
The Response to Oxidative Damage Correlates with Driver Mutations and Clinical Outcome in Patients with Myelofibrosis

 , , , , , , , , , , and add
Show full author list
, , , , , , , , , , and add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. Plasma Isolation
2.3. Human CD34+ Hematopoietic Stem/Progenitor Cells Purification
2.4. CD34+ Cells Culture Conditions
2.5. Detection of Intracellular ROS Levels
2.6. Annexin V/PI Staining
2.7. Measurement of SOD Activity
2.8. Measurement of 8-OHdG Levels
2.9. Measurement of TAC Levels
2.10. Measurement of L-Lactate Levels
2.11. Statistical Analysis
3. Results
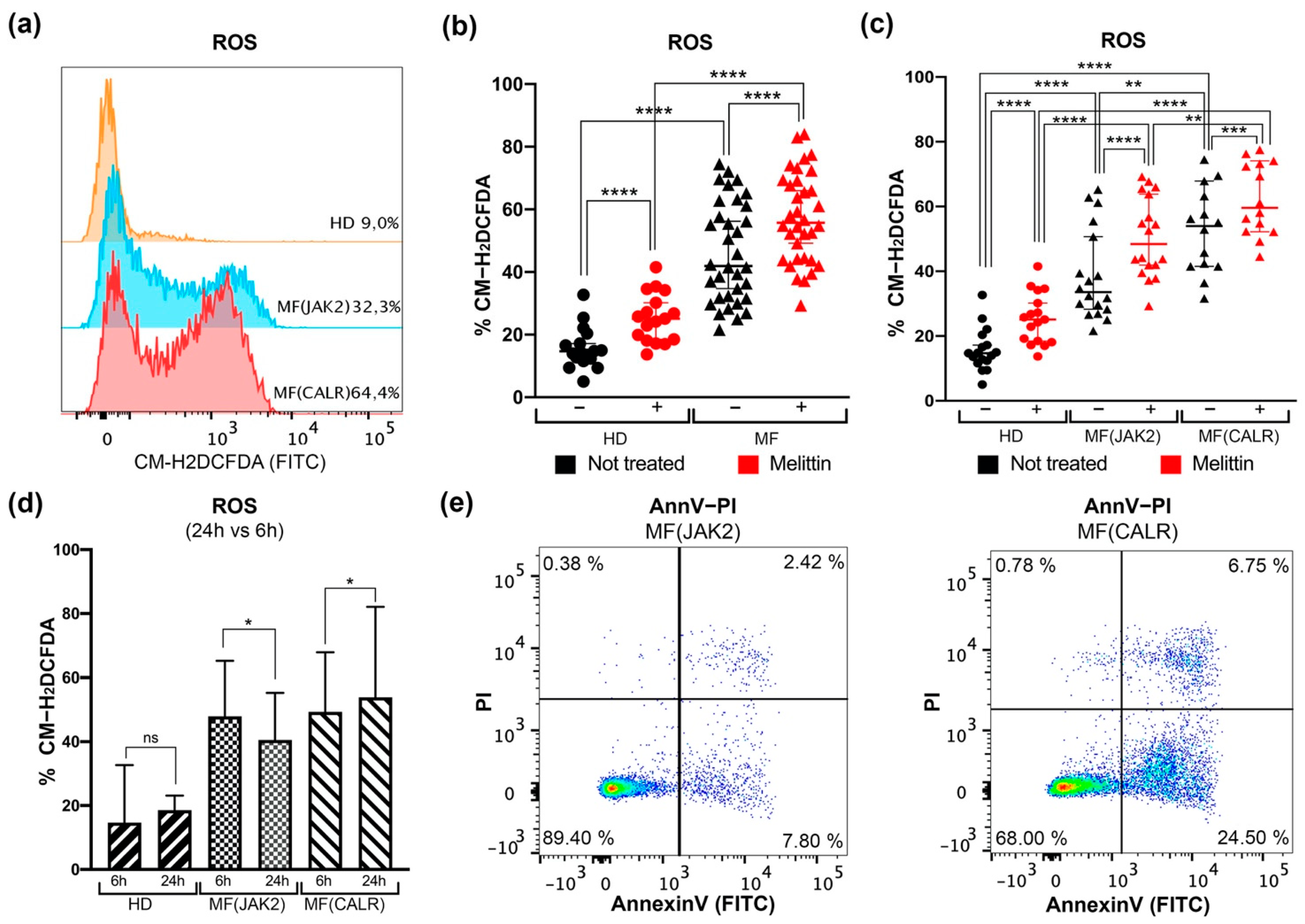
3.1. Intracellular ROS Quantification and Apoptosis Evaluation in CD34+ Cells from JAK2 or CALR-Mutated MF Patients
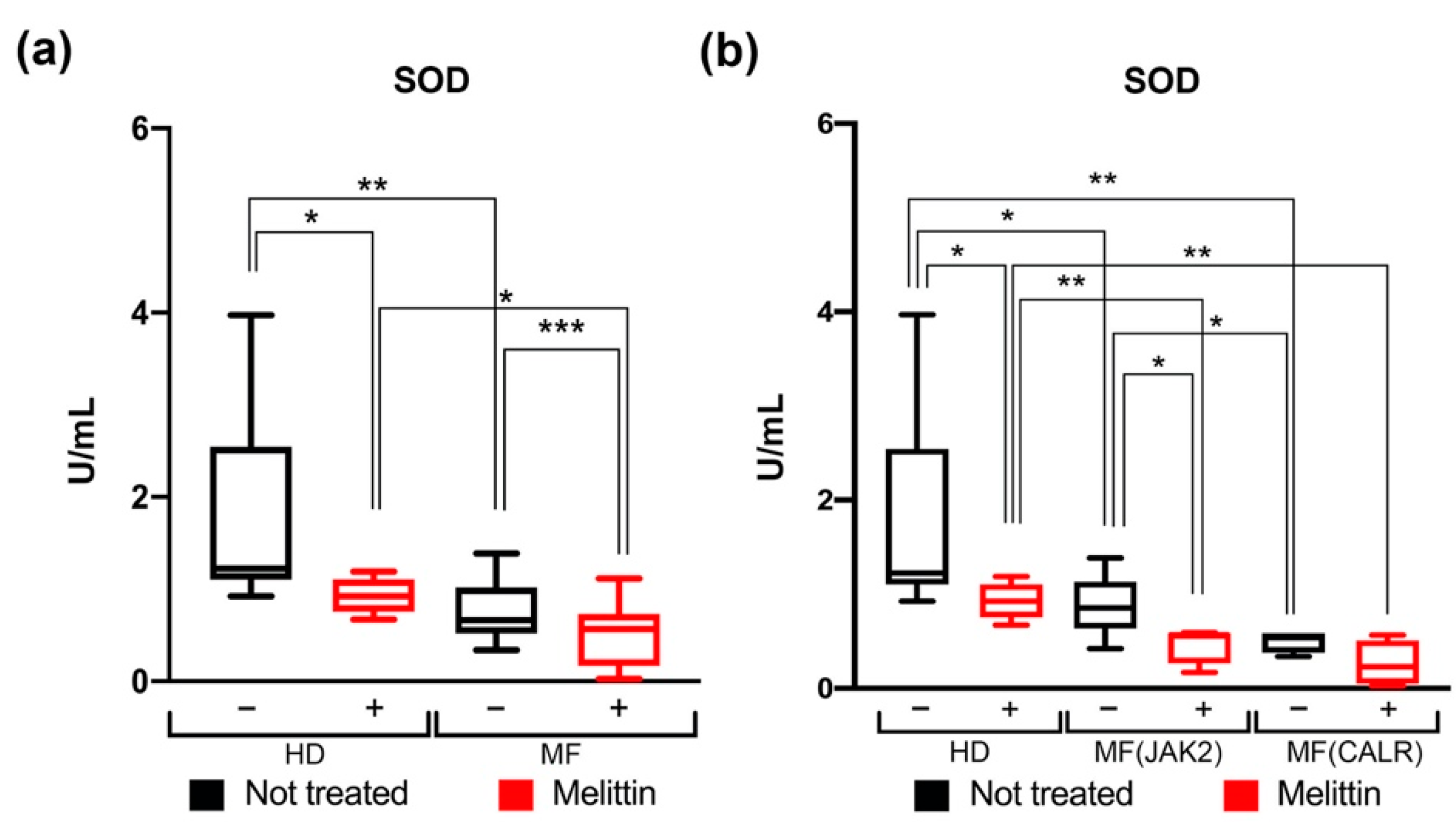
3.2. Modulation of SOD Activity in CD34+ Cells from JAK2- or CALR-Mutated MF Patients
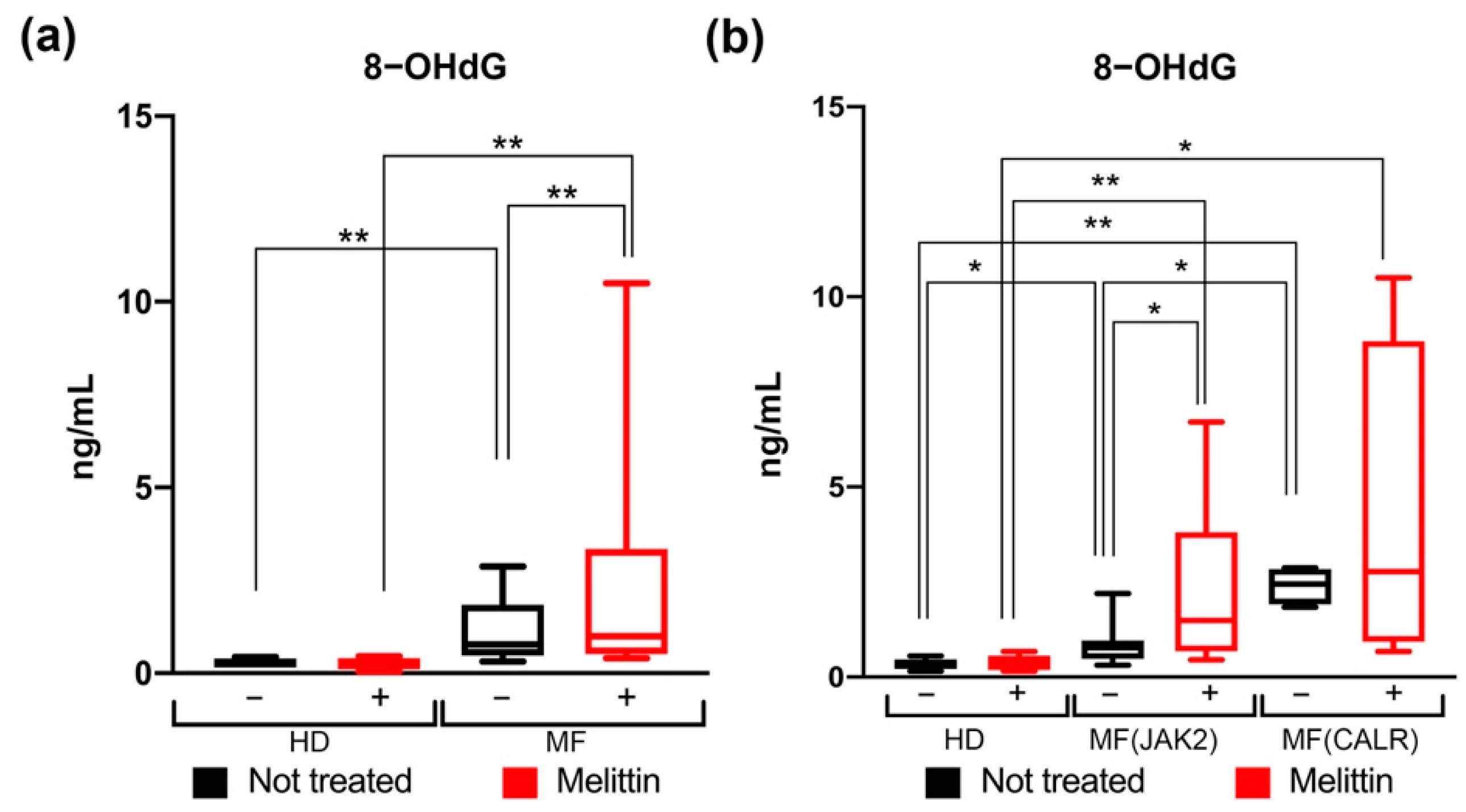
3.3. Quantification of Oxidative DNA Damage in CD34+ Cells from JAK2 and CALR-Mutated MF Patients
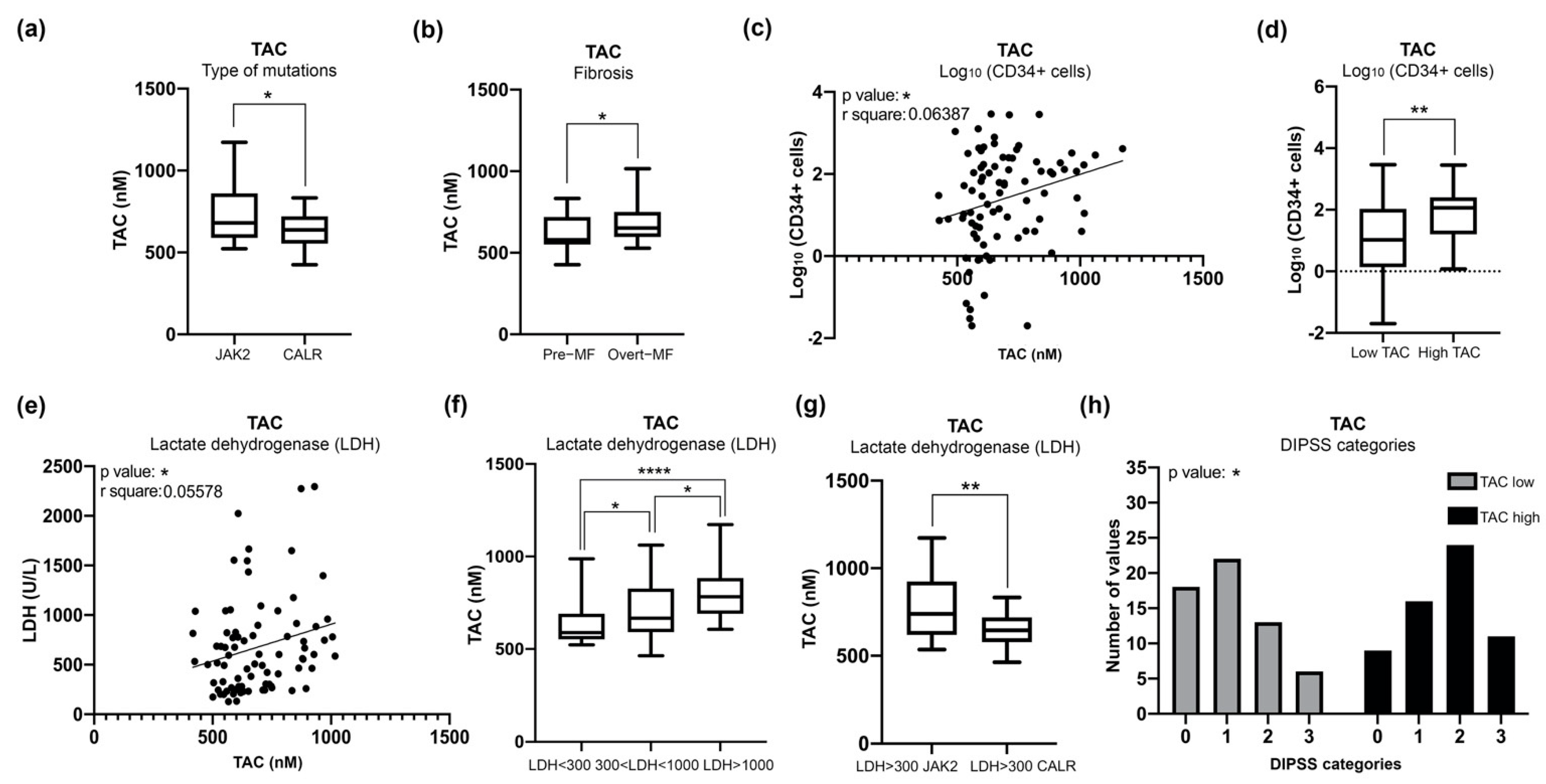
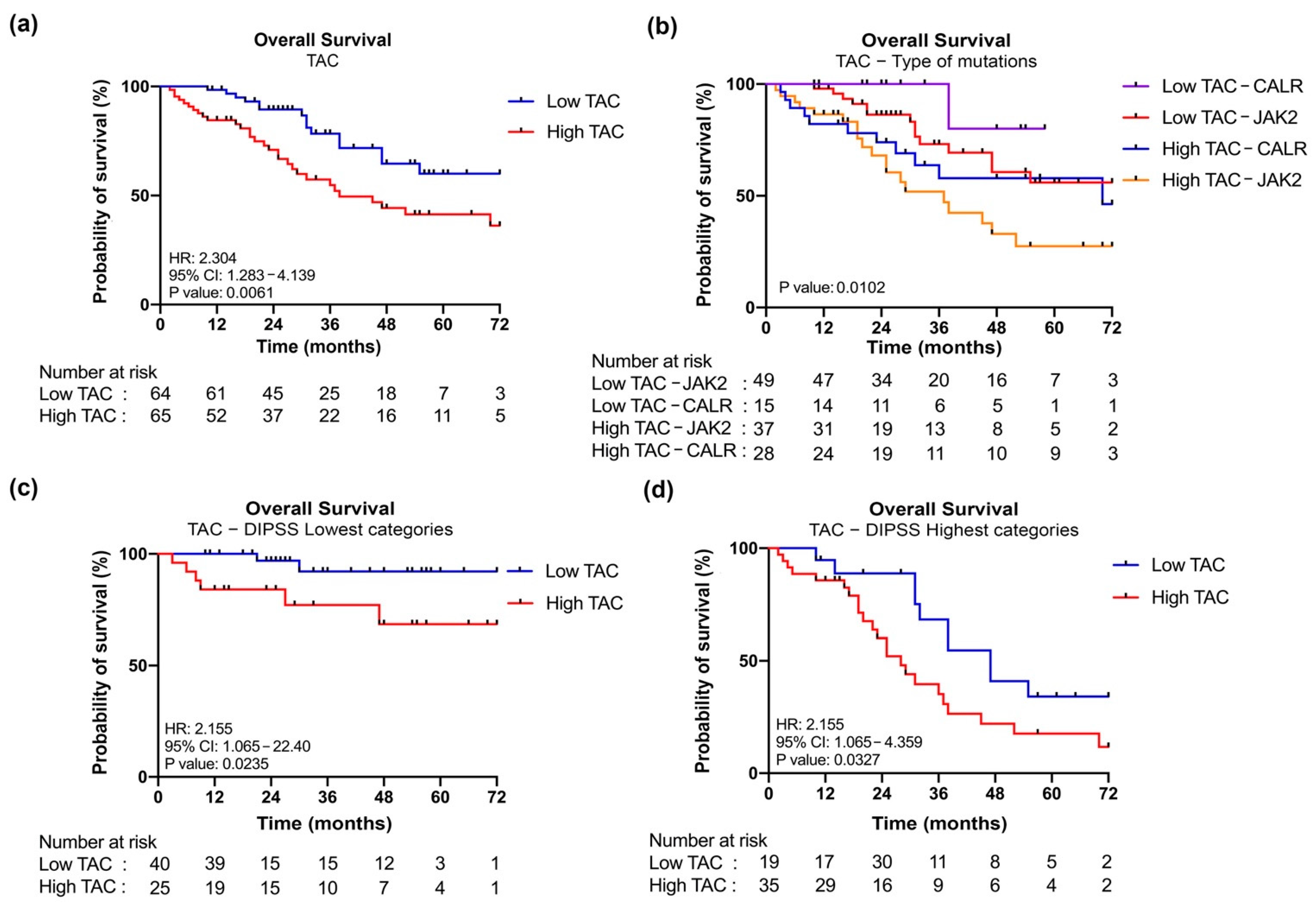
3.4. Increased Plasma Levels of TAC in MF Patients Correlate with Clinical Detrimental Features
3.5. JAK2-Mutant MF Patients with High Level of TAC Are Associated with a Poor OS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sies, H. Biochemistry of Oxidative Stress. Angew. Chem. Int. Ed. Engl. 1986, 25, 1058–1071. [Google Scholar] [CrossRef]
- Bjørn, M.E.; Hasselbalch, H.C. The Role of Reactive Oxygen Species in Myelofibrosis and Related Neoplasms. Mediat. Inflamm. 2015, 2015, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Allegra, A.; Pioggia, G.; Tonacci, A.; Casciaro, M.; Musolino, C.; Gangemi, S. Synergic Crosstalk between Inflammation, Oxidative Stress, and Genomic Alterations in BCR–ABL-Negative Myeloproliferative Neoplasm. Antioxidants 2020, 9, 1037. [Google Scholar] [CrossRef]
