
Contribution of the Nrf2 Pathway on Oxidative Damage and Mitochondrial Failure in Parkinson and Alzheimer’s Disease

{kind=link}
{kind=link}
Abstract
:1. Introduction
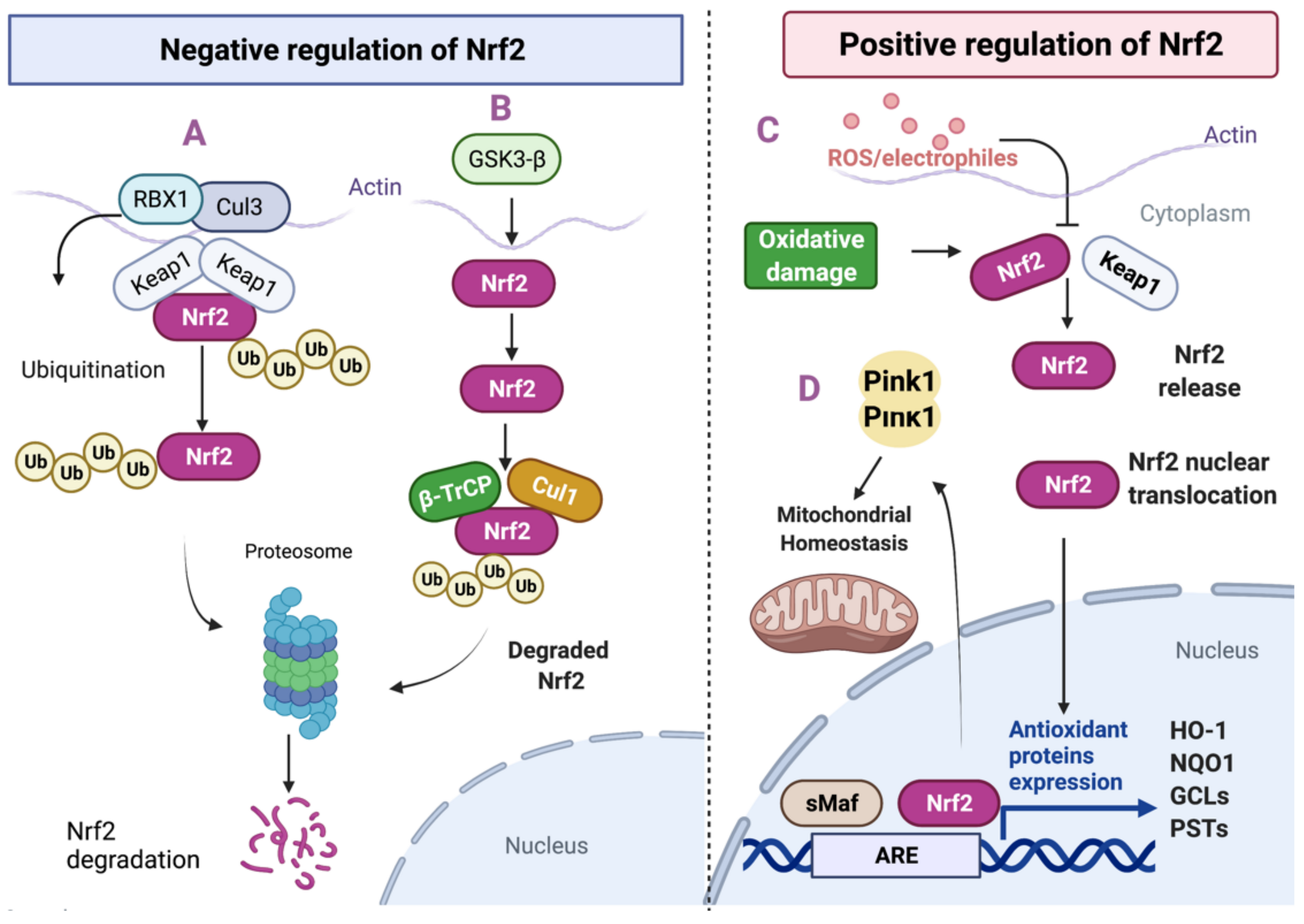
2. Nrf2 Pathway
2.1. Regulation of Nrf2 Pathway by Protein Stability
2.2. Transcriptional and Post-Transcriptional Regulation of Nrf2
2.3. Nrf2 and the Antioxidant Response in the Brain
2.4. Nrf2 and Mitochondrial Function in the Brain
2.4.1. Mitochondrial Bioenergetics
2.4.2. Mitochondrial Biogenesis
2.5. Nrf2 and Neuroinflammation
3. Neurodegeneration in PD
3.1. PD and ROS
3.2. PD and Mitochondrial Impairment
3.2.1. Mitochondrial Bioenergetics Defects
3.2.2. Mitophagy
3.3. Nrf2 Activation Prevents Neurodegeneration in PD
4. Neurodegeneration in AD
4.1. ROS in AD
4.2. AD and Mitochondrial Impairment
4.2.1. Mitochondrial Dynamics Defects
4.2.2. Mitochondrial Bioenergetics Defects
4.2.3. Mitochondrial Transport Defects
4.2.4. AD, Tau Pathology, and Mitochondrial Dysfunction
4.3. Nrf2 Activation Reduces Tau Pathology in AD
4.4. Nrf2 Activation Prevents Neurodegeneration and Mitochondrial Failure in AD
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osama, A.; Zhang, J.; Yao, J.; Yao, X.; Fang, J. Nrf2: A dark horse in Alzheimer’s disease treatment. Ageing Res. Rev. 2020, 64, 101206. [Google Scholar] [CrossRef] [PubMed]
- Joshi, G.; Johnson, J.A. The Nrf2-ARE Pathway: A Valuable Therapeutic Target for the Treatment of Neurodegenerative Diseases. Recent Patents CNS Drug Discov. 2012, 7, 218–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.A.; Johnson, J.A. Nrf2—A therapeutic target for the treatment of neurodegenerative diseases. Free Radic. Biol. Med. 2015, 88, 253–267. [Google Scholar] [CrossRef] [Green Version]
- Aruoma, O.I. Free radicals, oxidative stress, and antioxidants in human health and disease. J. Am. Oil Chem. Soc. 1998, 75, 199–212. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Akeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative Damage Is the Earliest Event in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Nickel, A.; Kohlhaas, M.; Maack, C. Mitochondrial reactive oxygen species production and elimination. J. Mol. Cell. Cardiol. 2014, 73, 26–33. [Google Scholar] [CrossRef]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2010, 1802, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Burchell, V.S.; Gandhi, S.; Deas, E.; Wood, N.W.; Abramov, A.Y.; Plun-Favreau, H. Targeting mitochondrial dysfunction in neurodegenerative disease: Part I. Expert Opin. Ther. Targets 2010, 14, 369–385. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free. Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Kirkwood, T.B.L. A systematic look at an old problem. Nature 2008, 451, 644–647. [Google Scholar] [CrossRef]
- Suh, J.H.; Shenvi, S.V.; Dixon, B.M.; Liu, H.; Jaiswal, A.K.; Liu, R.-M.; Hagen, T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA 2004, 101, 3381–3386. [Google Scholar] [CrossRef] [Green Version]
- Rebrin, I.; Kamzalov, S.; Sohal, R.S. Effects of age and caloric restriction on glutathione redox state in mice. Free. Radic. Biol. Med. 2003, 35, 626–635. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, H.; Tanji, K.; Wakabayashi, K.; Matsuura, S.; Itoh, K. Role of the Keap1/Nrf2 pathway in neurodegenerative diseases. Pathol. Int. 2015, 65, 210–219. [Google Scholar] [CrossRef]
- Wang, Q.; Li, W.-X.; Dai, S.-X.; Guo, Y.-C.; Han, F.-F.; Zheng, J.-J.; Li, G.-H.; Huang, J.-F. Meta-Analysis of Parkinson’s Disease and Alzheimer’s Disease Revealed Commonly Impaired Pathways and Dysregulation of NRF2-Dependent Genes. J. Alzheimer’s Dis. 2017, 56, 1525–1539. [Google Scholar] [CrossRef]
- Branca, C.; Ferreira, E.; Nguyen, T.-V.; Doyle, K.; Caccamo, A.; Oddo, S. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 4823–4835. [Google Scholar] [CrossRef]
- Schipperab, H.M.; Liberman, A.; Stopa, E.G. Neural Heme Oxygenase-1 Expression in Idiopathic Parkinson’s Disease. Exp. Neurol. 1998, 150, 60–68. [Google Scholar] [CrossRef]
- Rojo, A.I.; Innamorato, N.G.; Martín-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia 2010, 58, 588–598. [Google Scholar] [CrossRef]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 2020, 12, 1759091419899782. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; García-Yagüe, Á.J.; Scannevin, R.H.; Casarejos, M.J.; Kügler, S.; Rábano, A.; Cuadrado, A. Repurposing the NRF2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Douglas Engel, J.; Yamamoto, M. Nrf2–MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Chanas, S.A.; Henderson, C.J.; McMahon, M.; Sun, C.; Moffat, G.J.; Wolf, C.R.; Yamamoto, M. The Nrf2 transcription factor contributes both to the basal expression of glutathione S-transferases in mouse liver and to their induction by the chemopreventive synthetic antioxidants, butylated hydroxyanisole and ethoxyquin. Biochem. Soc. Trans. 2000, 28, 33–41. [Google Scholar] [CrossRef]
- Chan, J.Y.; Kwong, M. Impaired expression of glutathione synthetic enzyme genes in mice with targeted deletion of the Nrf2 basic-leucine zipper protein. Biochim. Biophys. Acta BBA Gene Struct. Express. 2000, 1517, 19–26. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Moinova, H.R.; Mulcahy, R.T. Up-Regulation of the Human γ-Glutamylcysteine Synthetase Regulatory Subunit Gene Involves Binding of Nrf-2 to an Electrophile Responsive Element. Biochem. Biophys. Res. Commun. 1999, 261, 661–668. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Itoh, K.; Tong, K.I.; Yamamoto, M. Molecular mechanism activating nrf2–keap1 pathway in regulation of adaptive response to electrophiles. Free Radic. Biol. Med. 2004, 36, 1208–1213. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB Protein Is an Adaptor That Bridges Nrf2 to a Cul3-Based E3 Ligase: Oxidative Stress Sensing by a Cul3-Keap1 Ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase To Regulate Proteasomal Degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, M.; Xiong, Y. BTB Protein Keap1 Targets Antioxidant Transcription Factor Nrf2 for Ubiquitination by the Cullin 3-Roc1 Ligase. Mol. Cell. Biol. 2005, 25, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Lo, S.-C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 Is a Redox-Regulated Substrate Adaptor Protein for a Cul3-Dependent Ubiquitin Ligase Complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [Green Version]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Yamamoto, M. Nrf2–Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv. Enzyme Regul. 2006, 46, 113–140. [Google Scholar] [CrossRef]
- McMahon, M.; Lamont, D.J.; Beattie, K.A.; Hayes, J.D. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc. Natl. Acad. Sci. USA 2010, 107, 18838–18843. [Google Scholar] [CrossRef] [Green Version]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen Synthase Kinase-3β Inhibits the Xenobiotic and Antioxidant Cell Response by Direct Phosphorylation and Nuclear Exclusion of the Transcription Factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef] [Green Version]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/β-TrCP Promotes Glycogen Synthase Kinase 3-Dependent Degradation of the Nrf2 Transcription Factor in a Keap1-Independent Manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Rojo, A.I.; De Sagarra, M.R.; Cuadrado, A. GSK-3β down-regulates the transcription factor Nrf2 after oxidant damage: Relevance to exposure of neuronal cells to oxidative stress. J. Neurochem. 2008, 105, 192–202. [Google Scholar] [CrossRef]
- Huang, H.-C.; Nguyen, T.; Pickett, C.B. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc. Natl. Acad. Sci. USA 2000, 97, 12475–12480. [Google Scholar] [CrossRef] [Green Version]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.K.; Jaiswal, A.K. GSK-3β Acts Upstream of Fyn Kinase in Regulation of Nuclear Export and Degradation of NF-E2 Related Factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef] [Green Version]
- Kanninen, K.; White, A.R.; Koistinaho, J.; Malm, T. Targeting Glycogen Synthase Kinase-3β for Therapeutic Benefit against Oxidative Stress in Alzheimer’s Disease: Involvement of the Nrf2-ARE Pathway. Int. J. Alzheimer’s Dis. 2011, 2011, 985085. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Sherratt, P.J.; Nioi, P.; Yang, C.S.; Pickett, C.B. Nrf2 Controls Constitutive and Inducible Expression of ARE-driven Genes through a Dynamic Pathway Involving Nucleocytoplasmic Shuttling by Keap1. J. Biol. Chem. 2005, 280, 32485–32492. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, N.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kang, M.-I.; Kobayashi, A.; Yamamoto, M.; Kensler, T.W.; Talalay, P. Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. USA 2004, 101, 2040–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apopa, P.L.; He, X.; Ma, Q. Phosphorylation of Nrf2 in the transcription activation domain by casein kinase 2 (CK2) is critical for the nuclear translocation and transcription activation function of Nrf2 in IMR-32 neuroblastoma cells. J. Biochem. Mol. Toxicol. 2008, 22, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Jia, Z.; Zhu, H. Regulation of Nrf2 Signaling. React. Oxyg. Species 2019, 8, 312–322. [Google Scholar] [CrossRef]
