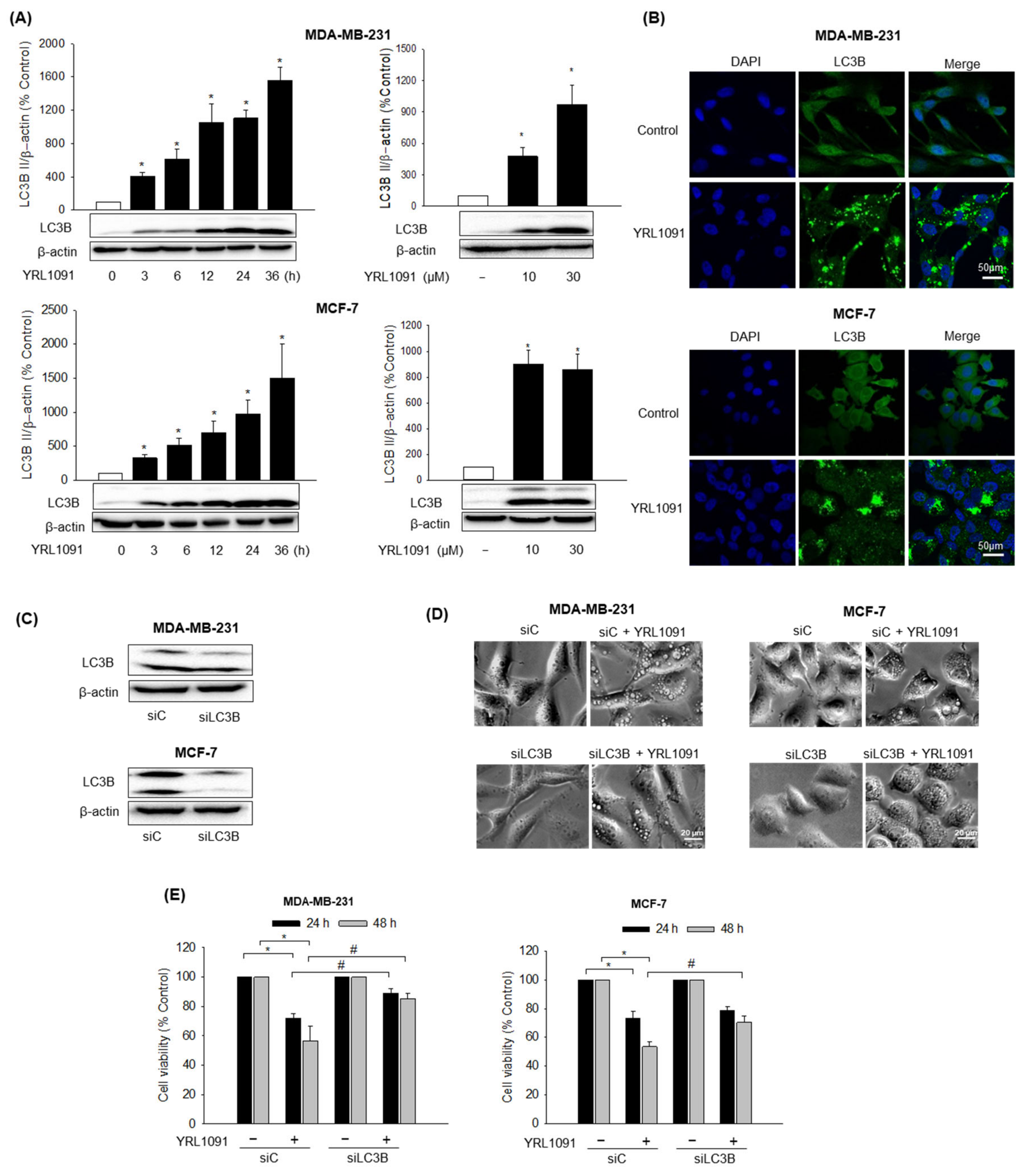
3.2.1. Role of LC3B in YRL1091-Induced Cytoplasmic Vacuolization and Cell Death
3.2.2. Role of MAPKs in YRL1091-Induced Cytoplasmic Vacuolization and Cell Death
3.2.3. Effect of YRL1091 on the Expression of Alix Protein
3.2.4. Effect of YRL1091 on the Activation of Caspase 9
3.3. YRL1091-Mediated Cytoplasmic Vacuolization and Cell Death Show Non-Autophagic and Non-Apoptotic Characteristics
3.3.1. Non-Autophagic Characteristics
3.3.2. Non-Apoptotic Characteristics
3.4. Generation of Intracellular ROS Contributes to YRL1091-Induced Cell Death and Cytoplasmic Vacuolization
3.5. YRL1091-Mediated Cell Death Requires Protein Synthesis
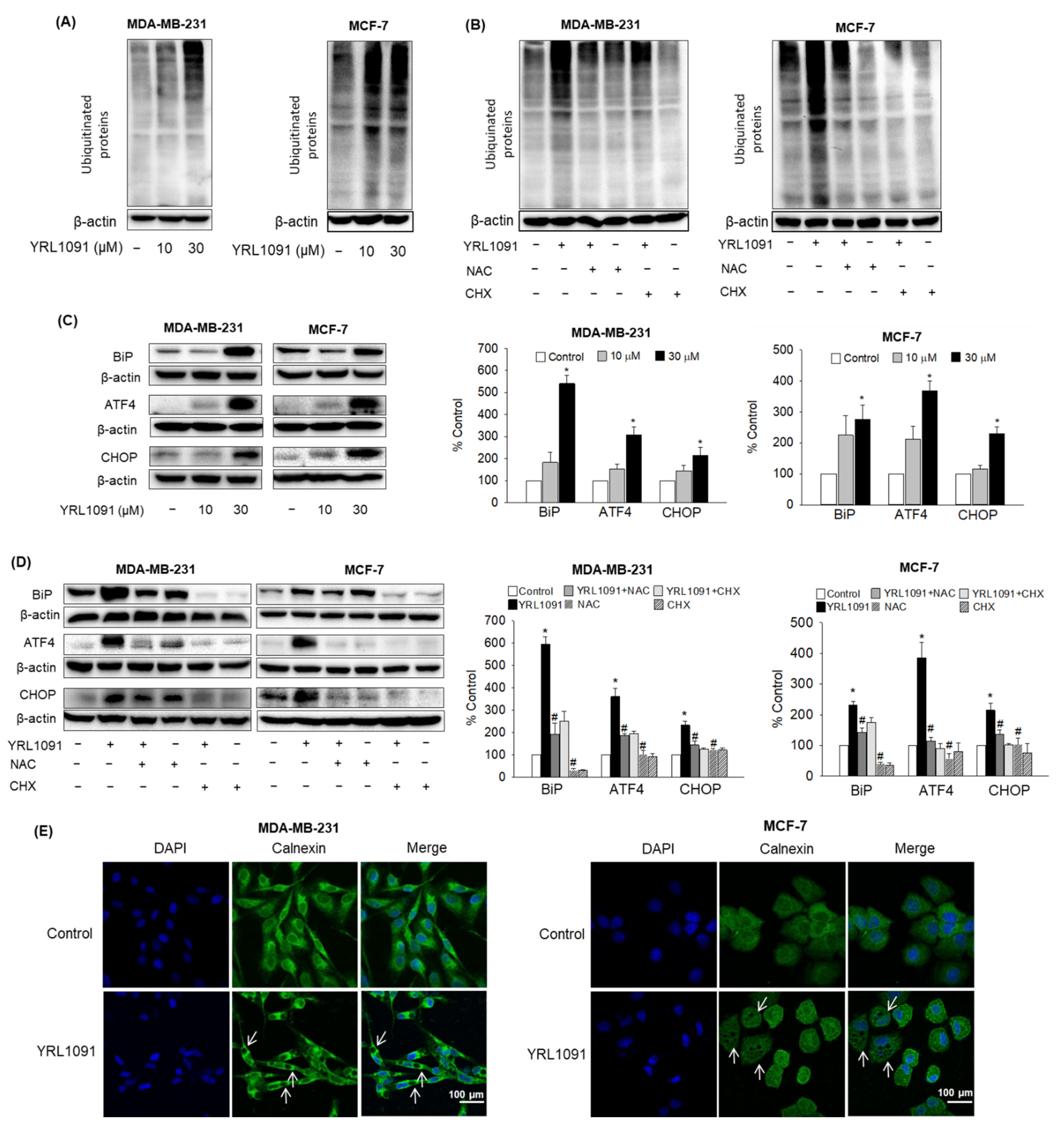
3.6. YRL1091-Induced Cytoplasmic Vacuoles Are Derived from ER Structure
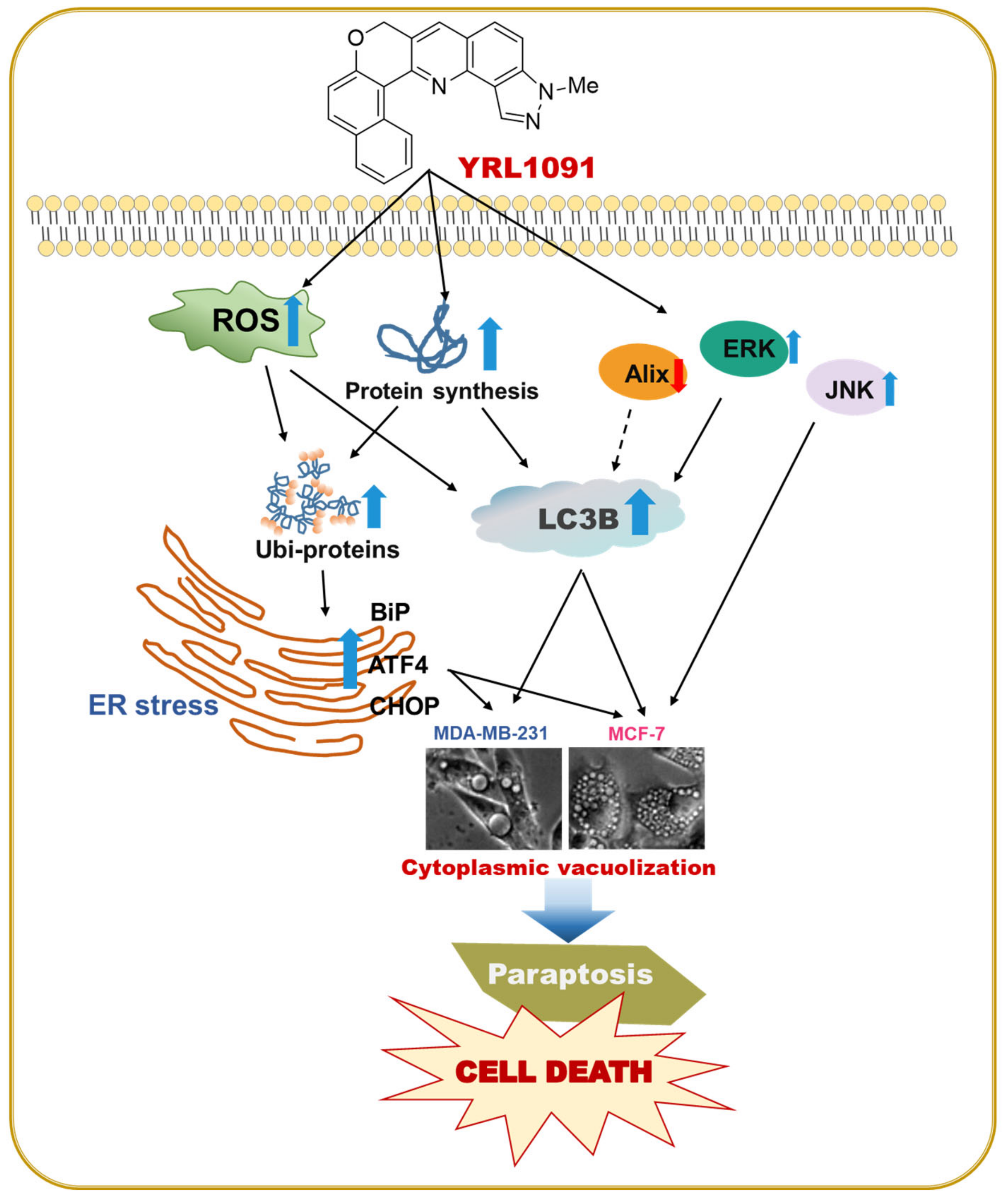
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Byler, S.; Goldgar, S.; Heerboth, S.; Leary, M.; Housman, G.; Moulton, K.; Sarkar, S. Genetic and epigenetic aspects of breast cancer progression and therapy. Anticancer Res. 2014, 34, 1071–1077. [Google Scholar]
- Luque-Bolivar, A.; Pérez-Mora, E.; Villegas, V.E.; Rondón-Lagos, M. Resistance and Overcoming Resistance in Breast Cancer. Breast Cancer 2020, 12, 211–229. [Google Scholar] [CrossRef]
- Khalili, M.; Radosevich, J.A. Paraptosis. In Apoptosis and Beyond; Radosevich, J.A., Ed.; Wiley: Hoboken, NJ, USA, 2018; pp. 343–366. [Google Scholar]
- Shubin, A.V.; Demidyuk, I.V.; Komissarov, A.A.; Rafieva, L.M.; Kostrov, S.V. Cytoplasmic vacuolization in cell death and survival. Oncotarget 2016, 7, 55863–55889. [Google Scholar] [CrossRef] [Green Version]
- Fontana, F.; Raimondi, M.; Marzagalli, M.; Di Domizio, A.; Limonta, P. The emerging role of paraptosis in tumor cell biology: Perspectives for cancer prevention and therapy with natural compounds. Biochim Biophys Acta Rev. Cancer 2020, 1873, 188338. [Google Scholar] [CrossRef]