- Tefferi, A.; Pardanani, A. Myeloproliferative Neoplasms: A Contemporary Review. JAMA Oncol. 2015, 1, 97. [Google Scholar] [CrossRef]
- Tefferi, A. Primary Myelofibrosis: 2021 Update on Diagnosis, Risk--stratification and Management. Am. J. Hematol. 2021, 96, 145–162. [Google Scholar] [CrossRef] [PubMed]
- On behalf of the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT); Barosi, G.; Mesa, R.A.; Thiele, J.; Cervantes, F.; Campbell, P.J.; Verstovsek, S.; Dupriez, B.; Levine, R.L.; Passamonti, F.; et al. Proposed Criteria for the Diagnosis of Post-Polycythemia Vera and Post-Essential Thrombocythemia Myelofibrosis: A Consensus Statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia 2008, 22, 437–438. [Google Scholar] [CrossRef] [Green Version]
- Vainchenker, W.; Kralovics, R. Genetic Basis and Molecular Pathophysiology of Classical Myeloproliferative Neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating Interleukin (IL)-8, IL-2R, IL-12, and IL-15 Levels Are Independently Prognostic in Primary Myelofibrosis: A Comprehensive Cytokine Profiling Study. JCO 2011, 29, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Yahata, T.; Takanashi, T.; Muguruma, Y.; Ibrahim, A.A.; Matsuzawa, H.; Uno, T.; Sheng, Y.; Onizuka, M.; Ito, M.; Kato, S.; et al. Accumulation of Oxidative DNA Damage Restricts the Self-Renewal Capacity of Human Hematopoietic Stem Cells. Blood 2011, 118, 2941–2950. [Google Scholar] [CrossRef]