- Miao, W.; Hu, L.; Scrivens, P.J.; Batist, G. Transcriptional Regulation of NF-E2 p45-related Factor (NRF2) Expression by the Aryl Hydrocarbon Receptor-Xenobiotic Response Element Signaling Pathway: Direct Cross-Talk between Phase I and II Drug-Metabolizing Enzymes. J. Biol. Chem. 2005, 280, 20340–20348. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Wakabayashi, N.; Misra, V.; Biswal, S.; Lee, G.H.; Agoston, E.S.; Yamamoto, M.; Kensler, T.W. NRF2 Modulates Aryl Hydrocarbon Receptor Signaling: Influence on Adipogenesis. Mol. Cell. Biol. 2007, 27, 7188–7197. [Google Scholar] [CrossRef] [Green Version]
- Nioi, P.; Hayes, J.D. Contribution of NAD(P)H:quinone oxidoreductase 1 to protection against carcinogenesis, and regulation of its gene by the Nrf2 basic-region leucine zipper and the arylhydrocarbon receptor basic helix-loop-helix transcription factors. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2004, 555, 149–171. [Google Scholar] [CrossRef]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.-C.; Li, S.-J.; Yang, C.-L.; Xue, R.-L.; Xi, Y.-Y.; Wang, L.; Zhao, Q.-L.; Li, D.-J. Sulforaphane Attenuates Muscle Inflammation in Dystrophin-deficient mdx Mice via NF-E2-related Factor 2 (Nrf2)-mediated Inhibition of NF-κB Signaling Pathway. J. Biol. Chem. 2015, 290, 17784–17795. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Li, H.; Liu, Q.; Liu, F.; Tang, L.; Li, C.; Yuan, Y.; Zhan, Y.; Xu, W.; Li, W.; et al. Nuclear factor p65 interacts with Keap1 to repress the Nrf2-ARE pathway. Cell. Signal. 2011, 23, 883–892. [Google Scholar] [CrossRef]
- Neymotin, A.; Calingasan, N.Y.; Wille, E.; Naseri, N.; Petri, S.; Damiano, M.; Liby, K.T.; Risingsong, R.; Sporn, M.; Beal, M.F.; et al. Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2011, 51, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995. [Google Scholar] [CrossRef] [Green Version]
- Motohashi, H.; Yamamoto, M. Nrf2–Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Pitoniak, A.; Bohmann, D. Mechanisms and functions of Nrf2 signaling in Drosophila. Free Radic. Biol. Med. 2015, 88, 302–313. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; D’Amora, D.R.; MacNeil, L.T.; Walhout, A.J.M.; Kubiseski, T.J. The Oxidative Stress Response in Caenorhabditis elegans Requires the GATA Transcription Factor ELT-3 and SKN-1/Nrf2. Genetics 2017, 206, 1909–1922. [Google Scholar] [CrossRef] [Green Version]
- Fuse, Y.; Kobayashi, M. Conservation of the Keap1-Nrf2 System: An Evolutionary Journey through Stressful Space and Time. Molecules 2017, 22, 436. [Google Scholar] [CrossRef]
- Meakin, P.J.; Chowdhry, S.; Sharma, R.S.; Ashford, F.B.; Walsh, S.V.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; Dillon, J.F.; Hayes, J.D.; Ashford, M.L.J. Susceptibility of Nrf2-Null Mice to Steatohepatitis and Cirrhosis upon Consumption of a High-Fat Diet Is Associated with Oxidative Stress, Perturbation of the Unfolded Protein Response, and Disturbance in the Expression of Metabolic Enzymes but Not with Insulin Resistance. Mol. Cell. Biol. 2014, 34, 3305–3320. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef]
- Lee, S.; Hur, E.-G.; Ryoo, I.-G.; Jung, K.-A.; Kwak, J.; Kwak, M.-K. Involvement of the Nrf2-proteasome pathway in the endoplasmic reticulum stress response in pancreatic β-cells. Toxicol. Appl. Pharmacol. 2012, 264, 431–438. [Google Scholar] [CrossRef]
- Chen, W.; Sun, Z.; Wang, X.-J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct Interaction between Nrf2 and p21Cip1/WAF1 Upregulates the Nrf2-Mediated Antioxidant Response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Gureev, A.P.; Sadovnikova, I.S.; Starkova, N.N.; Starkov, A.A.; Popov, V.N. p62-Nrf2-p62 Mitophagy Regulatory Loop as a Target for Preventive Therapy of Neurodegenerative Diseases. Brain Sci. 2020, 10, 847. [Google Scholar] [CrossRef]
- Kapeta, S.; Chondrogianni, N.; Gonos, E.S. Nuclear Erythroid Factor 2-mediated Proteasome Activation Delays Senescence in Human Fibroblasts. J. Biol. Chem. 2010, 285, 8171–8184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, M.-K.; Wakabayashi, N.; Greenlaw, J.L.; Yamamoto, M.; Kensler, T.W. Antioxidants Enhance Mammalian Proteasome Expression through the Keap1-Nrf2 Signaling Pathway. Mol. Cell. Biol. 2003, 23, 8786–8794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajares, M.; Cuadrado, A.; Rojo, A.I. Modulation of proteostasis by transcription factor NRF2 and impact in neurodegenerative diseases. Redox Biol. 2017, 11, 543–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jirousek, L.; Stárka, L. DCber das Vorkommen von Trithionen (1,2-dithiacyclopent-4-en-3-thione) in Brassicapflanzen. Naturwissenschaften 1958, 45, 386–387. [Google Scholar] [CrossRef]
- Kwak, M.-K.; Cho, J.-M.; Huang, B.; Shin, S.; Kensler, T.W. Role of increased expression of the proteasome in the protective effects of sulforaphane against hydrogen peroxide-mediated cytotoxicity in murine neuroblastoma cells. Free Radic. Biol. Med. 2007, 43, 809–817. [Google Scholar] [CrossRef]
- Cuadrado, A.; Kügler, S.; Lastres-Becker, I. Pharmacological targeting of GSK-3 and NRF2 provides neuroprotection in a preclinical model of tauopathy. Redox Biol. 2018, 14, 522–534. [Google Scholar] [CrossRef]
- Yang, Y.; Jiang, S.; Yan, J.; Li, Y.; Xin, Z.; Lin, Y.; Qu, Y. An overview of the molecular mechanisms and novel roles of Nrf2 in neurodegenerative disorders. Cytokine Growth Factor Rev. 2015, 26, 47–57. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta BBA Gen. Subj. 2015, 1850, 794–801. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial Role of Nrf2 in Regulating NADPH Generation and Consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-M.; Calkins, M.J.; Chan, K.; Kan, Y.W.; Johnson, J.A. Identification of the NF-E2-related Factor-2-dependent Genes Conferring Protection against Oxidative Stress in Primary Cortical Astrocytes Using Oligonucleotide Microarray Analysis. J. Biol. Chem. 2003, 278, 12029–12038. [Google Scholar] [CrossRef] [Green Version]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar]
- Jimenez-Blasco, D.; Santofimia-Castaño, P.; Gonzalez, A.; Almeida, A.; Bolaños, J.P. Astrocyte NMDA receptors’ activity sustains neuronal survival through a Cdk5–Nrf2 pathway. Cell Death Differ. 2015, 22, 1877–1889. [Google Scholar] [CrossRef] [Green Version]
- Wild, A.C.; Mulcahy, R.T. Regulation ofγ-glutamylcysteine synthetase subunit gene expression: Insights into transcriptional control of antioxidant defenses. Free Radic. Res. 2000, 32, 281–301. [Google Scholar] [CrossRef]
- Harvey, C.J.; Thimmulappa, R.K.; Singh, A.; Blake, D.J.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic. Biol. Med. 2009, 46, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [Green Version]
- Ryoo, I.-G.; Kwak, M.-K. Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol. Appl. Pharmacol. 2018, 359, 24–33. [Google Scholar] [CrossRef]
- Greco, T.; Shafer, J.; Fiskum, G. Sulforaphane inhibits mitochondrial permeability transition and oxidative stress. Free Radic. Biol. Med. 2011, 51, 2164–2171. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, N.; Izumi, H.; Miyamoto, R.; Kondo, H.; Tawara, A.; Sasaguri, Y.; Kohno, K. Quercetin Induces the Expression of Peroxiredoxins 3 and 5 via the Nrf2/NRF1 Transcription Pathway. Investig. Opthalmol. Vis. Sci. 2011, 52, 1055–1063. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Wu, Y.; Liu, J.; Shen, X.; Tong, F.; Xu, G.; Shen, R. GSK-3beta Inhibitor Induces Expression of Nrf2/TrxR2 Signaling Pathway to Protect against Renal Ischemia/Reperfusion Injury in Diabetic Rats. Kidney Blood Press. Res. 2016, 41, 937–946. [Google Scholar] [CrossRef]
- Lee, J.-M.; Shih, A.Y.; Murphy, T.H.; Johnson, J.A. NF-E2-related Factor-2 Mediates Neuroprotection against Mitochondrial Complex I Inhibitors and Increased Concentrations of Intracellular Calcium in Primary Cortical Neurons. J. Biol. Chem. 2003, 278, 37948–37956. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Szewczyk, A.; Wojtczak, L. Mitochondria as a pharmacological target. Pharmacol. Rev. 2002, 54, 101–127. [Google Scholar] [CrossRef] [Green Version]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [Green Version]
- Handy, D.E.; Loscalzo, J. Redox Regulation of Mitochondrial Function. Antioxid. Redox Signal. 2012, 16, 1323–1367. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Reactive oxygen species and signal transduction. IUBMB Life 2001, 52, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Fiskum, G.; Murphy, A.N.; Beal, M.F. Mitochondria in Neurodegeneration: Acute Ischemia and Chronic Neurodegenerative Diseases. J. Cereb. Blood Flow Metab. 1999, 19, 351–369. [Google Scholar] [CrossRef] [PubMed]