- Wang, Y.; Wen, X.; Zhang, N.; Wang, L.; Hao, D.; Jiang, X.; He, G. Small-molecule compounds target paraptosis to improve cancer therapy. Biomed Pharmacother 2019, 118, 109203. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.J.; Kim, E.H.; Lim, J.H.; Kwon, T.K.; Choi, K.S. Superoxide anion and proteasomal dysfunction contribute to curcumin-induced paraptosis of malignant breast cancer cells. Free Radic. Biol. Med. 2010, 48, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.J.; Lee, A.R.; Jeong, S.A.; Kim, Y.S.; Kim, J.Y.; Kwon, Y.J.; Choi, K.S. Release of Ca2+ from the endoplasmic reticulum and its subsequent influx into mitochondria trigger celastrol-induced paraptosis in cancer cells. Oncotarget 2014, 5, 6816–6831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, K.; De, S.; Das, S.; Mukherjee, S.; Sengupta Bandyopadhyay, S. Withaferin A Induces ROS-Mediated Paraptosis in Human Breast Cancer Cell-Lines MCF-7 and MDA-MB-231. PLoS ONE 2016, 11, e0168488. [Google Scholar] [CrossRef]
- Rolver, M.G.; Elingaard-Larsen, L.O.; Andersen, A.P.; Counillon, L.; Pedersen, S.F. Pyrazine ring-based Na(+)/H(+) exchanger (NHE) inhibitors potently inhibit cancer cell growth in 3D culture, independent of NHE1. Sci. Rep. 2020, 10, 5800. [Google Scholar] [CrossRef]
- Dam, J.; Ismail, Z.; Kurebwa, T.; Gangat, N.; Harmse, L.; Marques, H.M.; Lemmerer, A.; Bode, M.L.; de Koning, C.B. Synthesis of copper and zinc 2-(pyridin-2-yl)imidazo[1,2-a]pyridine complexes and their potential anticancer activity. Eur. J. Med. Chem. 2017, 126, 353–368. [Google Scholar] [CrossRef]
- Gaurav, A.; Gautam, V.; Singh, R. An overview on synthetic methodologies and biological activities of pyrazoloquinolines. Mini Rev. Med. Chem. 2010, 10, 1194–1210. [Google Scholar] [CrossRef]
- Opoku-Temeng, C.; Dayal, N.; Aflaki Sooreshjani, M.; Sintim, H.O. 3H-pyrazolo[4,3-f]quinoline haspin kinase inhibitors and anticancer properties. Bioorganic Chem. 2018, 78, 418–426. [Google Scholar] [CrossRef]