- Gloire, G.; Legrand-Poels, S.; Piette, J. NF-ΚB Activation by Reactive Oxygen Species: Fifteen Years Later. Biochem. Pharmacol. 2006, 72, 1493–1505. [Google Scholar] [CrossRef] [PubMed]
- Vener, C.; Novembrino, C.; Bamonti Catena, F.; Fracchiolla, N.S.; Gianelli, U.; Savi, F.; Radaelli, F.; Fermo, E.; Cortelezzi, A.; Lonati, S. Oxidative Stress Is Increased in Primary and Post−polycythemia Vera Myelofibrosis. Exp. Hematol. 2010, 38, 1058–1065. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.P.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating Mutation in the Tyrosine Kinase JAK2 in Polycythemia Vera, Essential Thrombocythemia, and Myeloid Metaplasia with Myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurtado-Nedelec, M.; Csillag-Grange, M.-J.; Boussetta, T.; Belambri, S.A.; Fay, M.; Cassinat, B.; Gougerot-Pocidalo, M.-A.; Dang, P.M.-C.; El-Benna, J. Increased Reactive Oxygen Species Production and P47phox Phosphorylation in Neutrophils from Myeloproliferative Disorders Patients with JAK2 (V617F) Mutation. Haematologica 2013, 98, 1517–1524. [Google Scholar] [CrossRef]
- Djikic, D.; Markovic, D.; Bogdanovic, A.; Mitrovic-Ajtic, O.; Suboticki, T.; Diklic, M.; Beleslin-Cokic, B.; Bjelica, S.; Kovacic, M.; P Cokic, V. Oxidative and Nitrosative Stress in Myeloproliferative Neoplasms: The Impact on the AKT/MTOR Signaling Pathway. J. BUON 2018, 23, 1481–1491. [Google Scholar]
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.-P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.-L.; Plo, I. A Role for Reactive Oxygen Species in JAK2V617F Myeloproliferative Neoplasm Progression. Leukemia 2013, 27, 2187–2195. [Google Scholar] [CrossRef] [Green Version]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR Mutations in Myeloproliferative Neoplasms with Nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef] [Green Version]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [Green Version]