- Niizuma, K.; Endo, H.; Chan, P.H. Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J. Neurochem. 2009, 109, 133–138. [Google Scholar] [CrossRef] [Green Version]
- Navarro, A.; Boveris, A. Brain mitochondrial dysfunction and oxidative damage in Parkinson’s disease. J. Bioenerg. Biomembr. 2009, 41, 517–521. [Google Scholar] [CrossRef]
- Lemasters, J.J.; Theruvath, T.P.; Zhong, Z.; Nieminen, A.-L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta BBA Bioenerg. 2009, 1787, 1395–1401. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P.; McStay, G.P.; Clarke, S.J. The permeability transition pore complex: Another view. Biochimie 2002, 84, 153–166. [Google Scholar] [CrossRef]
- Lo, S.-C.; Hannink, M. PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp. Cell Res. 2008, 314, 1789–1803. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, K.; Thaker, T.M.; Agnew, C.; Miller-Vedam, L.; Trenker, R.; Herrera, C.; Ingaramo, M.; Toso, D.; Frost, A.; Jura, N. Functional role of PGAM5 multimeric assemblies and their polymerization into filaments. Nat. Commun. 2019, 10, 531. [Google Scholar] [CrossRef] [Green Version]
- O’Mealey, G.B.; Plafker, K.S.; Berry, W.L.; Janknecht, R.; Chan, J.Y.; Plafker, S.M. A PGAM5-KEAP1-Nrf2 complex is required for stress-induced mitochondrial retrograde trafficking. J. Cell Sci. 2017, 130, 3467–3480. [Google Scholar] [CrossRef] [Green Version]
- Dinkova-Kostova, A.T.; Baird, L.; Holmström, K.M.; Meyer, C.J.; Abramov, A.Y. The spatiotemporal regulation of the Keap1–Nrf2 pathway and its importance in cellular bioenergetics. Biochem. Soc. Trans. 2015, 43, 602–610. [Google Scholar] [CrossRef] [Green Version]
- Esteras, N.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 activation in the treatment of neurodegenerative diseases: A focus on its role in mitochondrial bioenergetics and function. Biol. Chem. 2016, 397, 383–400. [Google Scholar] [CrossRef]
- Holmström, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The Multifaceted Role of Nrf2 in Mitochondrial Function. Curr. Opin. Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef] [Green Version]
- Ludtmann, M.H.R.; Angelova, P.R.; Zhang, Y.; Abramov, A.Y.; Dinkova-Kostova, A.T. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem. J. 2014, 457, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Holmström, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 Impacts Cellular Bioenergetics by Controlling Substrate Availability for Mitochondrial Respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef] [Green Version]
- Piantadosi, C.A.; Carraway, M.S.; Babiker, A.; Suliman, H.B. Heme Oxygenase-1 Regulates Cardiac Mitochondrial Biogenesis via Nrf2-Mediated Transcriptional Control of Nuclear Respiratory Factor-1. Circ. Res. 2008, 103, 1232–1240. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Lippai, M.; Lőw, P. The Role of the Selective Adaptor p62 and Ubiquitin-Like Proteins in Autophagy. BioMed Res. Int. 2014, 2014, 832704. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 Is a Target Gene for Transcription Factor NRF2 and Creates a Positive Feedback Loop by Inducing Antioxidant Response Element-driven Gene Transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, Y.; Waguri, S.; Sou, Y.-S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 Activates the Keap1-Nrf2 Pathway during Selective Autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [Green Version]
- Cherry, A.D.; Suliman, H.B.; Bartz, R.R.; Piantadosi, C.A. Peroxisome Proliferator-activated Receptor γ Co-activator 1-α as a Critical Co-activator of the Murine Hepatic Oxidative Stress Response and Mitochondrial Biogenesis in Staphylococcus aureus Sepsis. J. Biol. Chem. 2014, 289, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Min, K.-J.; Yang, M.-S.; Kim, S.-U.; Jou, I.; Joe, E.-H. Astrocytes Induce Hemeoxygenase-1 Expression in Microglia: A Feasible Mechanism for Preventing Excessive Brain Inflammation. J. Neurosci. 2006, 26, 1880–1887. [Google Scholar] [CrossRef] [Green Version]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. NRF2 and NF-κB interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019, 21, 101059. [Google Scholar] [CrossRef]
- Castro-Sánchez, S.; García-Yagüe, Á.J.; Kügler, S.; Lastres-Becker, I. CX3CR1-deficient microglia shows impaired signalling of the transcription factor NRF2: Implications in tauopathies. Redox Biol. 2019, 22, 101118. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory Response in the CNS: Friend or Foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef] [Green Version]
- Scuderi, S.A.; Ardizzone, A.; Paterniti, I.; Esposito, E.; Campolo, M. Antioxidant and Anti-Inflammatory Effect of Nrf2 Inducer Dimethyl Fumarate in Neurodegenerative Diseases. Antioxidants 2020, 9, 630. [Google Scholar] [CrossRef]
- Inui, M.; Ishida, Y.; Kimura, A.; Kuninaka, Y.; Mukaida, N.; Kondo, T. Protective Roles of CX3CR1-Mediated Signals in Toxin A-Induced Enteritis through the Induction of Heme Oxygenase-1 Expression. J. Immunol. 2011, 186, 423–431. [Google Scholar] [CrossRef] [Green Version]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Microglia and Aging: The Role of the TREM2–DAP12 and CX3CL1-CX3CR1 Axes. Int. J. Mol. Sci. 2018, 19, 318. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Nagalakshmi, D.; Sharma, K.K.; Ravichandiran, V. Natural antioxidants for neuroinflammatory disorders and possible involvement of Nrf2 pathway: A review. Heliyon 2021, 7, e06216. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.-Y.J.; Collado-Mateo, D.; et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s Disease: Biomarkers, Treatment, and Risk Factors. Front. Neurosci. 2018, 12, 612. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years On. J. Parkinsons Dis. 2017, 7, S51–S69. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Steur, E.N.H.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Iborra, S.F.; Cuadros, T.; Parent, A.; Romero-Gimenez, J.; Vila, M.; Perier, C. Defective mitochondrial protein import contributes to complex I-induced mitochondrial dysfunction and neurodegeneration in Parkinson’s disease. Cell Death Dis. 2018, 9, 1122. [Google Scholar] [CrossRef] [Green Version]
- Helley, M.P.; Pinnell, J.; Sportelli, C.; Tieu, K. Mitochondria: A Common Target for Genetic Mutations and Environmental Toxicants in Parkinson’s Disease. Front. Genet. 2017, 8, 177. [Google Scholar] [CrossRef]
- Devoto, V.M.P.; Falzone, T.L. Mitochondrial dynamics in Parkinson’s disease: A role for α-synuclein? Dis. Models Mech. 2017, 10, 1075–1087. [Google Scholar] [CrossRef] [Green Version]
- Grassi, D.; Howard, S.; Zhou, M.; Diaz-Perez, N.; Urban, N.T.; Guerrero-Given, D.; Kamasawa, N.; Volpicelli-Daley, L.A.; LoGrasso, P.; Lasmézas, C.I. Identification of a highly neurotoxic α-synuclein species inducing mitochondrial damage and mitophagy in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E2634–E2643. [Google Scholar] [CrossRef] [Green Version]
- Inamdar, N.; Arulmozhi, D.; Tandon, A.; Bodhankar, S. Parkinsons Disease: Genetics and Beyond. Curr. Neuropharmacol. 2007, 5, 99–113. [Google Scholar] [CrossRef]
- International Parkinson Disease Genomics Consortium. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet 2011, 377, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [Green Version]
- Alegre-Abarrategui, J.; Christian, H.; Lufino, M.M.P.; Mutihac, R.; Venda, L.L.; Ansorge, O.; Wade-Martins, R. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum. Mol. Genet. 2009, 18, 4022–4034. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Stevens, D.A.; Kang, S.-U.; Jiang, H.; Lee, Y.-I.; Ko, H.S.; Scarffe, L.A.; Umanah, G.E.; Kang, H.; Ham, S.; et al. PINK1 Primes Parkin-Mediated Ubiquitination of PARIS in Dopaminergic Neuronal Survival. Cell Rep. 2017, 18, 918–932. [Google Scholar] [CrossRef] [Green Version]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Mita, Y.; Kataoka, Y.; Saito, Y.; Kashi, T.; Hayashi, K.; Iwasaki, A.; Imanishi, T.; Miyasaka, T.; Noguchi, N. Distribution of oxidized DJ-1 in Parkinson’s disease-related sites in the brain and in the peripheral tissues: Effects of aging and a neurotoxin. Sci. Rep. 2018, 8, 12056. [Google Scholar] [CrossRef] [PubMed]
- Strobbe, D.; Robinson, A.A.; Harvey, K.; Rossi, L.; Ferraina, C.; De Biase, V.; Rodolfo, C.; Harvey, R.J.; Campanella, M. Distinct Mechanisms of Pathogenic DJ-1 Mutations in Mitochondrial Quality Control. Front. Mol. Neurosci. 2018, 11, 68. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Motherwell, M.S. The impact of reactive oxygen species and genetic mitochondrial mutations in Parkinson’s disease. Gene 2013, 532, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003, 53, S26–S38. [Google Scholar] [CrossRef] [PubMed]