- Spanò, V.; Parrino, B.; Carbone, A.; Montalbano, A.; Salvador, A.; Brun, P.; Vedaldi, D.; Diana, P.; Cirrincione, G.; Barraja, P. Pyrazolo[3,4-h]quinolines promising photosensitizing agents in the treatment of cancer. Eur. J. Med. Chem. 2015, 102, 334–351. [Google Scholar] [CrossRef]
- Azizmohammadi, M.; Khoobi, M.; Ramazani, A.; Emami, S.; Zarrin, A.; Firuzi, O.; Miri, R.; Shafiee, A. 2H-chromene derivatives bearing thiazolidine-2,4-dione, rhodanine or hydantoin moieties as potential anticancer agents. Eur. J. Med. Chem. 2013, 59, 15–22. [Google Scholar] [CrossRef]
- Pratap, R.; Ram, V.J. Natural and synthetic chromenes, fused chromenes, and versatility of dihydrobenzo[h]chromenes in organic synthesis. Chem. Rev. 2014, 114, 10476–10526. [Google Scholar] [CrossRef]
- Choi, M.; Hwang, Y.S.; Kumar, A.S.; Jo, H.; Jeong, Y.; Oh, Y.; Lee, J.; Yun, J.; Kim, Y.; Han, S.B.; et al. Design and synthesis of 3,4-dihydro-2H-benzo[h]chromene derivatives as potential NF-κB inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 2404–2407. [Google Scholar] [CrossRef]
- Sim, S.; Lee, S.; Ko, S.; Phuong Bui, B.; Linh Nguyen, P.; Cho, J.; Lee, K.; Kang, J.-S.; Jung, J.-K.; Lee, H. Design, synthesis, and biological evaluation of potent 1,2,3,4-tetrahydroisoquinoline derivatives as anticancer agents targeting NF-κB signaling pathway. Bioorg. Med. Chem. 2021, 46, 116371. [Google Scholar] [CrossRef] [PubMed]
- Arepalli, S.K.; Lee, C.; Sim, S.; Lee, K.; Jo, H.; Jun, K.Y.; Kwon, Y.; Kang, J.S.; Jung, J.K.; Lee, H. Development of 13H-benzo[f]chromeno[4,3-b][1,7]naphthyridines and their salts as potent cytotoxic agents and topoisomerase I/IIα inhibitors. Bioorg. Med. Chem. 2018, 26, 5181–5193. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Yu, P.; Tang, J. Characterization of Triple-Negative Breast Cancer MDA-MB-231 Cell Spheroid Model. Onco. Targets Ther. 2020, 13, 5395–5405. [Google Scholar] [CrossRef] [PubMed]