- Michalak, M.; Corbett, E.F.; Mesaeli, N.; Nakamura, K.; Opas, M. Calreticulin: One Protein, One Gene, Many Functions. Biochem. J. 1999, 344 Pt 2, 281–292. [Google Scholar] [CrossRef]
- Elf, S.; Abdelfattah, N.S.; Chen, E.; Perales-Paton, J.; Rosen, E.A.; Ko, A.; Peisker, F.; Florescu, N.; Giannini, S.; Wolach, O.; et al. Mutant Calreticulin Requires Both Its Mutant C-Terminus and the Thrombopoietin Receptor for Oncogenic Transformation. Cancer Discov. 2016, 6, 368–381. [Google Scholar] [CrossRef] [Green Version]
- Araki, M.; Yang, Y.; Masubuchi, N.; Hironaka, Y.; Takei, H.; Morishita, S.; Mizukami, Y.; Kan, S.; Shirane, S.; Edahiro, Y.; et al. Activation of the Thrombopoietin Receptor by Mutant Calreticulin in CALR-Mutant Myeloproliferative Neoplasms. Blood 2016, 127, 1307–1316. [Google Scholar] [CrossRef]
- Araki, M.; Yang, Y.; Imai, M.; Mizukami, Y.; Kihara, Y.; Sunami, Y.; Masubuchi, N.; Edahiro, Y.; Hironaka, Y.; Osaga, S.; et al. Homomultimerization of Mutant Calreticulin Is a Prerequisite for MPL Binding and Activation. Leukemia 2019, 33, 122–131. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, L.; Jin, L.; Yi, X.; Dang, E.; Yang, Y.; Li, C.; Gao, T. Oxidative Stress–Induced Calreticulin Expression and Translocation: New Insights into the Destruction of Melanocytes. J. Investig. Dermatol. 2014, 134, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihara, Y.; Urata, Y.; Goto, S.; Kondo, T. Role of Calreticulin in the Sensitivity of Myocardiac H9c2 Cells to Oxidative Stress Caused by Hydrogen Peroxide. Am. J. Physiol. Cell Physiol. 2006, 290, C208–C221. [Google Scholar] [CrossRef] [PubMed]