- Kolodkin, A.N.; Sharma, R.P.; Papa, M.; Kumar, V.; Peters, B.; Skupin, A.; Alberghina, L.; Balling, R.; Westerhoff, H.V.; Colangelo, A.M.; et al. ROS networks: Designs, aging, Parkinson’s disease and precision therapies. npj Syst. Biol. Appl. 2020, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Brennan, A.M.; Suh, S.W.; Won, S.J.; Narasimhan, P.; Kauppinen, T.M.; Lee, H.; Edling, Y.; Chan, P.H.; Swanson, R.A. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci. 2009, 12, 857–863. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H.; Jenner, P. Etiology and pathogenesis of Parkinson’s disease. Mov. Disord. 2011, 26, 1049–1055. [Google Scholar] [CrossRef]
- Jenner, P.; Olanow, C.W. The pathogenesis of cell death in Parkinson’s disease. Neurology 2006, 66, S24–S36. [Google Scholar] [CrossRef]
- Kwon, D.H.; Cha, H.-J.; Lee, H.; Hong, S.-H.; Park, C.; Park, S.-H.; Kim, G.-Y.; Kim, S.; Kim, H.-S.; Hwang, H.-J.; et al. Protective Effect of Glutathione against Oxidative Stress-induced Cytotoxicity in RAW 264.7 Macrophages through Activating the Nuclear Factor Erythroid 2-Related Factor-2/Heme Oxygenase-1 Pathway. Antioxidants 2019, 8, 82. [Google Scholar] [CrossRef] [Green Version]
- Smeyne, M.; Smeyne, R.J. Glutathione metabolism and Parkinson’s disease. Free Radic. Biol. Med. 2013, 62, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Pearce, R.K.B.; Owen, A.; Daniel, S.; Jenner, P.; Marsden, C.D. Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. J. Neural Transm. 1997, 104, 661–677. [Google Scholar] [CrossRef]
- Hsu, M.; Srinivas, B.; Kumar, J.; Subramanian, R.; Andersen, J. Glutathione depletion resulting in selective mitochondrial complex I inhibition in dopaminergic cells is via an NO-mediated pathway not involving peroxynitrite: Implications for Parkinson’s disease: Glutathione Affects Mitochondrial Complex I via NO Not ONOO. J. Neurochem. 2005, 92, 1091–1103. [Google Scholar] [CrossRef]
- Haas, R.H.; Nasirian, F.; Nakano, K.; Ward, D.; Pay, M.; Hill, R.; Shults, C.W. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson’s disease. Ann. Neurol. 1995, 37, 714–722. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Burbulla, L.F.; Song, P.; Savas, J.N.; Kiskinis, E.; Zhuang, X.; Krüger, R.; Surmeier, D.J.; Krainc, D.; Mazzulli, J.R.; Zampese, E.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [Green Version]
- Berman, S.B.; Hastings, T.G. Dopamine Oxidation Alters Mitochondrial Respiration and Induces Permeability Transition in Brain Mitochondria: Implications for Parkinson’s Disease. J. Neurochem. 2001, 73, 1127–1137. [Google Scholar] [CrossRef]
- Bosco, D.A.; Fowler, D.M.; Zhang, Q.; Nieva, J.; Powers, E.T.; Wentworth, P.; Lerner, R.A.; Kelly, J.W. Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate α-synuclein fibrilization. Nat. Chem. Biol. 2006, 2, 249–253. [Google Scholar] [CrossRef]
- Davis, G.C.; Williams, A.C.; Markey, S.P.; Ebert, M.H.; Caine, E.D.; Reichert, C.M.; Kopin, I.J. Chronic parkinsonism secondary to intravenous injection of meperidine analogues. Psychiatry Res. 1979, 1, 249–254. [Google Scholar] [CrossRef]
- Langston, J.; Ballard, P.; Tetrud, J.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [Green Version]
- Nicklas, W.J.; Vyas, I.; Heikkila, R.E. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 1985, 36, 2503–2508. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Salach, J.I.; Dadgar, J.; Singer, T.P. Inhibition of mitochondrial NADH dehydrogenase by pyridine derivatives and its possible relation to experimental and idiopathic parkinsonism. Biochem. Biophys. Res. Commun. 1986, 135, 269–275. [Google Scholar] [CrossRef]
- Chan, P.; DeLanney, L.E.; Irwin, I.; Langston, J.W.; Di Monte, D. Rapid ATP Loss Caused by 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine in Mouse Brain. J. Neurochem. 1991, 57, 348–351. [Google Scholar] [CrossRef]
- Drechsel, D.A.; Patel, M. Role of reactive oxygen species in the neurotoxicity of environmental agents implicated in Parkinson’s disease. Free Radic. Biol. Med. 2008, 44, 1873–1886. [Google Scholar] [CrossRef] [Green Version]
- Dauer, W.; Przedborski, S. Parkinson’s Disease: Mechanisms and Models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, A.; Michel, P.P.; Troadec, J.-D.; Mouatt-Prigent, A.; Faucheux, B.A.; Ruberg, M.; Agid, Y.; Hirsch, E.C. Is Bax a mitochondrial mediator in apoptotic death of dopaminergic neurons in Parkinson’s disease? J. Neurochem. 2001, 76, 1785–1793. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 333, 1269. [Google Scholar] [CrossRef]
- Janetzky, B.; Hauck, S.; Youdim, M.B.H.; Riederer, P.; Jellinger, K.; Pantucek, F.; Zöchling, R.; Boissl, K.W.; Reichmann, H. Unaltered aconitase activity, but decreased complex I activity in substantia nigra pars compacta of patients with Parkinson’s disease. Neurosci. Lett. 1994, 169, 126–128. [Google Scholar] [CrossRef]
- Smigrodzki, R.; Parks, J.; Parker, W.D. High frequency of mitochondrial complex I mutations in Parkinson’s disease and aging. Neurobiol. Aging 2004, 25, 1273–1281. [Google Scholar] [CrossRef]
- Parker, D.W.; Parks, J.K.; Swerdlow, R.H. Complex I Deficiency in Parkinson’s Disease Frontal Cortex. Neurobiol. Aging 2008, 25, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Bury, A.G.; Pyle, A.; Elson, J.L.; Greaves, L.; Morris, C.M.; Hudson, G.; Pienaar, I.S. Mitochondrial DNA changes in pedunculopontine cholinergic neurons in Parkinson disease. Ann. Neurol. 2017, 82, 1016–1021. [Google Scholar] [CrossRef]
- Flønes, I.H.; Fernandez-Vizarra, E.; Lykouri, M.; Brakedal, B.; Skeie, G.O.; Miletic, H.; Lilleng, P.K.; Alves, G.; Tysnes, O.-B.; Haugarvoll, K.; et al. Neuronal complex I deficiency occurs throughout the Parkinson’s disease brain, but is not associated with neurodegeneration or mitochondrial DNA damage. Acta Neuropathol. 2018, 135, 409–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virbasius, J.V.; Scarpulla, R.C. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: A potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 1309–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekstrand, M.I.; Terzioglu, M.; Hoffer, B.; Cullheim, S.; Mohammed, A.H.; Olson, L.; Larsson, N.-G.; Galter, D.; Zhu, S.; Hofstetter, C.; et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 1325–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.-X.; Yin, X.-M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [Green Version]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat. Cell Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef]
- Barazzuol, L.; Giamogante, F.; Brini, M.; Calì, T. PINK1/Parkin Mediated Mitophagy, Ca2+ Signalling, and ER–Mitochondria Contacts in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1772. [Google Scholar] [CrossRef] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Khymenets, O.; Ortuño, J.; Fitó, M.; Covas, M.I.; Farré, M.; De La Torre, R. Evaluation of RNA isolation procedures from human blood and its application for gene expression studies (Sod-1, Sod-2). Anal. Biochem. 2005, 347, 156–158. [Google Scholar] [CrossRef]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct Membrane Association Drives Mitochondrial Fission by the Parkinson Disease-associated Protein α-Synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [Green Version]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Shirihai, O.S.; Song, M.; Dorn, G.W., II. How Mitochondrial Dynamism Orchestrates Mitophagy. Circ. Res. 2015, 116, 1835–1849. [Google Scholar] [CrossRef] [Green Version]
- Stevens, D.A.; Lee, Y.; Kang, H.C.; Lee, B.D.; Lee, Y.-I.; Bower, A.; Jiang, H.; Kang, S.-U.; Andrabi, S.A.; Dawson, V.L.; et al. Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc. Natl. Acad. Sci. USA 2015, 112, 11696–11701. [Google Scholar] [CrossRef] [Green Version]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A Cold-Inducible Coactivator of Nuclear Receptors Linked to Adaptive Thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Castillo-Quan, J.I. Parkin’ control: Regulation of PGC-1α through PARIS in Parkinson’s disease. Dis. Models Mech. 2011, 4, 427–429. [Google Scholar] [CrossRef] [Green Version]
- Murata, H.; Takamatsu, H.; Liu, S.; Kataoka, K.; Huh, N.-H.; Sakaguchi, M. NRF2 Regulates PINK1 Expression under Oxidative Stress Conditions. PLoS ONE 2015, 10, e0142438. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, N.; Lu, B. Mechanisms and roles of mitophagy in neurodegenerative diseases. CNS Neurosci. Ther. 2019, 25, 859–875. [Google Scholar] [CrossRef]
- Dagda, R.K.; Gusdon, A.M.; Pien, I.; Strack, S.; Green, S.; Li, C.; Van Houten, B.; Cherra, S.J.; Chu, C.T. Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinson’s disease. Cell Death Differ. 2011, 18, 1914–1923. [Google Scholar] [CrossRef] [Green Version]
- Deas, E.; Wood, N.W.; Plun-Favreau, H. Mitophagy and Parkinson’s disease: The PINK1–parkin link. Biochim. Biophys. Acta BBA Mol. Cell Res. 2011, 1813, 623–633. [Google Scholar] [CrossRef] [Green Version]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial Dysfunction and Oxidative Damage in parkin-deficient Mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar] [CrossRef] [Green Version]