- Collignon, J.; Lousberg, L.; Schroeder, H.; Jerusalem, G. Triple-negative breast cancer: Treatment challenges and solutions. Breast Cancer 2016, 8, 93–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comşa, Ş.; Cîmpean, A.M.; Raica, M. The Story of MCF-7 Breast Cancer Cell Line: 40 years of Experience in Research. Anticancer Res. 2015, 35, 3147–3154. [Google Scholar]
- Do, H.T.T.; Cho, J. Involvement of the ERK/HIF-1α/EMT Pathway in XCL1-Induced Migration of MDA-MB-231 and SK-BR-3 Breast Cancer Cells. Int. J. Mol. Sci. 2020, 22, 89. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Park, M.K.; Park, J.H.; Lee, H.J.; Shin, D.H.; Kang, Y.; Lee, C.H.; Kong, G. Loss of the polycomb protein Mel-18 enhances the epithelial-mesenchymal transition by ZEB1 and ZEB2 expression through the downregulation of miR-205 in breast cancer. Oncogene 2014, 33, 1325–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orellana, E.A.; Kasinski, A.L. Sulforhodamine B (SRB) Assay in Cell Culture to Investigate Cell Proliferation. Bio-protocol 2016, 6, e1984. [Google Scholar] [CrossRef] [Green Version]
- Moniruzzaman, M.; Bose, S.; Kim, Y.M.; Chin, Y.W.; Cho, J. The ethyl acetate fraction from Physalis alkekengi inhibits LPS-induced pro-inflammatory mediators in BV2 cells and inflammatory pain in mice. J. Ethnopharmacol. 2016, 181, 26–36. [Google Scholar] [CrossRef]
- Nguyen, P.L.; Bui, B.P.; Lee, H.; Cho, J. A Novel 1,8-Naphthyridine-2-Carboxamide Derivative Attenuates Inflammatory Responses and Cell Migration in LPS-Treated BV2 Cells via the Suppression of ROS Generation and TLR4/Myd88/NF-κB Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 2527. [Google Scholar] [CrossRef]
- Nguyen, P.L.; Bui, B.P.; Duong, M.T.H.; Lee, K.; Ahn, H.C.; Cho, J. Suppression of LPS-Induced Inflammation and Cell Migration by Azelastine through Inhibition of JNK/NF-κB Pathway in BV2 Microglial Cells. Int. J. Mol. Sci. 2021, 22, 9061. [Google Scholar] [CrossRef]
