
Reactive Oxygen Species and Pressure Ulcer Formation after Traumatic Injury to Spinal Cord and Brain


Abstract
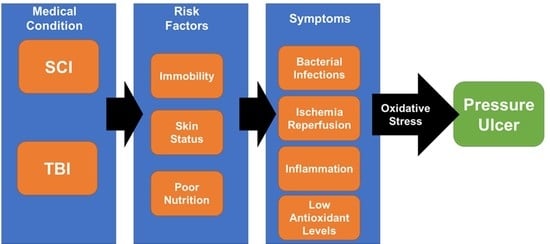
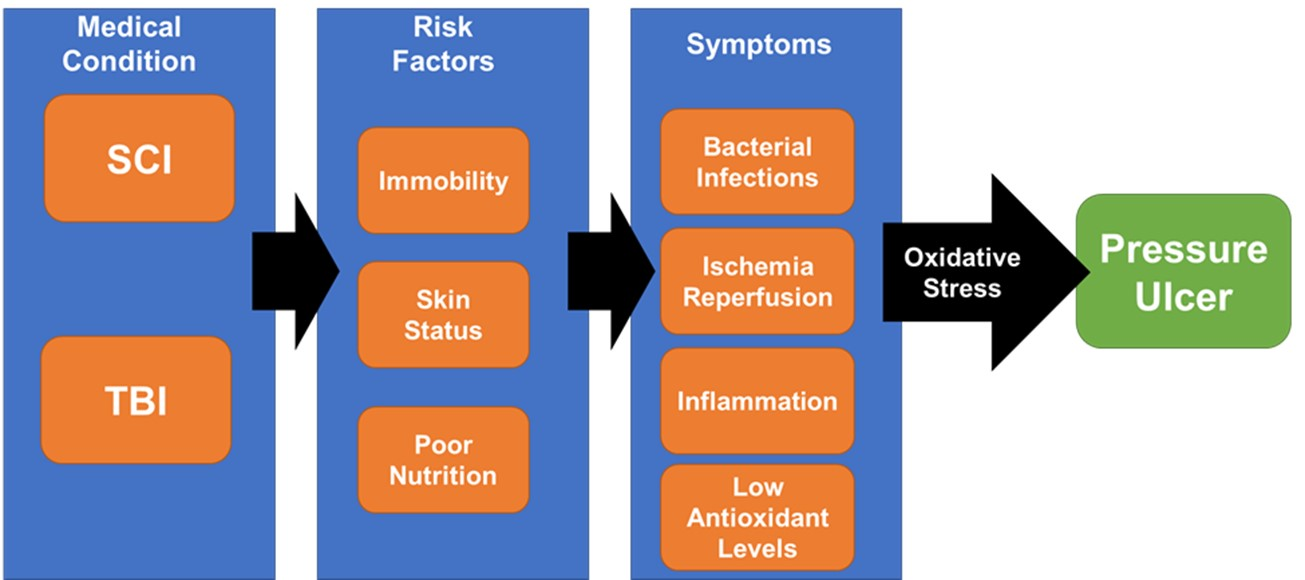
:
1. Introduction
2. Spinal Cord Injury and Pressure Ulcers
2.1. Underlying Mechanisms in the Development of Pressure Ulcers
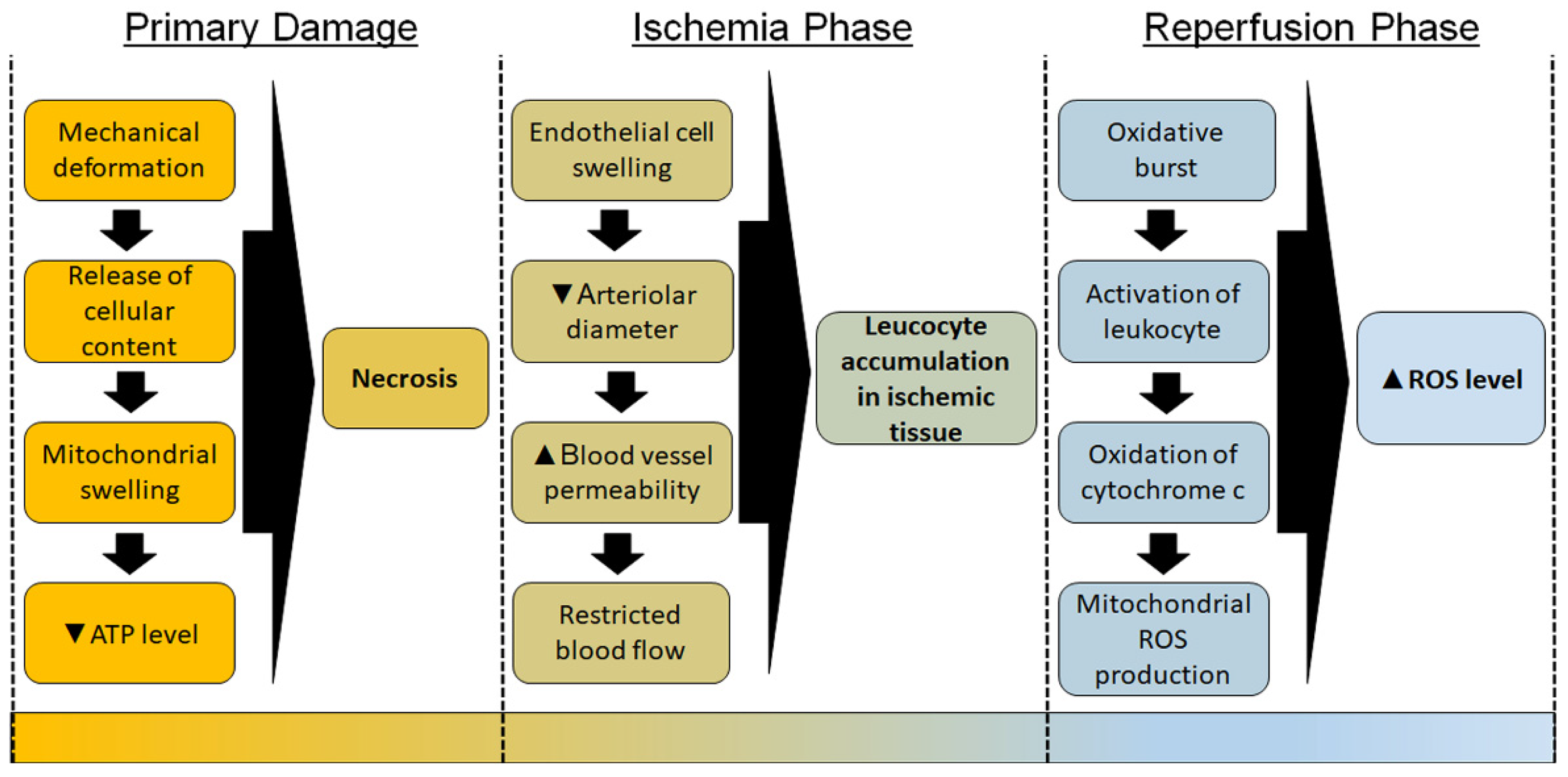
2.2. Role of Oxidative Stress in the Development of Pressure Ulcers
2.3. Impaired Healing of Pressure Ulcers
3. Brain Injury and Pressure Ulcers
4. Overview of Clinical Trials

{kind=link}
{kind=link}
| NCT # (Start–End) | Study Title (Phase) | # Enrolled/Completed (M/F) | Recruitment Status | Condition (SCI/TBI) | Treatment | Dose Frequency/Repetition/Duration | Country Co-Sponsor | Results | References |
|---|---|---|---|---|---|---|---|---|---|
| NCT01999816 (2008–2015) | Pressure ulcer prevention study in SCI (Phase 3) | 170/NP | Completed | SCI | Pressure Ulcer Prevention Program (PUPP) | One-time home intervention | USA University of Southern California | Rates of medically serious pressure injuries were not significantly different across treatment groups. | [112,113,114,115,116,117] |
| NCT01500174 (2007–2011) | Ultraviolet-C effectiveness in the management of pressure ulcers in people with spinal cord injury (NA) | 43/NP | Completed | SCI | Ultraviolet-C therapy |
UV-C-radiation 3-time/week until
| Canada Toronto Rehabilitation Institute | In stage 2 buttock ulcers, UVC significantly reduces the % area relative to baseline, but not in stage 3 or 4 ulcers. | [118,119] |
| NCT01572376 (2007–2010) | Autologous bone marrow stem cells in pressure ulcer treatment (Phases 1 and 2) | 30/22 (19/3) | Completed | SCI | Ulcers treated with bone marrow mononuclear cells (BMSCs) | One-time procedure | Spain Hospital Universitario Central de Asturias | Patients with (BMSC) treatment reduced 50% stay time in the hospital and also reduced the 75% of daily care requirement in comparison to the patients with conventional surgery. | [108] |
| NCT00763282 (2008–2014) | Self-management to prevent ulcers in veterans with SCI (spinal cord injury) (NA) | 143 (129/4) /78 | Completed | SCI | Telephone-based individual MI counseling and SM skills group | 8 coordinator-initiated calls over | USA US-DVA | No significant increases in skin behaviors between motivational interviewing (MI)/self-management (SM) and control group. High rates of skin worsening (51.7%) were observed in both groups. | [120] |
| NCT00624806 (2008–2015) | Developing a home telehealth program to manage pressure ulcers in SCI/D (NA) | 18 (M)/18 (M) | Completed | SCI | Daily or weekly (depending on treatment group) telephone calls to remind patients how they should prevent ulcers | Daily (56 calls) Weekly (8 calls) | USA US-DVA | The daily group had slightly more days of data than the weekly group. Both groups showed a similar number of days with triggers. More participants in the weekly call group experienced equipment issues, skin moisture issues, existing PU care, depression, and ongoing problems affecting self-management than in the daily call group. | [121] |
| NCT00101361 (2005–2013) | Oxandrolone to heal pressure ulcers (Phase 3) | 212 (201/2) /212 (201/2) | Terminated | SCI | Oxandrolone | 25 mg/day for 24-week | USA US-DVA | Oxandrolone had no significant benefit over placebo for healing. | [122] |
| NCT00047619 (2001–2008) | Enhancement of pressure healing with pulsatile lavage (Phase 2) | 28 (M)/28 (M) | Completed | SCI | Pulsatile lavage treatment | Once-daily for 3 weeks | USA US-DVA | A trend of improvement of pulsatile lavage therapy in terms of reducing ulcer length and depth in comparison to the Control group. | [123] |