- Flinn, L.; Mortiboys, H.; Volkmann, K.; Köster, R.W.; Ingham, P.W.; Bandmann, O. Complex I deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio). Brain 2009, 132, 1613–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanon, A.; Kalvakuri, S.; Rakovic, A.; Foco, L.; Guida, M.; Schwienbacher, C.; Serafin, A.; Rudolph, F.; Trilck, M.; Grünewald, A.; et al. Corrigendum: SLP-2 interacts with Parkin in mitochondria and prevents mitochondrial dysfunction in Parkin-deficient human iPSC-derived neurons and Drosophila. Hum. Mol. Genet. 2019, 28, 1225. [Google Scholar] [CrossRef] [PubMed]
- Cherra, S.J., III; Dagda, R.K.; Chu, C.T. Review: Autophagy and neurodegeneration: Survival at a cost? Neuropathol. Appl. Neurobiol. 2010, 36, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Plowey, E.D.; Dagda, R.K.; Hickey, R.W.; Cherra, S.J.; Clark, R.S.B. Autophagy in neurite injury and neurodegeneration: In vitro and in vivo models. Methods Enzymol. 2009, 453, 217–249. [Google Scholar]
- Lim, J.L.; Wilhelmus, M.M.M.; De Vries, H.E.; Drukarch, B.; Hoozemans, J.J.M.; Van Horssen, J. Antioxidative defense mechanisms controlled by Nrf2: State-of-the-art and clinical perspectives in neurodegenerative diseases. Arch. Toxicol. 2014, 88, 1773–1786. [Google Scholar] [CrossRef]
- Ahuja, M.; Kaidery, N.A.; Yang, L.; Calingasan, N.; Smirnova, N.; Gaisin, A.; Gaisina, I.N.; Gazaryan, I.; Hushpulian, D.M.; Kaddour-Djebbar, I.; et al. Distinct Nrf2 Signaling Mechanisms of Fumaric Acid Esters and Their Role in Neuroprotection against 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Experimental Parkinson’s-Like Disease. J. Neurosci. 2016, 36, 6332–6351. [Google Scholar] [CrossRef] [Green Version]
- Campolo, M.; Casili, G.; Lanza, M.; Filippone, A.; Paterniti, I.; Cuzzocrea, S.; Esposito, E. Multiple mechanisms of dimethyl fumarate in amyloid β-induced neurotoxicity in human neuronal cells. J. Cell. Mol. Med. 2018, 22, 1081–1094. [Google Scholar] [CrossRef]
- Onasanwo, S.A.; Velagapudi, R.; El-Bakoush, A.; Olajide, O.A. Inhibition of neuroinflammation in BV2 microglia by the biflavonoid kolaviron is dependent on the Nrf2/ARE antioxidant protective mechanism. Mol. Cell. Biochem. 2016, 414, 23–36. [Google Scholar] [CrossRef]
- Bowling, A.C.; Beal, M.F. Bioenergetic and oxidative stress in neurodegenerative diseases. Life Sci. 1995, 56, 1151–1171. [Google Scholar] [CrossRef]
- Browne, S.E.; Beal, M.F. Oxidative Damage in Huntington’s Disease Pathogenesis. Antioxidants Redox Signal. 2006, 8, 2061–2073. [Google Scholar] [CrossRef]
- De Vries, H.E.; Witte, M.; Hondius, D.; Rozemuller, A.J.M.; Drukarch, B.; Hoozemans, J.; van Horssen, J. Nrf2-induced antioxidant protection: A promising target to counteract ROS-mediated damage in neurodegenerative disease? Free Radic. Biol. Med. 2008, 45, 1375–1383. [Google Scholar] [CrossRef]
- Van Muiswinkel, F.L.; Kuiperij, H.B. The Nrf2-ARE Signalling Pathway: Promising Drug Target to Combat Oxidative Stress in Neurodegenerative Disorders. Curr. Drug Target CNS Neurol. Disord. 2005, 4, 267–281. [Google Scholar] [CrossRef]
- Clements, C.M.; McNally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P.-Y. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Jain, M.R.; Chen, C.; Yue, X.; Hebbar, V.; Zhou, R.; Kong, A.-N.T. Nrf2 Possesses a Redox-insensitive Nuclear Export Signal Overlapping with the Leucine Zipper Motif. J. Biol. Chem. 2005, 280, 28430–28438. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, D.; Thimmulappa, R.; Navas-Acien, A.; Sandford, A.; Elliott, M.; Singh, A.; Chen, L.; Zhuang, X.; Hogg, J.; Paré, P.; et al. Decline in NRF2-regulated Antioxidants in Chronic Obstructive Pulmonary Disease Lungs Due to Loss of Its Positive Regulator, DJ-1. Am. J. Respir. Crit. Care Med. 2008, 178, 592–604. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Freed, C.R. DJ-1 Up-regulates Glutathione Synthesis during Oxidative Stress and Inhibits A53T α-Synuclein Toxicity. J. Biol. Chem. 2005, 280, 43150–43158. [Google Scholar] [CrossRef] [Green Version]
- Dick, F.D.; De Palma, G.; Ahmadi, A.; Osborne, A.; Scott, N.W.; Prescott, G.J.; Bennett, J.; Semple, S.; Dick, S.; Mozzoni, P.; et al. Gene-environment interactions in parkinsonism and Parkinson’s disease: The Geoparkinson study. Occup. Environ. Med. 2007, 64, 673–680. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Khan, A.J.; Shah, P.P.; Shukla, R.; Khanna, V.K.; Parmar, D. Polymorphism in environment responsive genes and association with Parkinson disease. Mol. Cell. Biochem. 2008, 312, 131–138. [Google Scholar] [CrossRef]
- Petrillo, S.; Schirinzi, T.; Di Lazzaro, G.; D’Amico, J.; Colona, V.L.; Bertini, E.; Pierantozzi, M.; Mari, L.; Mercuri, N.B.; Piemonte, F.; et al. Systemic Activation of Nrf2 Pathway in Parkinson’s Disease. Mov. Disord. 2020, 35, 180–184. [Google Scholar] [CrossRef]
- Tufekci, K.U.; Bayin, E.C.; Genc, S.; Genc, K. The Nrf2/ARE Pathway: A Promising Target to Counteract Mitochondrial Dysfunction in Parkinson’s Disease. Parkinson’s Dis. 2011, 2011, 314082. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Hoeflich, K.P.; Woodgett, J.R. Glycogen Synthase Kinase-3: Properties, Functions, and Regulation. Chem. Rev. 2001, 101, 2527–2540. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.; Chen, Z.; Gao, J.; Shi, W.; Li, L.; Jiang, S.; Hu, H.; Liu, Z.; Xu, D.; Wu, L. The Key Roles of GSK-3β in Regulating Mitochondrial Activity. Cell. Physiol. Biochem. 2017, 44, 1445–1459. [Google Scholar] [CrossRef] [PubMed]
- Kwok, J.B.J.; Hallupp, M.; Loy, C.T.; Chan, D.K.Y.; Woo, J.; Mellick, G.D.; Buchanan, D.D.; Silburn, P.A.; Halliday, G.M.; Schofield, P.R. GSK3B polymorphisms alter transcription and splicing in Parkinson’s disease. Ann. Neurol. 2005, 58, 829–839. [Google Scholar] [CrossRef]
- Skibinski, G.; Hwang, V.; Ando, D.M.; Daub, A.; Lee, A.K.; Ravisankar, A.; Modan, S.; Finucane, M.M.; Shaby, B.A.; Finkbeiner, S. Nrf2 mitigates LRRK2- and α-synuclein–induced neurodegeneration by modulating proteostasis. Proc. Natl. Acad. Sci. USA 2017, 114, 1165–1170. [Google Scholar] [CrossRef] [Green Version]
- Jakel, R.J.; Townsend, J.A.; Kraft, A.D.; Johnson, J.A. Nrf2-mediated protection against 6-hydroxydopamine. Brain Res. 2007, 1144, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Williamson, T.P.; Johnson, D.A.; Johnson, J.A. Activation of the Nrf2-ARE pathway by siRNA knockdown of Keap1 reduces oxidative stress and provides partial protection from MPTP-mediated neurotoxicity. NeuroToxicology 2012, 33, 272–279. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.-C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar] [CrossRef] [Green Version]
- Seidl, S.E.; Potashkin, J.A. The Promise of Neuroprotective Agents in Parkinson’s Disease. Front. Neurol. 2011, 2, 68. [Google Scholar] [CrossRef] [Green Version]
- Coles, L.D.; Tuite, P.J.; Öz, G.; Mishra, U.R.; Kartha, R.V.; Sullivan, K.M.; Cloyd, J.C.; Terpstra, M. Repeated-Dose Oral N-Acetylcysteine in Parkinson’s Disease: Pharmacokinetics and Effect on Brain Glutathione and Oxidative Stress. J. Clin. Pharmacol. 2018, 58, 158–167. [Google Scholar] [CrossRef]
- Jazwa, A.; Rojo, A.I.; Innamorato, N.G.; Hesse, M.; Fernández-Ruiz, J.; Cuadrado, A. Pharmacological Targeting of the Transcription Factor Nrf2 at the Basal Ganglia Provides Disease Modifying Therapy for Experimental Parkinsonism. Antioxid. Redox Signal. 2011, 14, 2347–2360. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Beltrán, C.E.; Mukhopadhyay, P.; Horváth, B.; Rajesh, M.; Tapia, E.; García-Torres, I.; Pedraza-Chaverri, J.; Pacher, P. Sulforaphane, a natural constituent of broccoli, prevents cell death and inflammation in nephropathy. J. Nutr. Biochem. 2012, 23, 494–500. [Google Scholar] [CrossRef] [Green Version]
- Uddin, M.S.; Al Mamun, A.; Jakaria, M.; Thangapandiyan, S.; Ahmad, J.; Rahman, M.A.; Mathew, B.; Abdel-Daim, M.M.; Aleya, L. Emerging promise of sulforaphane-mediated Nrf2 signaling cascade against neurological disorders. Sci. Total Environ. 2020, 707, 135624. [Google Scholar] [CrossRef]
- Angeloni, C.; Malaguti, M.; Rizzo, B.; Barbalace, M.C.; Fabbri, D.; Hrelia, S. Neuroprotective Effect of Sulforaphane against Methylglyoxal Cytotoxicity. Chem. Res. Toxicol. 2015, 28, 1234–1245. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, J.; Fang, L.; Li, X.; Zhao, Y.; Shi, W.; An, L. Neuroprotective Effects of Sulforaphane on Cholinergic Neurons in Mice with Alzheimer’s Disease-Like Lesions. Int. J. Mol. Sci. 2014, 15, 14396–14410. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, T.; Leak, R.K.; Chen, J.; Zhang, F. Preventive and Protective Roles of Dietary Nrf2 Activators Against Central Nervous System Diseases. CNS Neurol. Disord. Drug Targets 2017, 16, 326–338. [Google Scholar] [CrossRef] [Green Version]
- Hong, F.; Freeman, A.M.L.; Liebler, D.C. Identification of Sensor Cysteines in Human Keap1 Modified by the Cancer Chemopreventive Agent Sulforaphane. Chem. Res. Toxicol. 2005, 18, 1917–1926. [Google Scholar] [CrossRef]