- Salati, S.; Genovese, E.; Carretta, C.; Zini, R.; Bartalucci, N.; Prudente, Z.; Pennucci, V.; Ruberti, S.; Rossi, C.; Rontauroli, S.; et al. Calreticulin Ins5 and Del52 Mutations Impair Unfolded Protein and Oxidative Stress Responses in K562 Cells Expressing CALR Mutants. Sci. Rep. 2019, 9, 10558. [Google Scholar] [CrossRef] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Fantini, S.; Rontauroli, S.; Sartini, S.; Mirabile, M.; Bianchi, E.; Badii, F.; Maccaferri, M.; Guglielmelli, P.; Ottone, T.; Palmieri, R.; et al. Increased Plasma Levels of LncRNAs LINC01268, GAS5 and MALAT1 Correlate with Negative Prognostic Factors in Myelofibrosis. Cancers 2021, 13, 4744. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.S.; Soon, P.S.; Marsh, D.J. Comparison of Methodologies to Detect Low Levels of Hemolysis in Serum for Accurate Assessment of Serum MicroRNAs. PLoS ONE 2016, 11, e0153200. [Google Scholar] [CrossRef]
- Bianchi, E.; Ruberti, S.; Rontauroli, S.; Guglielmelli, P.; Salati, S.; Rossi, C.; Zini, R.; Tagliafico, E.; Vannucchi, A.; Manfredini, R. Role of MiR-34a-5p in Hematopoietic Progenitor Cells Proliferation and Fate Decision: Novel Insights into the Pathogenesis of Primary Myelofibrosis. IJMS 2017, 18, 145. [Google Scholar] [CrossRef] [Green Version]
- Gajski, G.; Domijan, A.-M.; Žegura, B.; Štern, A.; Gerić, M.; Novak Jovanović, I.; Vrhovac, I.; Madunić, J.; Breljak, D.; Filipič, M.; et al. Melittin Induced Cytogenetic Damage, Oxidative Stress and Changes in Gene Expression in Human Peripheral Blood Lymphocytes. Toxicon 2016, 110, 56–67. [Google Scholar] [CrossRef]