- Wang, W.B.; Feng, L.X.; Yue, Q.X.; Wu, W.Y.; Guan, S.H.; Jiang, B.H.; Yang, M.; Liu, X.; Guo, D.A. Paraptosis accompanied by autophagy and apoptosis was induced by celastrol, a natural compound with influence on proteasome, ER stress and Hsp90. J. Cell Physiol. 2012, 227, 2196–2206. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Ren, J.; Wang, Y.; Su, J.Y.; Zhu, Y.M.; Chen, C.G.; Long, W.G.; Jiang, Q.; Li, J. Synergistic killing effect of paclitaxel and honokiol in non-small cell lung cancer cells through paraptosis induction. Cell Oncol. 2021, 44, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Kar, R.; Singha, P.K.; Venkatachalam, M.A.; Saikumar, P. A novel role for MAP1 LC3 in nonautophagic cytoplasmic vacuolation death of cancer cells. Oncogene 2009, 28, 2556–2568. [Google Scholar] [CrossRef] [Green Version]
- Sperandio, S.; Poksay, K.; de Belle, I.; Lafuente, M.J.; Liu, B.; Nasir, J.; Bredesen, D.E. Paraptosis: Mediation by MAP kinases and inhibition by AIP-1/Alix. Cell Death Differ. 2004, 11, 1066–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Polo, R.A.; Boya, P.; Pauleau, A.L.; Jalil, A.; Larochette, N.; Souquère, S.; Eskelinen, E.L.; Pierron, G.; Saftig, P.; Kroemer, G. The apoptosis/autophagy paradox: Autophagic vacuolization before apoptotic death. J. Cell. Sci. 2005, 118, 3091–3102. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Mizushima, N. LC3- and p62-based biochemical methods for the analysis of autophagy progression in mammalian cells. Methods 2015, 75, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Krolenko, S.A.; Adamyan, S.Y.; Belyaeva, T.N.; Mozhenok, T.P. Acridine orange accumulation in acid organelles of normal and vacuolated frog skeletal muscle fibres. Cell Biol. Int. 2006, 30, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, X.; Zhang, X.; Wang, L.; Cao, P.; Rajamanickam, V.; Wu, C.; Zhou, H.; Cai, Y.; Liang, G.; et al. Curcuminoid B63 induces ROS-mediated paraptosis-like cell death by targeting TrxR1 in gastric cells. Redox Biol. 2019, 21, 101061. [Google Scholar] [CrossRef]