| NCT00105859 (2005–2010) | Preventing pressure ulcers in veterans with spinal cord injury (SCI) (Phase—NA) | 278/NP | Terminated (recruitment issues) | SCI | Cognitive behavioral intervention | NP | USA US-DVA | NP | [124,125,126] |
| NCT02800915 (2017–2018) | Telemedicine makes the patient stay in hospital at home (NA) | 56/NP | Completed | SCI | Interdisciplinary outpatient follow-up via telemedicine | For 12-month or until the pressure ulcer heals | Norway Sunnaas Rehabilitation Hospital | NP | [127] |
| NCT # (Start–End) | Study Title (Phase) | # Enrolled/Completed (M/F) | Recruitment Status | Condition (SCI/TBI) | Treatment | Dose Frequency/repetition/duration | Country Co-Sponsor | Results | References |
|---|---|---|---|---|---|---|---|---|---|
| NCT01433159 (2011–2014) | Comparison of HP011-101 to standard care for stage I–II pressure ulcers in subjects with spinal cord injury (Phase 2) | 19 (M)/16 (M) | Terminated (business decision) | SCI | HP011-101 (Xenaderm Ointment) | Topical ointment applied daily (twice for 14 days) | USA Healthpoint | HP011-101 (Xenaderm Ointment) and Standard Care both showed an improved change in scores of the pressure ulcer scale for healing (PUSH) | NP |
| NCT02001558 (2013–2017) | Pressure ulcer healing with Microcyn (Phase 4) | 65 (55/10)/43 | Completed | SCI | Microcyn | Topical Microcyn sprayed on wound daily (twice for 24 weeks) | USA University of Alabama | Both microcyn and sterile saline (control) reduced ulcer size by more than half of baseline. The PUSH score improved slightly in both treatment groups. | NP |
| NCT01885962 (2012–2013) | Development and feasibility of an internet intervention for adults with spinal cord injury to prevent pressure ulcers (Phase—NA) | 19/NP | Completed | SCI | iSHIFTup: Internet skin health intervention for targeted ulcer prevention | NA | USA University of Virginia | NP | NP |
| NCT03317288 (2017–2019) | Alternating pressure overlay on weight-bearing tissue tolerance in people with spinal cord injury (Phase—NA) | 15/NP | Completed | SCI | Device: Dabir Air overlay | NP | USA University of Illinois | NP | NP |
| NCT02584426 (2017–2018) | Subcutaneous injection and ultrasonic dispersion of Cefazolin into chronic pelvic region pressure ulcers in persons with spinal cord injury (Phase—NA) | 20/NP | Unknown | SCI | Phonophoresis via ultrasonic distribution of Cefazolin | 1 Hypodermic antibiotic injection and Phonophoresis | USA James J. Peters Veterans Affairs Medical Center | NP | NP |
| NCT03220451 (2017–2020) | Use of adhesive elastic taping for the therapy of medium/severe pressure ulcers in spinal cord injured patients (Phase—NA) | 24/NP | Recruiting | SCI | Five-layer foam dressing on sacrum and installation of Heelmedix boot alternately from one foot to the other within 48 h after spinal surgery | NP | Canada Centre Integre Universitaire de Sante et Services Sociaux du Nord de l’ile de Montreal | NP | NP |
| NCT02894437 (2016–2018) | A qualitative study of the preventive organization of the pelvic bedsores injured spinal cord (QUALIPREPS) (NA) | 45/NP | Unknown | SCI | Conceptual framework for work established to prevent pelvic pressure ulcers | NP | France Nantes University Hospital | NP | NP |
| NCT03469141 (2018–2021) | Interactive telehealth for pressure ulcer prevention after SCI (NA) | 100/NP | Recruiting | SCI | Biofeedback via smartphone app | Run biofeedback scan daily for 4 weeks | USA Rancho Research Institute, Inc | NP | NP |