- Shang, G.; Tang, X.; Gao, P.; Guo, F.; Liu, H.; Zhao, Z.; Chen, Q.; Jiang, T.; Zhang, N.; Li, H. Sulforaphane attenuation of experimental diabetic nephropathy involves GSK-3 beta/Fyn/Nrf2 signaling pathway. J. Nutr. Biochem. 2015, 26, 596–606. [Google Scholar] [CrossRef]
- Han, J.M.; Lee, Y.J.; Lee, S.Y.; Kim, E.M.; Moon, Y.; Kim, H.W.; Hwang, O. Protective Effect of Sulforaphane against Dopaminergic Cell Death. J. Pharmacol. Exp. Ther. 2007, 321, 249–256. [Google Scholar] [CrossRef]
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 2013, 100, 30–47. [Google Scholar] [CrossRef] [Green Version]
- Siebert, A.; Desai, V.; Chandrasekaran, K.; Fiskum, G.; Jafri, M.S. Nrf2 activators provide neuroprotection against 6-hydroxydopamine toxicity in rat organotypic nigrostriatal cocultures. J. Neurosci. Res. 2009, 87, 1659–1669. [Google Scholar] [CrossRef]
- Tarozzi, A.; Angeloni, C.; Malaguti, M.; Morroni, F.; Hrelia, S.; Hrelia, P. Sulforaphane as a Potential Protective Phytochemical against Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2013, 2013, 415078. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Smith, M.A. Alzheimer Disease. Int. Rev. Neurobiol. 1998, 42, 1–54. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.-G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroy, K.; Yilmaz, Z.; Brion, J.-P. Increased level of active GSK-3β in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 2007, 33, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Hernández, F.; de Barreda, E.G.; Fuster-Matanzo, A.; Lucas, J.J.; Avila, J. GSK3: A possible link between beta amyloid peptide and tau protein. Exp. Neurol. 2010, 223, 322–325. [Google Scholar] [CrossRef]
- Takashima, A. GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 309–317. [Google Scholar] [CrossRef]
- Lucas, J.J.; Hernández, F.; Gómez-Ramos, P.; Morán, M.A.; Hen, R.; Avila, J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 2001, 20, 27–39. [Google Scholar] [CrossRef]
- Markesbery, W.R. Oxidative Stress Hypothesis in Alzheimer’s Disease. Free Radic. Biol. Med. 1997, 23, 134–147. [Google Scholar] [CrossRef]
- Christen, Y. Oxidative stress and Alzheimer disease. Am. J. Clin. Nutr. 2000, 71, 621S–629S. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D. Oxidative Stress, Amyloid-β Peptide, and Altered Key Molecular Pathways in the Pathogenesis and Progression of Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1345–1367. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhong, C. Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef]
- Xie, H.; Hou, S.; Jiang, J.; Sekutowicz, M.; Kelly, J.; Bacskai, B.J. Rapid cell death is preceded by amyloid plaque-mediated oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 7904–7909. [Google Scholar] [CrossRef] [Green Version]
- Siegel, S.J.; Bieschke, J.; Powers, E.T.; Kelly, J.W. The Oxidative Stress Metabolite 4-Hydroxynonenal Promotes Alzheimer Protofibril Formation. Biochemistry 2007, 46, 1503–1510. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Hensley, K.; Cole, P.; Subramaniam, R.; Aksenov, M.; Aksenova, M.; Bummer, P.M.; Haley, B.E.; Carney, J.M. Oxidatively Induced Structural Alteration of Glutamine Synthetase Assessed by Analysis of Spin Label Incorporation Kinetics: Relevance to Alzheimer’s Disease. J. Neurochem. 2002, 68, 2451–2457. [Google Scholar] [CrossRef]
- Aksenov, M.Y.; Aksenova, M.V.; Butterfield, D.A.; Geddes, J.W.; Markesbery, W.R. Protein oxidation in the brain in Alzheimer’s disease. Neuroscience 2001, 103, 373–383. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [Green Version]
- Trushina, E.; Nemutlu, E.; Dzeja, P.P.; Poduslo, J.F.; Zhang, S.; Christensen, T.; Camp, J.; Mesa, J.; Siddiqui, A.; Tamura, Y.; et al. Defects in Mitochondrial Dynamics and Metabolomic Signatures of Evolving Energetic Stress in Mouse Models of Familial Alzheimer’s Disease. PLoS ONE 2012, 7, e32737. [Google Scholar] [CrossRef] [Green Version]
- Valla, J.; Yaari, R.; Wolf, A.B.; Kusne, Y.; Beach, T.G.; Roher, A.E.; Corneveaux, J.J.; Huentelman, M.J.; Caselli, R.J.; Reiman, E.M. Reduced Posterior Cingulate Mitochondrial Activity in Expired Young Adult Carriers of the APOE ε4 Allele, the Major Late-Onset Alzheimer’s Susceptibility Gene. J. Alzheimer’s Dis. 2010, 22, 307–313. [Google Scholar] [CrossRef] [Green Version]
- Trushina, E.; Dutta, T.; Persson, X.-M.T.; Mielke, M.M.; Petersen, R.C. Identification of Altered Metabolic Pathways in Plasma and CSF in Mild Cognitive Impairment and Alzheimer’s Disease Using Metabolomics. PLoS ONE 2013, 8, e63644. [Google Scholar] [CrossRef] [Green Version]
- Price, J.L.; McKeel, D.W., Jr.; Buckles, V.D.; Roe, C.M.; Xiong, C.; Grundman, M.; Hansen, L.A.; Petersen, R.C.; Parisi, J.E.; Dickson, D.W.; et al. Neuropathology of nondemented aging: Presumptive evidence for preclinical Alzheimer disease. Neurobiol. Aging 2009, 30, 1026–1036. [Google Scholar] [CrossRef] [Green Version]
- Beal, M.F. Mitochondria take center stage in aging and neurodegeneration. Ann. Neurol. 2005, 58, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2009, 109, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detmer, S.A.; Chan, D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondria: Dynamic Organelles in Disease, Aging, and Development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–518. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of Fusion Results in Mitochondrial Heterogeneity and Dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Song, J.; Tan, M.; Albers, K.M.; Jia, J. Mitochondrial fission proteins in peripheral blood lymphocytes are potential biomarkers for Alzheimer’s disease. Eur. J. Neurol. 2012, 19, 1015–1022. [Google Scholar] [CrossRef]
- Cho, D.-H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-Nitrosylation of Drp1 Mediates β-Amyloid-Related Mitochondrial Fission and Neuronal Injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Perry, G.; Smith, M.A.; Wang, X. Abnormal Mitochondrial Dynamics in the Pathogenesis of Alzheimer’s Disease. J. Alzheimer’s Dis. 2013, 33, S253–S262. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-β overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Choi, H.; Min, J.-S.; Kim, B.; Lee, S.-R.; Yun, J.W.; Choi, M.-S.; Chang, K.-T.; Lee, D.-S. Loss of mitofusin 2 links beta-amyloid-mediated mitochondrial fragmentation and Cdk5-induced oxidative stress in neuron cells. J. Neurochem. 2015, 132, 687–702. [Google Scholar] [CrossRef] [Green Version]
- Castellani, R.; Hirai, K.; Aliev, G.; Drew, K.L.; Nunomura, A.; Takeda, A.; Cash, A.D.; Obrenovich, M.E.; Perry, G.; Smith, M.A. Role of mitochondrial dysfunction in Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 357–360. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative Stress and Neurodegenerative Diseases: A Review of Upstream and Downstream Antioxidant Therapeutic Options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of Amyloid Precursor Protein in the Mitochondrial Import Channels of Human Alzheimer’s Disease Brain Is Associated with Mitochondrial Dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [Green Version]
- Casley, C.S.; Canevari, L.; Land, J.M.; Clark, J.B.; Sharpe, M.A. β-Amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 2002, 80, 91–100. [Google Scholar] [CrossRef]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD Directly Links Aβ to Mitochondrial Toxicity in Alzheimer’s Disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, S.M.; Santana, I.; Swerdlow, R.H.; Oliveira, C.R. Mitochondria dysfunction of Alzheimer’s disease cybrids enhances Aβ toxicity. J. Neurochem. 2004, 89, 1417–1426. [Google Scholar] [CrossRef] [Green Version]
- Gordon, B.A.; Blazey, T.M.; Su, Y.; Hari-Raj, A.; Dincer, A.; Flores, S.; Christensen, J.; McDade, E.; Wang, G.; Xiong, C.; et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: A longitudinal study. Lancet Neurol. 2018, 17, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Kapogiannis, D.; Mattson, M.P. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurol. 2011, 10, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Sonntag, K.-C.; Ryu, W.-I.; Amirault, K.M.; Healy, R.A.; Siegel, A.J.; McPhie, D.L.; Forester, B.; Cohen, B.M. Late-onset Alzheimer’s disease is associated with inherent changes in bioenergetics profiles. Sci. Rep. 2017, 7, 14038. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, C.; Cardoso, S.; Correia, S.C.; Santos, R.X.; Santos, M.S.; Baldeiras, I.; Oliveira, C.R.; Moreira, P.I. Metabolic Alterations Induced by Sucrose Intake and Alzheimer’s Disease Promote Similar Brain Mitochondrial Abnormalities. Diabetes 2012, 61, 1234–1242. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Z.-H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [Green Version]
- Stokin, G.B.; Lillo, C.; Falzone, T.L.; Brusch, R.G.; Rockenstein, E.; Mount, S.L.; Raman, R.; Davies, P.; Masliah, E.; Williams, D.S.; et al. Axonopathy and Transport Deficits Early in the Pathogenesis of Alzheimer’s Disease. Science 2005, 307, 1282–1288. [Google Scholar] [CrossRef]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef]