- Shiau, J.Y.; Nakagawa-Goto, K.; Lee, K.H.; Shyur, L.F. Phytoagent deoxyelephantopin derivative inhibits triple negative breast cancer cell activity by inducing oxidative stress-mediated paraptosis-like cell death. Oncotarget 2017, 8, 56942–56958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessel, D. Apoptosis, Paraptosis and Autophagy: Death and Survival Pathways Associated with Photodynamic Therapy. Photochem. Photobiol. 2019, 95, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Weerasinghe, P.; Buja, L.M. Oncosis: An important non-apoptotic mode of cell death. Exp. Mol. Pathol. 2012, 93, 302–308. [Google Scholar] [CrossRef]
- Hitomi, J.; Christofferson, D.E.; Ng, A.; Yao, J.; Degterev, A.; Xavier, R.J.; Yuan, J. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 2008, 135, 1311–1323. [Google Scholar] [CrossRef] [Green Version]
- Runwal, G.; Stamatakou, E.; Siddiqi, F.H.; Puri, C.; Zhu, Y.; Rubinsztein, D.C. LC3-positive structures are prominent in autophagy-deficient cells. Sci. Rep. 2019, 9, 10147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y. Chapter 13—Role of Autophagy in Cancer Therapy. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Hayat, M.A., Ed.; Academic Press: San Diego, CA, USA, 2016; pp. 231–251. [Google Scholar]
- Nedungadi, D.; Binoy, A.; Pandurangan, N.; Pal, S.; Nair, B.G.; Mishra, N. 6-Shogaol induces caspase-independent paraptosis in cancer cells via proteasomal inhibition. Exp. Cell Res. 2018, 364, 243–251. [Google Scholar] [CrossRef]
- Binoy, A.; Nedungadi, D.; Katiyar, N.; Bose, C.; Shankarappa, S.A.; Nair, B.G.; Mishra, N. Plumbagin induces paraptosis in cancer cells by disrupting the sulfhydryl homeostasis and proteasomal function. Chem. Biol. Interact. 2019, 310, 108733. [Google Scholar] [CrossRef]