| NCT01943201 (2010–2012) | Low friction bedsheet (NA) | 20/NP | Completed | SCI | Low-friction bed sheet | Daily for 5 nights | Switzerland Swiss Paraplegic Centre Nottwil | NP | NP |
| NCT03048357 (2016–2017) | Effectiveness of freedom bed compared to manual turning in the prevention of pressure injuries in persons with limited mobility due to traumatic brain injury and/or spinal cord injury (NA) | 8/NP | Unknown | TBI and/or SCI | Freedom bed | Daily for 6 months | USA Northeast Center for Rehabilitation and Brain Injury | NP | NP |
| NCT04402398 (2019–2020) | Psychometric properties of a mobile application (NA) | 59/NP | Completed | SCI | imitoMeasure (smartphone app that measures wound size) | NP | France University Hospital, Montpellier | NP | NP |
| NCT04266808 (2020–2022) | Interactive telehealth for wheelchair users (NA) | 50/NP | Not yet recruiting | SCI | Interactive telehealth monitoring and biofeedback system on phone application and wheelchair | Used for one year | USA Rancho Research Institute, Inc. | NP | NP |
| NCT02876666 (2017–2017) | Spinal cord injury virtual coach RCT (NA) | 40/NP | Completed | SCI | SCI virtual coach interaction | Once-daily for 2 months | USA Boston University | NP | NP |
| NCT01834417 (2013–2020) | Preliminary study leading to prevention of pressure ulcers by the use of an on-board device: Ergonomic assessment of wheelchair-seat pressures in spinal cord injured (SCI) patients (PRESDIE) (NA) | 90/NP | Completed | SCI | On-board device (on wheelchair): TexiMat | Use for 4 weeks | France Nantes University Hospital | NP | NP |
| NCT02412046 (2015–2018) | Quantification of the pressure threshold related to tissue injury in bed-ridden paraplegics (NA) | 21/NP | Terminated (Departure of the Ph.D. in charge of the study) | SCI | Muscle biopsy after lying on air mattress | One-time procedure | France University Hospital, Montpellier | NP | NP |
| NCT03114345 (2017–2020) | Correlation between pressure differences and micro-vascularization changes in bedridden paraplegic patient (NA) | 4/NP | Terminated (Departure of the Ph.D. in charge of the study) | SCI | XSensor | XSensor by bed for 1 h and O2C applied for 1 min; one-time procedure | France University Hospital, Montpellier | NP | NP |
| NCT04309864 (2020–2024) | CMAP refinement for pressure injury prevention (NA) | 46/NP | Recruiting | SCI | Comprehensive Mobile Assessment of Pressure (CMAP mobile app) | In-hospital: Use during initial rehabilitation In-home: Use for 2 weeks during daily routine | USA VA Office of Research and Development | NP | NP |
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kruger, E.A.; Pires, M.; Ngann, Y.; Sterling, M.; Rubayi, S. Comprehensive Management of Pressure Ulcers in Spinal Cord Injury: Current Concepts and Future Trends. J. Spinal Cord Med. 2013, 36, 572–585. [Google Scholar] [CrossRef] [Green Version]
- Brem, H.; Maggi, J.; Nierman, D.; Rolnitzky, L.; Bell, D.; Rennert, R.; Golinko, M.; Yan, A.; Lyder, C.; Vladeck, B. High Cost of Stage IV Pressure Ulcers. Am. J. Surg. 2010, 200, 473–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grigorian, A.; Sugimoto, M.; Joe, V.; Schubl, S.; Lekawa, M.; Dolich, M.; Kuncir, E.; Barrios, C.; Nahmias, J. Pressure Ulcer in Trauma Patients: A Higher Spinal Cord Injury Level Leads to Higher Risk. J. Am. Coll. Clin. Wound Spec. 2018, 9, 24–31.e1. [Google Scholar] [CrossRef]
- Hughes, R.G. (Ed.) Patient Safety and Quality: An Evidence-Based Handbook for Nurses; Advances in Patient Safety; Agency for Healthcare Research and Quality: Rockville, MD, USA, 2008.