- Shibutani, S.; Takeshita, M.; Grollman, A.P. Insertion of Specific Bases during DNA Synthesis Past the Oxidation-Damaged Base 8-OxodG. Nature 1991, 349, 431–434. [Google Scholar] [CrossRef]
- Serafini, M.; Villano, D.; Spera, G.; Pellegrini, N. Redox Molecules and Cancer Prevention: The Importance of Understanding the Role of the Antioxidant Network. Nutr. Cancer 2006, 56, 232–240. [Google Scholar] [CrossRef]
- Lettieri-Barbato, D.; Tomei, F.; Sancini, A.; Morabito, G.; Serafini, M. Effect of Plant Foods and Beverages on Plasma Non-Enzymatic Antioxidant Capacity in Human Subjects: A Meta-Analysis. Br. J. Nutr. 2013, 109, 1544–1556. [Google Scholar] [CrossRef] [Green Version]
- Peluso, I.; Cavaliere, A.; Palmery, M. Plasma Total Antioxidant Capacity and Peroxidation Biomarkers in Psoriasis. J. Biomed. Sci. 2016, 23, 52. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, F.; Cervantes, F.; Vannucchi, A.M.; Morra, E.; Rumi, E.; Pereira, A.; Guglielmelli, P.; Pungolino, E.; Caramella, M.; Maffioli, M.; et al. A Dynamic Prognostic Model to Predict Survival in Primary Myelofibrosis: A Study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010, 115, 1703–1708. [Google Scholar] [CrossRef]
- Tefferi, A. Myelofibrosis with Myeloid Metaplasia. N. Engl. J. Med. 2000, 342, 1255–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. MPL515 Mutations in Myeloproliferative and Other Myeloid Disorders: A Study of 1182 Patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, C.; Ugo, V.; Le Couédic, J.-P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A Unique Clonal JAK2 Mutation Leading to Constitutive Signalling Causes Polycythaemia Vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Levine, R.L.; Pardanani, A.; Tefferi, A.; Gilliland, D.G. Role of JAK2 in the Pathogenesis and Therapy of Myeloproliferative Disorders. Nat. Rev. Cancer 2007, 7, 673–683. [Google Scholar] [CrossRef]