- Sperandio, S.; de Belle, I.; Bredesen, D.E. An alternative, nonapoptotic form of programmed cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 14376–14381. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [Green Version]
- Ram, B.M.; Ramakrishna, G. Endoplasmic reticulum vacuolation and unfolded protein response leading to paraptosis like cell death in cyclosporine A treated cancer cervix cells is mediated by cyclophilin B inhibition. Biochim. Biophys. Acta 2014, 1843, 2497–2512. [Google Scholar] [CrossRef] [Green Version]
- Sang, J.; Li, W.; Diao, H.J.; Fan, R.Z.; Huang, J.L.; Gan, L.; Zou, M.F.; Tang, G.H.; Yin, S. Jolkinolide B targets thioredoxin and glutathione systems to induce ROS-mediated paraptosis and apoptosis in bladder cancer cells. Cancer Lett. 2021, 509, 13–25. [Google Scholar] [CrossRef]
- Nguyen, H.G.; Conn, C.S.; Kye, Y.; Xue, L.; Forester, C.M.; Cowan, J.E.; Hsieh, A.C.; Cunningham, J.T.; Truillet, C.; Tameire, F.; et al. Development of a stress response therapy targeting aggressive prostate cancer. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Kim, I.Y.; Saha, S.; Choi, K.S. Paraptosis in the anti-cancer arsenal of natural products. Pharmacol. Ther. 2016, 162, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Kopito, R.R.; Christianson, J.C. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb. Perspect. Biol. 2013, 5, 13185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemus, L.; Goder, V. Regulation of Endoplasmic Reticulum-Associated Protein Degradation (ERAD) by Ubiquitin. Cells 2014, 3, 824–847. [Google Scholar] [CrossRef] [PubMed]
- Suh, D.H.; Kim, M.K.; Kim, H.S.; Chung, H.H.; Song, Y.S. Unfolded protein response to autophagy as a promising druggable target for anticancer therapy. Ann. N.Y. Acad. Sci. 2012, 1271, 20–32. [Google Scholar] [CrossRef] [Green Version]