- Baumgarten, M.; Margolis, D.; Doorn, C.V.; Gruber-Baldini, A.L.; Hebel, J.R.; Zimmerman, S.; Magaziner, J. Black/White Differences in Pressure Ulcer Incidence in Nursing Home Residents. J. Am. Geriatr. Soc. 2004, 52, 1293–1298. [Google Scholar] [CrossRef]
- Ham, H.W.; Schoonhoven, L.; Schuurmans, M.J.; Leenen, L.P.H. Pressure Ulcer Development in Trauma Patients with Suspected Spinal Injury; the Influence of Risk Factors Present in the Emergency Department. Int. Emerg. Nurs. 2017, 30, 13–19. [Google Scholar] [CrossRef]
- Garber, S.L.; Rintala, D.H.; Hart, K.A.; Fuhrer, M.J. Pressure Ulcer Risk in Spinal Cord Injury: Predictors of Ulcer Status over 3 Years. Arch. Phys. Med. Rehabil. 2000, 81, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Osis, S.L.; Diccini, S. Incidence and Risk Factors Associated with Pressure Injury in Patients with Traumatic Brain Injury. Int. J. Nurs. Pract. 2020, 26, e12821. [Google Scholar] [CrossRef] [PubMed]
- Teasdale, G.; Jennett, B. Assessment of Coma and Impaired Consciousness. A Practical Scale. Lancet 1974, 2, 81–84. [Google Scholar] [CrossRef]
- Dhandapani, M.; Dhandapani, S.; Agarwal, M.; Mahapatra, A.K. Pressure Ulcer in Patients with Severe Traumatic Brain Injury: Significant Factors and Association with Neurological Outcome. J. Clin. Nurs. 2014, 23, 1114–1119. [Google Scholar] [CrossRef]
- Paker, N.; Soy, D.; Kesiktaş, N.; Nur Bardak, A.; Erbil, M.; Ersoy, S.; Ylmaz, H. Reasons for Rehospitalization in Patients with Spinal Cord Injury: 5 Years’ Experience. Int. J. Rehabil. Res. 2006, 29, 71–76. [Google Scholar] [CrossRef]
- Kottner, J.; Cuddigan, J.; Carville, K.; Balzer, K.; Berlowitz, D.; Law, S.; Litchford, M.; Mitchell, P.; Moore, Z.; Pittman, J.; et al. Prevention and Treatment of Pressure Ulcers/Injuries: The Protocol for the Second Update of the International Clinical Practice Guideline 2019. J. Tissue Viability 2019, 28, 51–58. [Google Scholar] [CrossRef]
- Coleman, S.; Nixon, J.; Keen, J.; Wilson, L.; McGinnis, E.; Dealey, C.; Stubbs, N.; Farrin, A.; Dowding, D.; Schols, J.M.; et al. A New Pressure Ulcer Conceptual Framework. J. Adv. Nurs. 2014, 70, 2222–2234. [Google Scholar] [CrossRef]
- Bouten, C.V.; Oomens, C.W.; Baaijens, F.P.; Bader, D.L. The Etiology of Pressure Ulcers: Skin Deep or Muscle Bound? Arch. Phys. Med. Rehabil. 2003, 84, 616–619. [Google Scholar] [CrossRef]
- Gawlitta, D.; Oomens, C.W.J.; Bader, D.L.; Baaijens, F.P.T.; Bouten, C.V.C. Temporal Differences in the Influence of Ischemic Factors and Deformation on the Metabolism of Engineered Skeletal Muscle. J. Appl. Physiol. 2007, 103, 464–473. [Google Scholar] [CrossRef] [Green Version]
- Gefen, A.; van Nierop, B.; Bader, D.L.; Oomens, C.W. Strain-Time Cell-Death Threshold for Skeletal Muscle in a Tissue-Engineered Model System for Deep Tissue Injury. J. Biomech. 2008, 41, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Stekelenburg, A.; Strijkers, G.J.; Parusel, H.; Bader, D.L.; Nicolay, K.; Oomens, C.W. Role of Ischemia and Deformation in the Onset of Compression-Induced Deep Tissue Injury: MRI-Based Studies in a Rat Model. J. Appl. Physiol. 2007, 102, 2002–2011. [Google Scholar] [CrossRef] [Green Version]
- Bader, D.L.; Barnhill, R.L.; Ryan, T.J. Effect of Externally Applied Skin Surface Forces on Tissue Vasculature. Arch. Phys. Med. Rehabil. 1986, 67, 807–811. [Google Scholar] [PubMed]
- Dinsdale, S.M. Decubitus Ulcers: Role of Pressure and Friction in Causation. Arch. Phys. Med. Rehabil. 1974, 55, 147–152. [Google Scholar] [PubMed]
- Kosiak, M. Etiology of Decubitus Ulcers. Arch. Phys. Med. Rehabil. 1961, 42, 19–29. [Google Scholar]
- Peirce, S.M.; Skalak, T.C.; Rodeheaver, G.T. Ischemia-Reperfusion Injury in Chronic Pressure Ulcer Formation: A Skin Model in the Rat. Wound Repair Regen. 2000, 8, 68–76. [Google Scholar] [CrossRef]
- Tsuji, S.; Ichioka, S.; Sekiya, N.; Nakatsuka, T. Analysis of Ischemia-Reperfusion Injury in a Microcirculatory Model of Pressure Ulcers. Wound Repair Regen. 2005, 13, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.E.; Seale, J. Lymphatic Clearance during Compressive Loading. Lymphology 1981, 14, 161–166. [Google Scholar]
- Reddy, N.P.; Cochran, G.V.B.; Krouskop, T.A. Interstitial Fluid Flow as a Factor in Decubitus Ulcer Formation. J. Biomech. 1981, 14, 879–881. [Google Scholar] [CrossRef]
- Barton, A.A.; Barton, M. The Medical Management of Pressure Sores. Queens Nurs. J. 1973, 16, 148–149. [Google Scholar]
- Gutteridge, J.M.C.; Halliwell, B. Mini-Review: Oxidative Stress, Redox Stress or Redox Success? Biochem. Biophys. Res. Commun. 2018, 502, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. The Chemistry of Free Radicals. Toxicol. Ind. Health 1993, 9, 1–21. [Google Scholar] [CrossRef]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Halliwell, B. Oxidants and Human Disease: Some New Concepts. FASEB J. 1987, 1, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Wood, J.D.; Coffey, M.J.; Jones, O.T.G. The Functional Expression of P47-Phox and P67-Phox May Contribute to the Generation of Superoxide by an NADPH Oxidase-like System in Human Fibroblasts. FEBS Lett. 1994, 355, 178–182. [Google Scholar] [CrossRef] [Green Version]
- Lo, Y.Y.C.; Cruz, T.F. Involvement of Reactive Oxygen Species in Cytokine and Growth Factor Induction of C-Fos Expression in Chondrocytes. J. Biol. Chem. 1995, 270, 11727–11730. [Google Scholar] [CrossRef] [Green Version]