- Waris, G.; Ahsan, H. Reactive Oxygen Species: Role in the Development of Cancer and Various Chronic Conditions. J. Carcinog. 2006, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Bartosz, G. Total Antioxidant Capacity. In Advances in Clinical Chemistry; Elsevier: Amsterdam, The Netherlands, 2003; Volume 37, pp. 219–292. ISBN 978-0-12-010337-9. [Google Scholar]
- Philp, A.; Macdonald, A.L.; Watt, P.W. Lactate—A Signal Coordinating Cell and Systemic Function. J. Exp. Biol. 2005, 208, 4561–4575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, G.A. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018, 27, 757–785. [Google Scholar] [CrossRef] [Green Version]
- Lushchak, V.I. Free Radicals, Reactive Oxygen Species, Oxidative Stress and Its Classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef]
- Testa, U.; Labbaye, C.; Castelli, G.; Pelosi, E. Oxidative Stress and Hypoxia in Normal and Leukemic Stem Cells. Exp. Hematol. 2016, 44, 540–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in Cancer: Initiators, Amplifiers or an Achilles’ Heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Beer, P.A.; Campbell, P.J.; Green, A.R. Comparison of Different Criteria for the Diagnosis of Primary Myelofibrosis Reveals Limited Clinical Utility for Measurement of Serum Lactate Dehydrogenase. Haematologica 2010, 95, 1960–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison | Overall Survival | |
|---|---|---|
| Hazard Ratio (95% CI) | p | |
| Low TAC–JAK2 vs. High TAC–JAK2 | 2.487 (1.218–5.079) | 0.0068 |
| Low TAC–CALR vs. High TAC–CALR | 5.240 (1.562–17.58) | 0.0694 |