- Ma, H.; Cai, Q.; Lu, W.; Sheng, Z.-H.; Mochida, S. KIF5B Motor Adaptor Syntabulin Maintains Synaptic Transmission in Sympathetic Neurons. J. Neurosci. 2009, 29, 13019–13029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Q.; Davis, M.L.; Sheng, Z.-H. Regulation of axonal mitochondrial transport and its impact on synaptic transmission. Neurosci. Res. 2011, 70, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tammineni, P.; Ye, X.; Feng, T.; Aikal, D.; Cai, Q. Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons. eLife 2017, 6, e21776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H.; McWeeney, S.; Park, B.S.; Manczak, M.; Gutala, R.V.; Partovi, D.; Jung, Y.; Yau, V.; Searles, R.; Mori, M.; et al. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: Up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum. Mol. Genet. 2004, 13, 1225–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, L.; Latypova, X.; Terro, F. Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem. Int. 2011, 58, 458–471. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 22–35. [Google Scholar] [CrossRef]
- Alonso, A.D.C.; Grundke-Iqbal, I.; Barra, H.S.; Iqbal, K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: Sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. USA 1997, 94, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Gendron, T.F.; Petrucelli, L. The role of tau in neurodegeneration. Mol. Neurodegener. 2009, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Tapia-Rojas, C.; Cabezas-Opazo, F.; Deaton, C.A.; Vergara, E.H.; Johnson, G.V.W.; Quintanilla, R.A. It’s all about tau. Prog. Neurobiol. 2019, 175, 54–76. [Google Scholar] [CrossRef]
- Cente, M.; Filipcik, P.; Pevalova, M.; Novak, M. Expression of a truncated tau protein induces oxidative stress in a rodent model of tauopathy. Eur. J. Neurosci. 2006, 24, 1085–1090. [Google Scholar] [CrossRef]
- Schulz, K.L.; Eckert, A.; Rhein, V.; Mai, S.; Haase, W.; Reichert, A.S.; Jendrach, M.; Müller, W.E.; Leuner, K. A New Link to Mitochondrial Impairment in Tauopathies. Mol. Neurobiol. 2012, 46, 205–216. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Atlante, A.; Florenzano, F.; Capsoni, S.; Bussani, R.; Mercanti, D.; Calissano, P. Interaction between NH2-tau fragment and Aβ in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol. Aging 2012, 33, 833.e1–833.e25. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Calissano, P.; Florenzano, F.; Atlante, A.; Ciotti, M.T.; Mongiardi, M.P.; Bussani, R.; Nicolin, V.; Nori, S.L.; et al. AD-linked, toxic NH2 human tau affects the quality control of mitochondria in neurons. Neurobiol. Dis. 2014, 62, 489–507. [Google Scholar] [CrossRef]
- Fasulo, L.; Ugolini, G.; Cattaneo, A. Apoptotic effect of caspase-3 cleaved tau in hippocampal neurons and its potentiation by tau FTDP-mutation N279K. J. Alzheimer’s Dis. 2005, 7, 3–13. [Google Scholar] [CrossRef]
- Lopes, S.; Teplytska, L.; Vaz-Silva, J.; Dioli, C.; Trindade, R.; Morais, M.; Webhofer, C.; Maccarrone, G.; Almeida, O.F.X.; Turck, C.W.; et al. Tau Deletion Prevents Stress-Induced Dendritic Atrophy in Prefrontal Cortex: Role of Synaptic Mitochondria. Cereb. Cortex 2017, 27, 2580–2591. [Google Scholar] [CrossRef] [Green Version]
- Jara, C.; Aránguiz, A.; Cerpa, W.; Tapia-Rojas, C.; Quintanilla, R.A. Genetic ablation of tau improves mitochondrial function and cognitive abilities in the hippocampus. Redox Biol. 2018, 18, 279–294. [Google Scholar] [CrossRef]
- Gamblin, T.C.; Chen, F.; Zambrano, A.; Abraha, A.; Lagalwar, S.; Guillozet, A.L.; Lu, M.; Fu, Y.; Garcia-Sierra, F.; Lapointe, N.; et al. Caspase cleavage of tau: Linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 10032–10037. [Google Scholar] [CrossRef] [Green Version]
- Rissman, R.A.; Poon, W.W.; Blurton-Jones, M.; Oddo, S.; Torp, R.; Vitek, M.P.; LaFerla, F.M.; Rohn, T.T.; Cotman, C.W. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J. Clin. Investig. 2004, 114, 121–130. [Google Scholar] [CrossRef] [Green Version]
- García-Sierra, F.; Mondragón-Rodríguez, S.; Basurto-Islas, G. Truncation of Tau Protein and its Pathological Significance in Alzheimer’s Disease. J. Alzheimer’s Dis. 2008, 14, 401–409. [Google Scholar] [CrossRef]
- Chung, C.-W.; Song, Y.-H.; Kim, I.-K.; Yoon, W.-J.; Ryu, B.-R.; Jo, D.-G.; Woo, H.-N.; Kwon, Y.-K.; Kim, H.-H.; Gwag, B.-J.; et al. Proapoptotic Effects of Tau Cleavage Product Generated by Caspase-3. Neurobiol. Dis. 2001, 8, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Quintanilla, R.A.; Matthews-Roberson, T.A.; Dolan, P.J.; Johnson, G.V.W. Caspase-cleaved Tau Expression Induces Mitochondrial Dysfunction in Immortalized Cortical Neurons: Implications for the Pathogenesis of Alzheimer Disease. J. Biol. Chem. 2009, 284, 18754–18766. [Google Scholar] [CrossRef] [Green Version]
- Pérez, M.J.; Vergara-Pulgar, K.; Jara, C.; Cabezas-Opazo, F.; Quintanilla, R.A. Caspase-Cleaved Tau Impairs Mitochondrial Dynamics in Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 1004–1018. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Dolan, P.J.; Jin, Y.N.; Johnson, G.V.W. Truncated tau and Aβ cooperatively impair mitochondria in primary neurons. Neurobiol. Aging 2012, 33, 619.e25–619.e35. [Google Scholar] [CrossRef] [Green Version]
- Quintanilla, R.A.; von Bernhardi, R.; Godoy, J.A.; Inestrosa, N.C.; Johnson, G.V.W. Phosphorylated tau potentiates Aβ-induced mitochondrial damage in mature neurons. Neurobiol. Dis. 2014, 71, 260–269. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Tapia-Monsalves, C.; Vergara, E.H.; Pérez, M.J.; Aranguiz, A. Truncated Tau Induces Mitochondrial Transport Failure Through the Impairment of TRAK2 Protein and Bioenergetics Decline in Neuronal Cells. Front. Cell. Neurosci. 2020, 14, 175. [Google Scholar] [CrossRef]
- Brickley, K.; Stephenson, F.A. Trafficking Kinesin Protein (TRAK)-mediated Transport of Mitochondria in Axons of Hippocampal Neurons. J. Biol. Chem. 2011, 286, 18079–18092. [Google Scholar] [CrossRef] [Green Version]
- Bahn, G.; Jo, D.-G. Therapeutic Approaches to Alzheimer’s Disease Through Modulation of NRF2. NeuroMol. Med. 2019, 21, 1–11. [Google Scholar] [CrossRef]
- Jo, C.; Gundemir, S.; Pritchard, S.; Jin, Y.N.; Rahman, I.; Johnson, G.V.W. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014, 5, 3496. [Google Scholar] [CrossRef]
- Fujita, K.-I.; Maeda, D.; Xiao, Q.; Srinivasula, S.M. Nrf2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc. Natl. Acad. Sci. USA 2011, 108, 1427–1432. [Google Scholar] [CrossRef] [Green Version]
- Tanji, K.; Maruyama, A.; Odagiri, S.; Mori, F.; Itoh, K.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Keap1 Is Localized in Neuronal and Glial Cytoplasmic Inclusions in Various Neurodegenerative Diseases. J. Neuropathol. Exp. Neurol. 2013, 72, 18–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanji, K.; Miki, Y.; Kakita, A.; Takahashi, H.; Wakabayashi, K.; Ozaki, T.; Maruyama, A.; Yoshida, H.; Mimura, J.; Matsumiya, T.; et al. Phosphorylation of serine 349 of p62 in Alzheimer’s disease brain. Acta Neuropathol. Commun. 2014, 2, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Wang, W.; Liu, R.; Huang, H.; Zhang, R.; Sun, L. Effect of p62 on tau hyperphosphorylation in a rat model of Alzheimer’s disease. Neural Regen. Res. 2012, 7, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Pajares, M.; García-Yagüe, A.J.; Buendia, I.; Van Leuven, F.; Yamamoto, M.; López, M.G.; Cuadrado, A. Deficiency in the transcription factor NRF2 worsens inflammatory parameters in a mouse model with combined tauopathy and amyloidopathy. Redox Biol. 2018, 18, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Eftekharzadeh, B.; Maghsoudi, N.; Khodagholi, F. Stabilization of transcription factor Nrf2 by tBHQ prevents oxidative stress-induced amyloid β formation in NT2N neurons. Biochimie 2010, 92, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in Neurodegenerative Diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Chalovich, E.M.; Zhu, J.-H.; Caltagarone, J.; Bowser, R.; Chu, C.T. Functional Repression of cAMP Response Element in 6-Hydroxydopamine-treated Neuronal Cells. J. Biol. Chem. 2006, 281, 17870–17881. [Google Scholar] [CrossRef] [Green Version]
- Kerr, F.; Sofola-Adesakin, O.; Ivanov, D.K.; Gatliff, J.; Gomez Perez-Nievas, B.; Bertrand, H.C.; Martinez, P.; Callard, R.; Snoeren, I.; Cochemé, H.M.; et al. Direct Keap1-Nrf2 disruption as a potential therapeutic target for Alzheimer’s disease. PLoS Genet. 2017, 13, e1006593. [Google Scholar] [CrossRef] [Green Version]
- Gabbita, S.P.; Lovell, M.A.; Markesbery, W.R. Increased Nuclear DNA Oxidation in the Brain in Alzheimer’s Disease. J. Neurochem. 1998, 71, 2034–2040. [Google Scholar] [CrossRef] [Green Version]
- Hooper, C.; Markevich, V.; Plattner, F.; Killick, R.; Schofield, E.; Engel, T.; Hernández, F.; Anderton, B.; Rosenblum, K.; Bliss, T.; et al. Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur. J. Neurosci. 2007, 25, 81–86. [Google Scholar] [CrossRef]