- Meier, B.; Radeke, H.H.; Selle, S.; Younes, M.; Sies, H.; Resch, K.; Habermehl, G.G. Human Fibroblasts Release Reactive Oxygen Species in Response to Interleukin-1 or Tumour Necrosis Factor-Alpha. Biochem. J. 1989, 263, 539–545. [Google Scholar] [CrossRef]
- Suzuki, Y.J.; Ford, G.D. Redox Regulation of Signal Transduction in Cardiac and Smooth Muscle. J. Mol. Cell. Cardiol. 1999, 31, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Zweier, J.L.; Broderick, R.; Kuppusamy, P.; Thompson-Gorman, S.; Lutty, G.A. Determination of the Mechanism of Free Radical Generation in Human Aortic Endothelial Cells Exposed to Anoxia and Reoxygenation. J. Biol. Chem. 1994, 269, 24156–24162. [Google Scholar] [CrossRef]
- Babior, B.M. Phagocytes and Oxidative Stress. Am. J. Med. 2000, 109, 33–44. [Google Scholar] [CrossRef]
- McCord, J.M. Oxygen-Derived Free Radicals in Postischemic Tissue Injury. N. Engl. J. Med. 1985, 312, 159–163. [Google Scholar] [CrossRef] [PubMed]
- D’Autréaux, B.; Toledano, M.B. ROS as Signalling Molecules: Mechanisms That Generate Specificity in ROS Homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Kehrer, J.P. The Haber–Weiss Reaction and Mechanisms of Toxicity. Toxicology 2000, 149, 43–50. [Google Scholar] [CrossRef]
- Chevion, M. A Site-Specific Mechanism for Free Radical Induced Biological Damage: The Essential Role of Redox-Active Transition Metals. Free Radic. Biol. Med. 1988, 5, 27–37. [Google Scholar] [CrossRef]
- Thornberry, N.A. Caspases: Enemies Within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef]
- Halliwell, B.; Dizdaroglu, M. The Measurement of Oxidative Damage to DNA by HPLC and GC/MS Techniques. Free Radic. Res. Commun. 1992, 16, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.; Leeuwenburgh, C. Aging and the Role of Reactive Nitrogen Species. Ann. N. Y. Acad. Sci. 2002, 959, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.; Singel, D.; Loscalzo, J. Biochemistry of Nitric Oxide and Its Redox-Activated Forms. Science 1992, 258, 1898–1902. [Google Scholar] [CrossRef] [PubMed]
- Snyder, S.H. No Endothelial NO. Nature 1995, 377, 196–197. [Google Scholar] [CrossRef]
- Wink, D.A.; Miranda, K.M.; Espey, M.G.; Pluta, R.M.; Hewett, S.J.; Colton, C.; Vitek, M.; Feelisch, M.; Grisham, M.B. Mechanisms of the Antioxidant Effects of Nitric Oxide. Antioxid. Redox Signal. 2001, 3, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Hille, R.; Hall, J.; Basu, P. The Mononuclear Molybdenum Enzymes. Chem. Rev. 2014, 114, 3963–4038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide Metabolism in Mammalian Organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef]
- Granger, D.N. Role of Xanthine Oxidase and Granulocytes in Ischemia-Reperfusion Injury. Am. J. Physiol. Heart Circ. Physiol. 1988, 255, H1269–H1275. [Google Scholar] [CrossRef]
- Rees, R.; Smith, D.; Li, T.D.; Cashmer, B.; Garner, W.; Punch, J.; Smith, D.J. The Role of Xanthine Oxidase and Xanthine Dehydrogenase in Skin Ischemia. J. Surg. Res. 1994, 56, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Shindo, Y.; Witt, E.; Han, D.; Epstein, W.; Packer, L. Enzymic and Non-Enzymic Antioxidants in Epidermis and Dermis of Human Skin. J. Investig. Dermatol. 1994, 102, 122–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreher, F.; Maibach, H. Protective Effects of Topical Antioxidants in Humans. In Oxidants and Antioxidants in Cutaneous Biology; Karger: Basel, Switzerland, 2001; Volume 29, pp. 157–164. [Google Scholar] [CrossRef]
- Nabi, Z.; Tavakkol, A.; Dobke, M.; Polefka, T.G. Bioconversion of Vitamin E Acetate in Human Skin. In Oxidants and Antioxidants in Cutaneous Biology; Karger: Basel, Switzerland, 2001; Volume 29, pp. 175–186. [Google Scholar] [CrossRef]
- Haslett, C. Resolution of Acute Inflammation and the Role of Apoptosis in the Tissue Fate of Granulocytes. Clin. Sci. 1992, 83, 639–648. [Google Scholar] [CrossRef] [Green Version]
- Hourmant, M.; Vasse, N.; le Mauff, B.; Soulillou, J.P. The Role of Adhesion Molecules in Ischaemia-Reperfusion Injury of Renal Transplants. Nephrol. Dial. Transplant. 1997, 12, 2485–2487. [Google Scholar] [CrossRef] [PubMed]
- Perry, M.A.; Granger, D.N. Role of CD11/CD18 in Shear Rate-Dependent Leukocyte-Endothelial Cell Interactions in Cat Mesenteric Venules. J. Clin. Investig. 1991, 87, 1798–1804. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Asako, H.; Kubes, P.; Jennings, S.; Grisham, M.B.; Granger, D.N. Neutrophil-Derived Oxidants Promote Leukocyte Adherence in Postcapillary Venules. Microvasc. Res. 1991, 42, 125–138. [Google Scholar] [CrossRef]
- Salvemini, D.; Cuzzocrea, S. Superoxide, Superoxide Dismutase and Ischemic Injury. Curr. Opin. Investig. Drugs 2002, 3, 886–895. [Google Scholar]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Goode, H.F.; Webster, N.R.; Howdle, P.D.; Walker, B.E. Nitric Oxide Production by Human Peripheral Blood Polymorphonuclear Leucocytes. Clin. Sci. 1994, 86, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Su, Z. Peroxynitrite Is Not a Major Mediator of Endothelial Cell Injury by Activated Neutrophils in Vitro. Cardiovasc. Res. 1998, 39, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Lo, S.K.; Janakidevi, K.; Lai, L.; Malik, A.B. Hydrogen Peroxide-Induced Increase in Endothelial Adhesiveness Is Dependent on ICAM-1 Activation. Am. J. Physiol. Lung Cell. Mol. Physiol. 1993, 264, L406–L412. [Google Scholar] [CrossRef]
- Patel, K.D.; Zimmerman, G.A.; Prescott, S.M.; McEver, R.P.; McIntyre, T.M. Oxygen Radicals Induce Human Endothelial Cells to Express GMP-140 and Bind Neutrophils. J. Cell Biol. 1991, 112, 749–759. [Google Scholar] [CrossRef] [Green Version]