| Low TAC–JAK2 vs. Low TAC–CALR | 0.2756 (0.07882–0.9635) | 0.1758 |
| High TAC–JAK2 vs. High TAC–CALR | 0.6290 (0.3073–1.288) | 0.2132 |
| Low TAC–JAK2 vs. High TAC–CALR | 1.440 (0.6341–3.271) | 0.3563 |
| Low TAC–CALR vs. High TAC–JAK2 | 9.265 (3.642–23.57) | 0.0076 |
| Clinical Features | Overall Survival | |
|---|---|---|
| Hazard Ratio (95% CI) | p | |
| Presence of JAK2V17F | 3.8620 (1.32–11.30) | 0.01365 |
| Overt MF | 0.5023 (0.19–1.36) | 0.17510 |
| Circulating CD34+ cells (Log10) | 1.7490 (1.05–2.90) | 0.03063 |
| LDH > 1000 nM | 2.2210 (0.77–6.39) | 0.13910 |
| High TAC classification | 2.8910 (1.18–7.1) | 0.006838 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Genovese, E.; Mirabile, M.; Rontauroli, S.; Sartini, S.; Fantini, S.; Tavernari, L.; Maccaferri, M.; Guglielmelli, P.; Bianchi, E.; Parenti, S.; et al. The Response to Oxidative Damage Correlates with Driver Mutations and Clinical Outcome in Patients with Myelofibrosis. Antioxidants 2022, 11, 113. https://doi.org/10.3390/antiox11010113
Genovese E, Mirabile M, Rontauroli S, Sartini S, Fantini S, Tavernari L, Maccaferri M, Guglielmelli P, Bianchi E, Parenti S, et al. The Response to Oxidative Damage Correlates with Driver Mutations and Clinical Outcome in Patients with Myelofibrosis. Antioxidants. 2022; 11(1):113. https://doi.org/10.3390/antiox11010113
Chicago/Turabian StyleGenovese, Elena, Margherita Mirabile, Sebastiano Rontauroli, Stefano Sartini, Sebastian Fantini, Lara Tavernari, Monica Maccaferri, Paola Guglielmelli, Elisa Bianchi, Sandra Parenti, and et al. 2022. "The Response to Oxidative Damage Correlates with Driver Mutations and Clinical Outcome in Patients with Myelofibrosis" Antioxidants 11, no. 1: 113. https://doi.org/10.3390/antiox11010113