- Praticò, D.; Clark, C.M.; Liun, F.; Rokach, J.; Lee, V.Y.-M.; Trojanowski, J.Q. Increase of Brain Oxidative Stress in Mild Cognitive Impairment: A Possible Predictor of Alzheimer Disease. Arch. Neurol. 2002, 59, 972–976. [Google Scholar] [CrossRef] [Green Version]
- Engelhart, M.J.; Geerlings, M.I.; Ruitenberg, A.; Van Swieten, J.C.; Hofman, A.; Witteman, J.C.M.; Breteler, M.M.B. Dietary Intake of Antioxidants and Risk of Alzheimer Disease. JAMA 2002, 287, 3223–3229. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Kader, R.; Hauptmann, S.; Keil, U.; Scherping, I.; Leuner, K.; Eckert, A.; Müller, W.E. Stabilization of mitochondrial function by Ginkgo biloba extract (EGb 761). Pharmacol. Res. 2007, 56, 493–502. [Google Scholar] [CrossRef]
- Upadhyay, S.; Dixit, M. Role of Polyphenols and Other Phytochemicals on Molecular Signaling. Oxid. Med. Cell. Longev. 2015, 2015, 504253. [Google Scholar] [CrossRef]
- Kosuge, Y. Neuroprotective mechanisms of S-allyl-L-cysteine in neurological disease (Review). Exp. Ther. Med. 2020, 19, 1565–1569. [Google Scholar] [CrossRef]
- Kanninen, K.; Heikkinen, R.; Malm, T.; Rolova, T.; Kuhmonen, S.; Leinonen, H.; Ylä-Herttuala, S.; Tanila, H.; Levonen, A.-L.; Koistinaho, M.; et al. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 16505–16510. [Google Scholar] [CrossRef] [Green Version]
- Park, H.-M.; Kim, J.-A.; Kwak, M.-K. Protection against amyloid beta cytotoxicity by sulforaphane: Role of the proteasome. Arch. Pharm. Res. 2009, 32, 109–115. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, R.; Zhan, Z.; Li, X.; Zhou, F.; Xing, A.; Jiang, C.; Chen, Y.; An, L. Beneficial Effects of Sulforaphane Treatment in Alzheimer’s Disease May Be Mediated through Reduced HDAC1/3 and Increased P75NTR Expression. Front. Aging Neurosci. 2017, 9, 121. [Google Scholar] [CrossRef] [Green Version]
- Zheng, K.; Dai, X.; Xiao, N.; Wu, X.; Wei, Z.; Fang, W.; Zhu, Y.; Zhang, J.; Chen, X. Curcumin Ameliorates Memory Decline via Inhibiting BACE1 Expression and β-Amyloid Pathology in 5×FAD Transgenic Mice. Mol. Neurobiol. 2017, 54, 1967–1977. [Google Scholar] [CrossRef]
- Gibellini, L.; Bianchini, E.; De Biasi, S.; Nasi, M.; Cossarizza, A.; Pinti, M. Natural Compounds Modulating Mitochondrial Functions. Evid. Based Complement. Altern. Med. 2015, 2015, 527209. [Google Scholar] [CrossRef] [Green Version]
- Cole, G.M.; Yang, F.; Lim, G.P.; Cummings, J.L.; Masterman, D.L.; Frautschy, S.A. A Rationale for Curcuminoids for the Prevention or Treatment of Alzheimers Disease. Curr. Med. Chem. Immunol. Endocr. Metab. Agents 2003, 3, 15–25. [Google Scholar] [CrossRef]
- Alisi, I.O.; Uzairu, A.; Abechi, S.E.; Idris, S.O. Evaluation of the antioxidant properties of curcumin derivatives by genetic function algorithm. J. Adv. Res. 2018, 12, 47–54. [Google Scholar] [CrossRef]
- Hasan, W.; Kori, R.K.; Thakre, K.; Yadav, R.S.; Jat, D. Synthesis, characterization and efficacy of mitochondrial targeted delivery of TPP-curcumin in rotenone-induced toxicity. DARU J. Pharm. Sci. 2019, 27, 557–570. [Google Scholar] [CrossRef]
- Jayaraj, R.L.; Tamilselvam, K.; Manivasagam, T.; Elangovan, N. Neuroprotective Effect of CNB-001, a Novel Pyrazole Derivative of Curcumin on Biochemical and Apoptotic Markers Against Rotenone-Induced SK-N-SH Cellular Model of Parkinson’s Disease. J. Mol. Neurosci. 2013, 51, 863–870. [Google Scholar] [CrossRef]
- Bagheri, H.; Ghasemi, F.; Barreto, G.E.; Rafiee, R.; Sathyapalan, T.; Sahebkar, A. Effects of curcumin on mitochondria in neurodegenerative diseases. BioFactors 2020, 46, 5–20. [Google Scholar] [CrossRef]
- Albrecht, P.; Bouchachia, I.; Goebels, N.; Henke, N.; Hofstetter, H.H.; Issberner, A.; Kovacs, Z.; Lewerenz, J.; Lisak, D.; Maher, P.; et al. Effects of dimethyl fumarate on neuroprotection and immunomodulation. J. Neuroinflamm. 2012, 9, 163. [Google Scholar] [CrossRef] [Green Version]
- Bomprezzi, R. Dimethyl fumarate in the treatment of relapsing–remitting multiple sclerosis: An overview. Ther. Adv. Neurol. Disord. 2015, 8, 20–30. [Google Scholar] [CrossRef] [Green Version]
- Paraiso, H.C.; Kuo, P.-C.; Curfman, E.T.; Moon, H.J.; Sweazey, R.D.; Yen, J.-H.; Chang, F.-L.; Yu, I.-C. Dimethyl fumarate attenuates reactive microglia and long-term memory deficits following systemic immune challenge. J. Neuroinflamm. 2018, 15, 100. [Google Scholar] [CrossRef]
- Wang, Y.; Santa–Cruz, K.; DeCarli, C.; Johnson, J.A. NAD(P)H:quinone oxidoreductase activity is increased in hippocampal pyramidal neurons of patients with alzheimer’s disease. Neurobiol. Aging 2000, 21, 525–531. [Google Scholar] [CrossRef]
- Kume, T.; Suenaga, A.; Izumi, Y.; Akaike, A. Protective Effect of Dimethyl Fumarate on an Oxidative Stress Model Induced by Sodium Nitroprusside in Mice. Biol. Pharm. Bull. 2016, 39, 1055–1059. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, G.; Jasoliya, M.; Sahdeo, S.; Saccà, F.; Pane, C.; Filla, A.; Marsili, A.; Puorro, G.; Lanzillo, R.; Brescia Morra, V.; et al. Dimethyl fumarate mediates Nrf2-dependent mitochondrial biogenesis in mice and humans. Hum. Mol. Genet. 2017, 26, 2864–2873. [Google Scholar] [CrossRef]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.-J.; Tanaka, T.; Tung, Y.-C.; Braak, E.; Iqbal, K.; Grundke-Iqbal, I. Distribution, Levels, and Activity of Glycogen Synthase Kinase-3 in the Alzheimer Disease Brain. J. Neuropathol. Exp. Neurol. 1997, 56, 70–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hye, A.; Kerr, F.; Archer, N.; Foy, C.; Poppe, M.; Brown, R.; Hamilton, G.; Powell, J.; Anderton, B.; Lovestone, S. Glycogen synthase kinase-3 is increased in white cells early in Alzheimer’s disease. Neurosci. Lett. 2004, 373, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, I.; Michalska, P.; Tenti, G.; Cores, Á.; Buendia, I.; Rojo, A.I.; Georgakopoulos, N.D.; Hernández-Guijo, J.M.; Ramos, M.T.; Wells, G.; et al. Discovery of the first dual GSK3β inhibitor/Nrf2 inducer. A new multitarget therapeutic strategy for Alzheimer’s disease. Sci. Rep. 2017, 7, 45701. [Google Scholar] [CrossRef] [PubMed]
- Farr, S.A.; Ripley, J.L.; Sultana, R.; Zhang, Z.; Niehoff, M.L.; Platt, T.L.; Murphy, M.P.; Morley, J.E.; Kumar, V.; Butterfield, D.A. Antisense oligonucleotide against GSK-3β in brain of SAMP8 mice improves learning and memory and decreases oxidative stress: Involvement of transcription factor Nrf2 and implications for Alzheimer disease. Free Radic. Biol. Med. 2014, 67, 387–395. [Google Scholar] [CrossRef] [Green Version]
- Malm, T.M.; Iivonen, H.; Goldsteins, G.; Keksa-Goldsteine, V.; Ahtoniemi, T.; Kanninen, K.; Salminen, A.; Auriola, S.; Van Groen, T.; Tanila, H.; et al. Pyrrolidine Dithiocarbamate Activates Akt and Improves Spatial Learning in APP/PS1 Mice without Affecting β-Amyloid Burden. J. Neurosci. 2007, 27, 3712–3721. [Google Scholar] [CrossRef] [Green Version]
- Kang, K.W.; Lee, S.J.; Park, J.W.; Kim, S.G. Phosphatidylinositol 3-Kinase Regulates Nuclear Translocation of NF-E2-Related Factor 2 through Actin Rearrangement in Response to Oxidative Stress. Mol. Pharmacol. 2002, 62, 1001–1010. [Google Scholar] [CrossRef]
- Mercado-Gómez, O.; Hernández-Fonseca, K.; Villavicencio-Queijeiro, A.; Massieu, L.; Chimal-Monroy, J.; Arias, C. Inhibition of Wnt and PI3K Signaling Modulates GSK-3β Activity and Induces Morphological Changes in Cortical Neurons: Role of Tau Phosphorylation. Neurochem. Res. 2008, 33, 1599–1609. [Google Scholar] [CrossRef]
- Zhu, G.; Wang, X.; Wu, S.; Li, X.; Li, Q. Neuroprotective Effects of Puerarin on 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine induced Parkinson’s Disease Model in Mice. Phytotherapy Res. 2014, 28, 179–186. [Google Scholar] [CrossRef]
- Zhou, Y.; Xie, N.; Li, L.; Zou, Y.; Zhang, X.; Dong, M. Puerarin alleviates cognitive impairment and oxidative stress in APP/PS1 transgenic mice. Int. J. Neuropsychopharmacol. 2014, 17, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Abeti, R.; Baccaro, A.; Esteras, N.; Giunti, P. Novel Nrf2-Inducer Prevents Mitochondrial Defects and Oxidative Stress in Friedreich’s Ataxia Models. Front. Cell. Neurosci. 2018, 12, 188. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villavicencio Tejo, F.; Quintanilla, R.A. Contribution of the Nrf2 Pathway on Oxidative Damage and Mitochondrial Failure in Parkinson and Alzheimer’s Disease. Antioxidants 2021, 10, 1069. https://doi.org/10.3390/antiox10071069
Villavicencio Tejo F, Quintanilla RA. Contribution of the Nrf2 Pathway on Oxidative Damage and Mitochondrial Failure in Parkinson and Alzheimer’s Disease. Antioxidants. 2021; 10(7):1069. https://doi.org/10.3390/antiox10071069
Chicago/Turabian StyleVillavicencio Tejo, Francisca, and Rodrigo A Quintanilla. 2021. "Contribution of the Nrf2 Pathway on Oxidative Damage and Mitochondrial Failure in Parkinson and Alzheimer’s Disease" Antioxidants 10, no. 7: 1069. https://doi.org/10.3390/antiox10071069