- Bogdan, C. Nitric Oxide and the Immune Response. Nat. Immunol. 2001, 2, 907–916. [Google Scholar] [CrossRef]
- Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Inside the Neutrophil Phagosome: Oxidants, Myeloperoxidase, and Bacterial Killing. Blood 1998, 92, 3007–3017. [Google Scholar] [CrossRef] [PubMed]
- Shingu, M.; Nobunaga, M. Chemotactic Activity Generated in Human Serum from the Fifth Component of Complement by Hydrogen Peroxide. Am. J. Pathol. 1984, 117, 201–206. [Google Scholar]
- Sisley, A.C.; Desai, T.; Harig, J.M.; Gewertz, B.L. Neutrophil Depletion Attenuates Human Intestinal Reperfusion Injury. J. Surg. Res. 1994, 57, 192–196. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A. The Role of Antioxidants in the Chemistry of Oxidative Stress: A Review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Marzella, L.; Jesudass, R.R.; Manson, P.N.; Myers, R.A.M.; Bulkley, G.B. Functional and Structural Evaluation of the Vasculature of Skin Flaps after Ischemia and Reperfusion. Plast. Reconstr. Surg. 1988, 81, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N.; Kvietys, P.R. Reperfusion Injury and Reactive Oxygen Species: The Evolution of a Concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strålin, P.; Marklund, S.L. Effects of Oxidative Stress on Expression of Extracellular Superoxide Dismutase, CuZn-Superoxide Dismutase and Mn-Superoxide Dismutase in Human Dermal Fibroblasts. Biochem. J. 1994, 298, 347–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepasi Tehrani, H.; Moosavi-Movahedi, A.A. Catalase and Its Mysteries. Prog. Biophys. Mol. Biol. 2018, 140, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.M.; Kugelman, A.; Iwamoto, T.; Tian, L.; Forman, H.J. Quinone-Induced Oxidative Stress Elevates Glutathione and Induces Gamma-Glutamylcysteine Synthetase Activity in Rat Lung Epithelial L2 Cells. J. Biol. Chem. 1994, 269, 26512–26517. [Google Scholar] [CrossRef]
- Fridovich, I.; Freeman, B. Antioxidant Defenses in the Lung. Annu. Rev. Physiol. 1986, 48, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Speer, R.E.; Karuppagounder, S.S.; Basso, M.; Sleiman, S.F.; Kumar, A.; Brand, D.; Smirnova, N.; Gazaryan, I.; Khim, S.J.; Ratan, R.R. Hypoxia-Inducible Factor Prolyl Hydroxylases as Targets for Neuroprotection by “Antioxidant” Metal Chelators: From Ferroptosis to Stroke. Free Radic. Biol. Med. 2013, 62, 26–36. [Google Scholar] [CrossRef]
- Karver, C.L.; Wade, S.L.; Cassedy, A.; Taylor, H.G.; Stancin, T.; Yeates, K.O.; Walz, N.C. Age at Injury and Long-Term Behavior Problems After Traumatic Brain Injury in Young Children. Rehabil. Psychol. 2012, 57, 256–265. [Google Scholar] [CrossRef]
- Sies, H. Oxidative Stress: A Concept in Redox Biology and Medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marczin, N.; El-Habashi, N.; Hoare, G.S.; Bundy, R.E.; Yacoub, M. Antioxidants in Myocardial Ischemia–Reperfusion Injury: Therapeutic Potential and Basic Mechanisms. Arch. Biochem. Biophys. 2003, 420, 222–236. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.M. Caspases: The Executioners of Apoptosis. Biochem. J. 1997, 326, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasahara, Y.; Iwai, K.; Yachie, A.; Ohta, K.; Konno, A.; Seki, H.; Miyawaki, T.; Taniguchi, N. Involvement of Reactive Oxygen Intermediates in Spontaneous and CD95(Fas/APO-1)—Mediated Apoptosis of Neutrophils. Blood J. Am. Soc. Hematol. 1997, 89, 1748–1753. [Google Scholar]
- Lundqvist-Gustafsson, H.; Bengtsson, T. Activation of the Granule Pool of the NADPH Oxidase Accelerates Apoptosis in Human Neutrophils. J. Leukoc. Biol. 1999, 65, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.F.; Donovan, M.; Cotter, T.G. Regulation and Measurement of Oxidative Stress in Apoptosis. J. Immunol. Methods 2002, 265, 49–72. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, J.T.; Ross, D.; Cohen, G.M. A Role for Metals and Free Radicals in the Induction of Apoptosis in Thymocytes. FEBS Lett. 1994, 352, 58–62. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, R.S.; Cotter, T.G. Apoptosis or Necrosis: Intracellular Levels of Glutathione Influence Mode of Cell Death. Biochem. Pharmacol. 1994, 48, 675–681. [Google Scholar] [CrossRef]
- Oda, T.; Sadakata, N.; Komatsu, N.; Muramatsu, T. Specific Efflux of Glutathione from the Basolateral Membrane Domain in Polarized MDCK Cells during Ricin-Induced Apoptosis. J. Biochem. 1999, 126, 7. [Google Scholar] [CrossRef]
- Creagh, E.M.; Carmody, R.J.; Cotter, T.G. Heat Shock Protein 70 Inhibits Caspase-Dependent and -Independent Apoptosis in Jurkat T Cells. Exp. Cell Res. 2000, 257, 58–66. [Google Scholar] [CrossRef]
- Hampton, M.B.; Orrenius, S. Dual Regulation of Caspase Activity by Hydrogen Peroxide: Implications for Apoptosis. FEBS Lett. 1997, 414, 552–556. [Google Scholar] [CrossRef] [Green Version]
- Leist, M.; Single, B.; Naumann, H.; Fava, E.; Simon, B.; Kühnle, S.; Nicotera, P. Inhibition of Mitochondrial ATP Generation by Nitric Oxide Switches Apoptosis to Necrosis. Exp. Cell Res. 1999, 249, 396–403. [Google Scholar] [CrossRef]