- Mimnaugh, E.G.; Xu, W.; Vos, M.; Yuan, X.; Isaacs, J.S.; Bisht, K.S.; Gius, D.; Neckers, L. Simultaneous inhibition of hsp 90 and the proteasome promotes protein ubiquitination, causes endoplasmic reticulum-derived cytosolic vacuolization, and enhances antitumor activity. Mol. Cancer Ther. 2004, 3, 551–566. [Google Scholar]
- Fontana, F.; Moretti, R.M.; Raimondi, M.; Marzagalli, M.; Beretta, G.; Procacci, P.; Sartori, P.; Montagnani Marelli, M.; Limonta, P. δ-Tocotrienol induces apoptosis, involving endoplasmic reticulum stress and autophagy, and paraptosis in prostate cancer cells. Cell Prolif. 2019, 52, e12576. [Google Scholar] [CrossRef]
- Kennedy, D.; Samali, A.; Jäger, R. Methods for studying ER stress and UPR markers in human cells. Methods Mol. Biol. 2015, 1292, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—from basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, A.A.; Li, K.T.; Fayyaz, S.; Chang, Y.T.; Ismail, M.; Liaw, C.C.; Yuan, S.S.; Tang, J.Y.; Chang, H.W. Anticancer drugs for the modulation of endoplasmic reticulum stress and oxidative stress. Tumour Biol. 2015, 36, 5743–5752. [Google Scholar] [CrossRef] [PubMed] [Green Version]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, P.L.; Lee, C.H.; Lee, H.; Cho, J. Induction of Paraptotic Cell Death in Breast Cancer Cells by a Novel Pyrazolo[3,4-h]quinoline Derivative through ROS Production and Endoplasmic Reticulum Stress. Antioxidants 2022, 11, 117. https://doi.org/10.3390/antiox11010117
Nguyen PL, Lee CH, Lee H, Cho J. Induction of Paraptotic Cell Death in Breast Cancer Cells by a Novel Pyrazolo[3,4-h]quinoline Derivative through ROS Production and Endoplasmic Reticulum Stress. Antioxidants. 2022; 11(1):117. https://doi.org/10.3390/antiox11010117
Chicago/Turabian StyleNguyen, Phuong Linh, Chang Hoon Lee, Heesoon Lee, and Jungsook Cho. 2022. "Induction of Paraptotic Cell Death in Breast Cancer Cells by a Novel Pyrazolo[3,4-h]quinoline Derivative through ROS Production and Endoplasmic Reticulum Stress" Antioxidants 11, no. 1: 117. https://doi.org/10.3390/antiox11010117