- Samali, A.; Nordgren, H.; Zhivotovsky, B.; Peterson, E.; Orrenius, S. A Comparative Study of Apoptosis and Necrosis in HepG2 Cells: Oxidant-Induced Caspase Inactivation Leads to Necrosis. Biochem. Biophys. Res. Commun. 1999, 255, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Amar, D.; Fleisher, M.; Pantuck, C.B.; Shamoon, H.; Zhang, H.; Roistacher, N.; Leung, D.H.Y.; Ginsburg, I.; Smiley, R.M. Persistent Alterations of the Autonomic Nervous System after Noncardiac Surgery. Anesthesiology 1998, 89, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, I. Could Synergistic Interactions among Reactive Oxygen Species, Proteinases, Membrane-Perforating Enzymes, Hydrolases, Microbial Hemolysins and Cytokines Be the Main Cause of Tissue Damage in Infectious and Inflammatory Conditions? Med. Hypotheses 1998, 51, 337–346. [Google Scholar] [CrossRef]
- Ginsburg, I. The Role of Bacteriolysis in the Pathophysiology of Inflammation, Infection and Post-Infectious Sequelae. APMIS 2002, 110, 753–770. [Google Scholar] [CrossRef] [PubMed]
- Durai, P.C.; Thappa, D.M.; Kumari, R.; Malathi, M. Aging in Elderly: Chronological Versus Photoaging. Indian J. Derm. 2012, 57, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Kozakiewicz, M.; Kornatowski, M.; Krzywińska, O.; Kędziora-Kornatowska, K. Changes in the Blood Antioxidant Defense of Advanced Age People. Clin. Interv. Aging 2019, 14, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Rhie, G.; Shin, M.H.; Seo, J.Y.; Choi, W.W.; Cho, K.H.; Kim, K.H.; Park, K.C.; Eun, H.C.; Chung, J.H. Aging- and Photoaging-Dependent Changes of Enzymic and Nonenzymic Antioxidants in the Epidermis and Dermis of Human Skin In Vivo. J. Investig. Derm. 2001, 117, 1212–1217. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P.; Mody, V.C.; Carlson, J.L.; Lynn, M.J.; Sternberg, P. Redox Analysis of Human Plasma Allows Separation of Pro-Oxidant Events of Aging from Decline in Antioxidant Defenses. Free Radic. Biol. Med. 2002, 33, 1290–1300. [Google Scholar] [CrossRef]
- Casimiro, C.; García-de-Lorenzo, A.; Usán, L. Prevalence of Decubitus Ulcer and Associated Risk Factors in an Institutionalized Spanish Elderly Population. Nutrition 2002, 18, 408–414. [Google Scholar] [CrossRef]
- Shukla, A.; Rasik, A.M.; Patnaik, G.K. Depletion of Reduced Glutathione, Ascorbic Acid, Vitamin E and Antioxidant Defence Enzymes in a Healing Cutaneous Wound. Free Radic. Res. 1997, 26, 93–101. [Google Scholar] [CrossRef]
- Khodr, B.; Khalil, Z. Modulation of Inflammation by Reactive Oxygen Species: Implications for Aging and Tissue Repair. Free Radic. Biol. Med. 2001, 30, 1–8. [Google Scholar] [CrossRef]
- Bliss, M.R. Pressure Injuries: Causes and Prevention. Hosp. Med. 1998, 59, 841–844. [Google Scholar] [PubMed]
- Aquilani, R.; Boschi, F.; Contardi, A.; Pistarini, C.; Achilli, M.P.; Fizzotti, G.; Moroni, S.; Catapano, M.; Verri, M.; Pastoris, O. Energy Expenditure and Nutritional Adequacy of Rehabilitation Paraplegics with Asymptomatic Bacteriuria and Pressure Sores. Spinal Cord 2001, 39, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Cruse, J.M.; Lewis, R.E.; Dilioglou, S.; Roe, D.L.; Wallace, W.F.; Chen, R.S. Review of Immune Function, Healing of Pressure Ulcers, and Nutritional Status in Patients with Spinal Cord Injury. J. Spinal Cord Med. 2000, 23, 129–135. [Google Scholar] [CrossRef]
- Evans, P.; Halliwell, B. Micronutrients: Oxidant/Antioxidant Status. Br. J. Nutr. 2001, 85, S67–S74. [Google Scholar] [CrossRef] [PubMed]
- Mervis, J.S.; Phillips, T.J. Pressure Ulcers: Pathophysiology, Epidemiology, Risk Factors, and Presentation. J. Am. Acad. Dermatol. 2019, 81, 881–890. [Google Scholar] [CrossRef]
- Bergstrom, N.; Demuth, P.J.; Braden, B.J. A Clinical Trial of the Braden Scale for Predicting Pressure Sore Risk. Nurs. Clin. N. Am. 1987, 22, 417–428. [Google Scholar]
- Montalcini, T.; Moraca, M.; Ferro, Y.; Romeo, S.; Serra, S.; Raso, M.G.; Rossi, F.; Sannita, W.G.; Dolce, G.; Pujia, A. Nutritional Parameters Predicting Pressure Ulcers and Short-Term Mortality in Patients with Minimal Conscious State as a Result of Traumatic and Non-Traumatic Acquired Brain Injury. J. Transl. Med. 2015, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachs, M.B.; Wolffbrandt, M.M.; Poulsen, I. Prevention of Pressure Ulcers in Patients Undergoing Subacute Rehabilitation after Severe Brain Injury: An Observational Study. J. Clin. Nurs. 2018, 27, 2776–2784. [Google Scholar] [CrossRef]
- Kahveci, K.; Dinçer, M.; Doger, C.; Yaricı, A.K. Traumatic Brain Injury and Palliative Care: A Retrospective Analysis of 49 Patients Receiving Palliative Care during 2013–2016 in Turkey. Neural Regen. Res. 2017, 12, 77–83. [Google Scholar] [CrossRef]
- Sarasúa, J.G.; López, S.P.; Viejo, M.Á.; Basterrechea, M.P.; Rodríguez, A.F.; Gutiérrez, A.F.; Gala, J.G.; Menéndez, Y.M.; Augusto, D.E.; Arias, A.P.; et al. Treatment of Pressure Ulcers with Autologous Bone Marrow Nuclear Cells in Patients with Spinal Cord Injury. J. Spinal Cord Med. 2011, 34, 301–307. [Google Scholar] [CrossRef] [Green Version]
- Ornellas, F.M.; Ornellas, D.S.; Martini, S.V.; Castiglione, R.C.; Ventura, G.M.; Rocco, P.R.; Gutfilen, B.; de Souza, S.A.; Takiya, C.M.; Morales, M.M. Bone Marrow–Derived Mononuclear Cell Therapy Accelerates Renal Ischemia-Reperfusion Injury Recovery by Modulating Inflammatory, Antioxidant and Apoptotic Related Molecules. Cell Physiol. Biochem. 2017, 41, 1736–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cereda, E.; Klersy, C.; Serioli, M.; Crespi, A.; D’Andrea, F.; Perna, S. A Nutritional Formula Enriched With Arginine, Zinc, and Antioxidants for the Healing of Pressure Ulcers A Randomized Trial. Ann. Intern. Med. 2015, 162, 167–174. [Google Scholar] [CrossRef]
- Theilla, M.; Schwartz, B.; Cohen, J.; Shapiro, H.; Anbar, R.; Singer, P. Impact of a Nutritional Formula Enriched in Fish Oil and Micronutrients on Pressure Ulcers in Critical Care Patients. Am. J. Crit. Care 2012, 21, e102–e109. [Google Scholar] [CrossRef] [PubMed]
- Pyatak, E.A.; Blanche, E.I.; Garber, S.L.; Diaz, J.; Blanchard, J.; Florindez, L.; Clark, F.A. Conducting Intervention Research among Underserved Populations: Lessons Learned and Recommendations for Researchers. Arch. Phys. Med. Rehabil. 2013, 94, 1190–1198. [Google Scholar] [CrossRef] [Green Version]
- Blanche, E.I.; Fogelberg, D.; Diaz, J.; Carlson, M.; Clark, F. Manualization of Occupational Therapy Interventions: Illustrations from the Pressure Ulcer Prevention Research Program. Am. J. Occup. Ther. 2011, 65, 711–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaishampayan, A.; Clark, F.; Carlson, M.; Blanche, E.I. Individualization of a Manualized Pressure Ulcer Prevention Program: Targeting Risky Life Circumstances Through a Community-Based Intervention for People with Spinal Cord Injury. Adv. Ski. Wound Care 2011, 24, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Fogelberg, D.; Atkins, M.; Blanche, E.I.; Carlson, M.; Clark, F. Decisions and Dilemmas in Everyday Life: Daily Use of Wheelchairs by Individuals with Spinal Cord Injury and the Impact on Pressure Ulcer Risk. Top. Spinal Cord Inj. Rehabil. 2009, 15, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Clark, F.; Pyatak, E.A.; Carlson, M.; Blanche, E.I.; Vigen, C.; Hay, J.; Mallinson, T.; Blanchard, J.; Unger, J.B.; Garber, S.L.; et al. Implementing Trials of Complex Interventions in Community Settings: The USC—Rancho Los Amigos Pressure Ulcer Prevention Study (PUPS). Clin. Trials 2014, 11, 218–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, M.; Vigen, C.L.P.; Rubayi, S.; Blanche, E.I.; Blanchard, J.; Atkins, M.; Bates-Jensen, B.; Garber, S.L.; Pyatak, E.A.; Diaz, J.; et al. Lifestyle Intervention for Adults with Spinal Cord Injury: Results of the USC–RLANRC Pressure Ulcer Prevention Study. J. Spinal Cord Med. 2019, 42, 2–19. [Google Scholar] [CrossRef]
- Nussbaum, E.L.; Biemann, I.; Mustard, B. Comparison of Ultrasound/Ultraviolet-C and Laser for Treatment of Pressure Ulcers in Patients With Spinal Cord Injury. Phys. Ther. 1994, 74, 812–823. [Google Scholar] [CrossRef] [Green Version]
- Nussbaum, E.L.; Flett, H.; Hitzig, S.L.; McGillivray, C.; Leber, D.; Morris, H.; Jing, F. Ultraviolet-C Irradiation in the Management of Pressure Ulcers in People With Spinal Cord Injury: A Randomized, Placebo-Controlled Trial. Arch. Phys. Med. Rehabil. 2013, 94, 650–659. [Google Scholar] [CrossRef]
- Guihan, M.; Bombardier, C.H.; Ehde, D.M.; Rapacki, L.M.; Rogers, T.J.; Bates-Jensen, B.; Thomas, F.P.; Parachuri, R.; Holmes, S.A. Comparing Multicomponent Interventions to Improve Skin Care Behaviors and Prevent Recurrence in Veterans Hospitalized for Severe Pressure Ulcers. Arch. Phys. Med. Rehabil. 2014, 95, 1246–1253.e3. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.; Guihan, M.; Frick, C.; Gill, C.M.; Ho, C.H. What’s Happening Now! Telehealth Management of Spinal Cord Injury/Disorders. J. Spinal Cord Med. 2011, 34, 322–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauman, W.A.; Spungen, A.M.; Collins, J.F.; Raisch, D.W.; Ho, C.; Deitrick, G.A.; Nemchausky, B.A.; Goetz, L.L.; Park, J.S.; Schwartz, M.; et al. The Effect of Oxandrolone on the Healing of Chronic Pressure Ulcers in Persons With Spinal Cord Injury: A Randomized Trial. Ann. Intern. Med. 2013, 158, 718. [Google Scholar] [CrossRef]
- Ho, C.H.; Bensitel, T.; Wang, X.; Bogie, K.M. Pulsatile Lavage for the Enhancement of Pressure Ulcer Healing: A Randomized Controlled Trial. Phys. Ther. 2012, 92, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Guihan, M.; Garber, S.L.; Bombardier, C.H.; Durazo-Arizu, R.; Goldstein, B.; Holmes, S.A. Lessons Learned While Conducting Research on Prevention of Pressure Ulcers in Veterans With Spinal Cord Injury. Arch. Phys. Med. Rehabil. 2007, 88, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Guihan, M.; Garber, S.L.; Bombardier, C.H.; Goldstein, B.; Holmes, S.A.; Cao, L. Predictors of Pressure Ulcer Recurrence in Veterans With Spinal Cord Injury. J. Spinal Cord Med. 2008, 31, 551–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guihan, M.; Bombardier, C.H. Potentially Modifiable Risk Factors among Veterans with Spinal Cord Injury Hospitalized for Severe Pressure Ulcers: A Descriptive Study. J. Spinal Cord Med. 2012, 35, 240–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irgens, I.; Hoff, J.M.; Sørli, H.; Haugland, H.; Stanghelle, J.K.; Rekand, T. Hospital Based Care at Home; Study Protocol for a Mixed Epidemiological and Randomized Controlled Trial. Trials 2019, 20. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, S.; Theis, T.; Tschang, M.; Nagaraj, V.; Berthiaume, F. Reactive Oxygen Species and Pressure Ulcer Formation after Traumatic Injury to Spinal Cord and Brain. Antioxidants 2021, 10, 1013. https://doi.org/10.3390/antiox10071013
Kumar S, Theis T, Tschang M, Nagaraj V, Berthiaume F. Reactive Oxygen Species and Pressure Ulcer Formation after Traumatic Injury to Spinal Cord and Brain. Antioxidants. 2021; 10(7):1013. https://doi.org/10.3390/antiox10071013
Chicago/Turabian StyleKumar, Suneel, Thomas Theis, Monica Tschang, Vini Nagaraj, and Francois Berthiaume. 2021. "Reactive Oxygen Species and Pressure Ulcer Formation after Traumatic Injury to Spinal Cord and Brain" Antioxidants 10, no. 7: 1013. https://doi.org/10.3390/antiox10071013