
CDDO-Me Attenuates Astroglial Autophagy via Nrf2-, ERK1/2-SP1- and Src-CK2-PTEN-PI3K/AKT-Mediated Signaling Pathways in the Hippocampus of Chronic Epilepsy Rats

Abstract
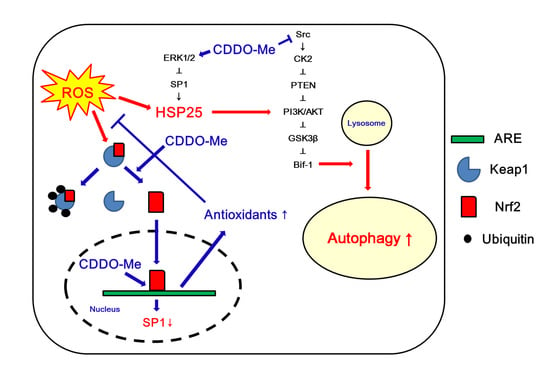
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Chemicals
2.2. Epilepsy Model
2.3. Electrode Implantation, CDDO-Me Trials and Quantification of Seizure Activity
2.4. Western Blots
2.5. Immunohistochemistry, Cell Counts and Measurement of Fluorescent Intensity
2.6. Data Analysis
3. Results
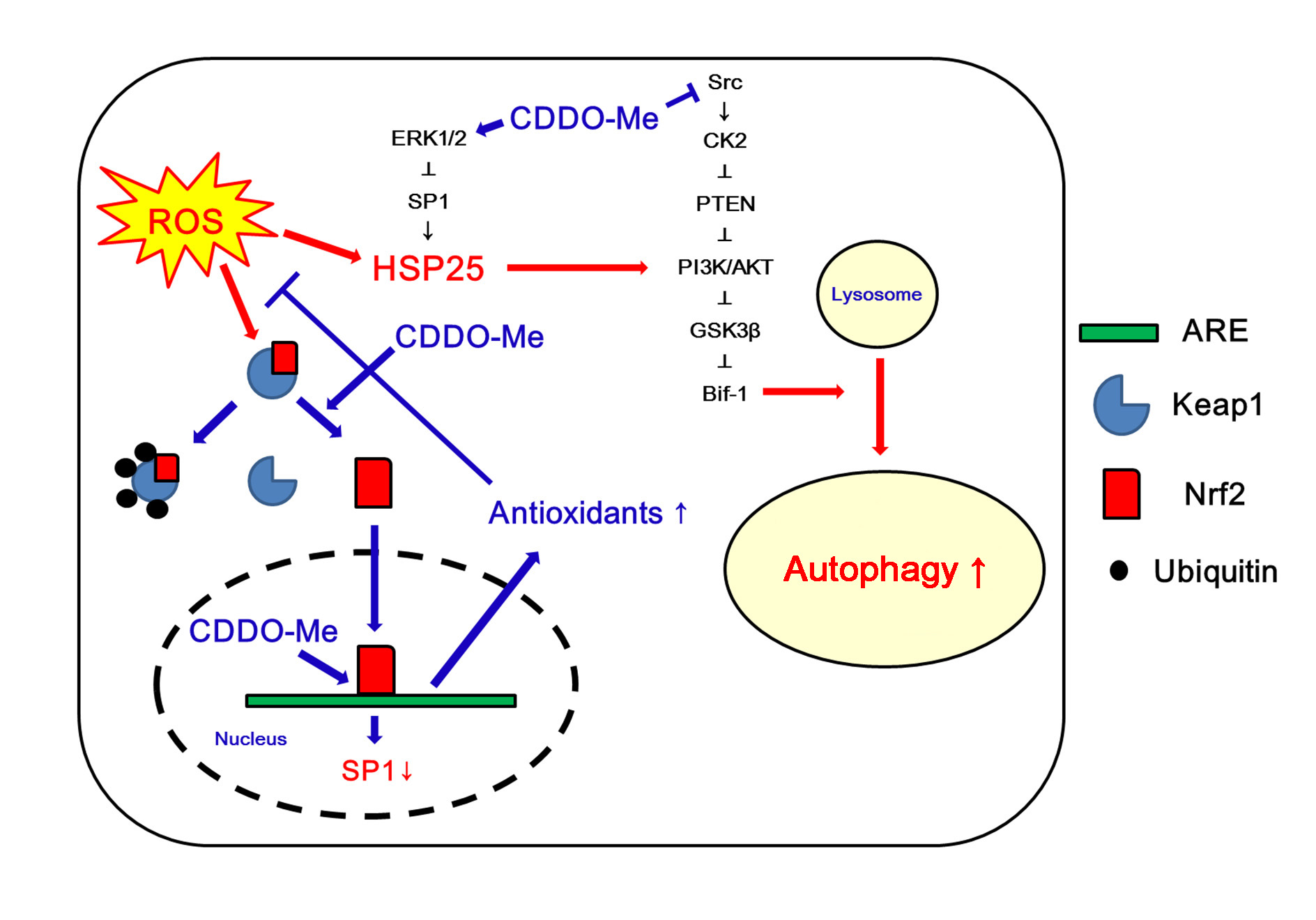
3.1. The Nrf2 Protein Level and Its Nuclear Translocation Are Reduced in Clasmatodendritic Astrocytes
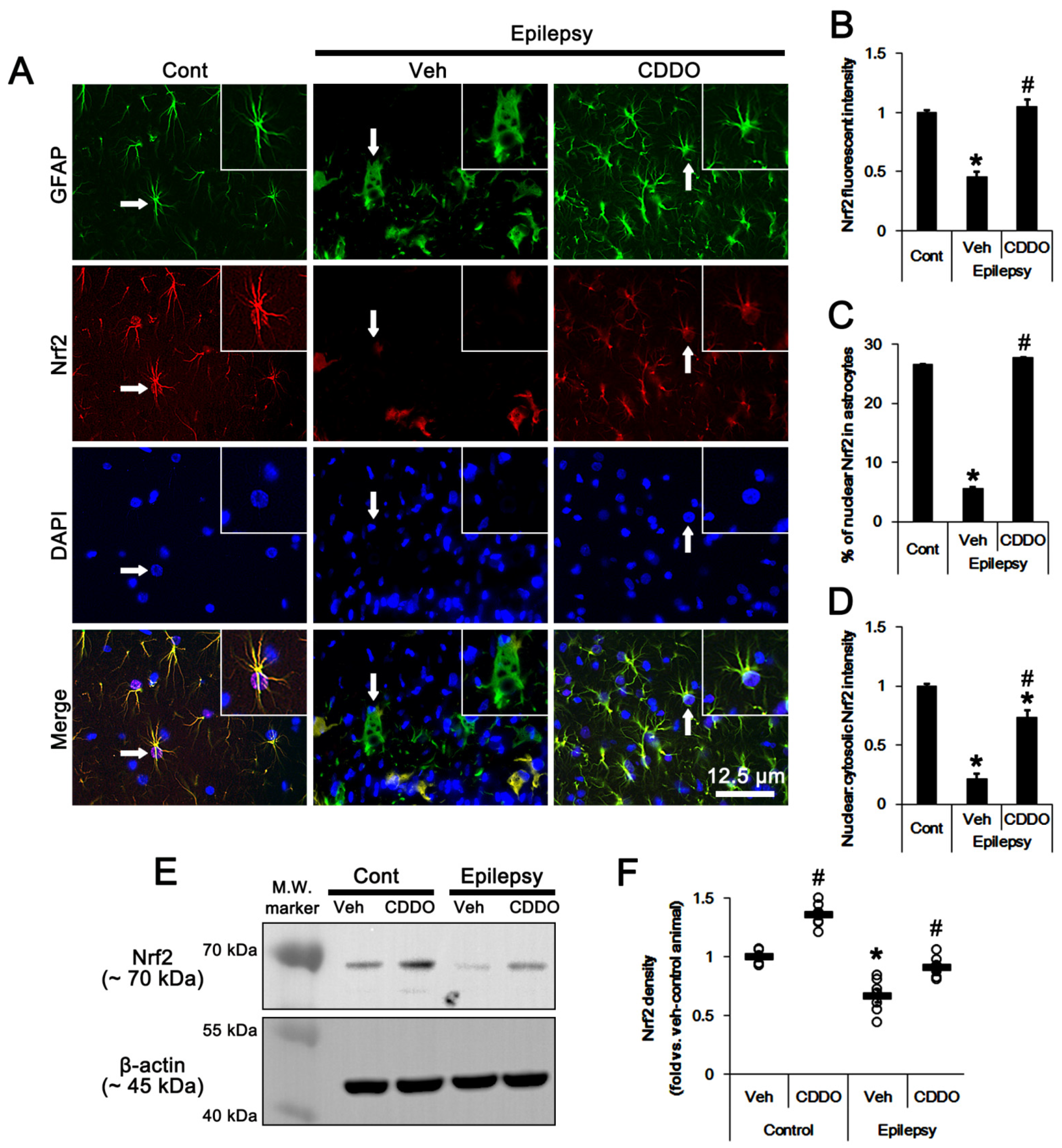
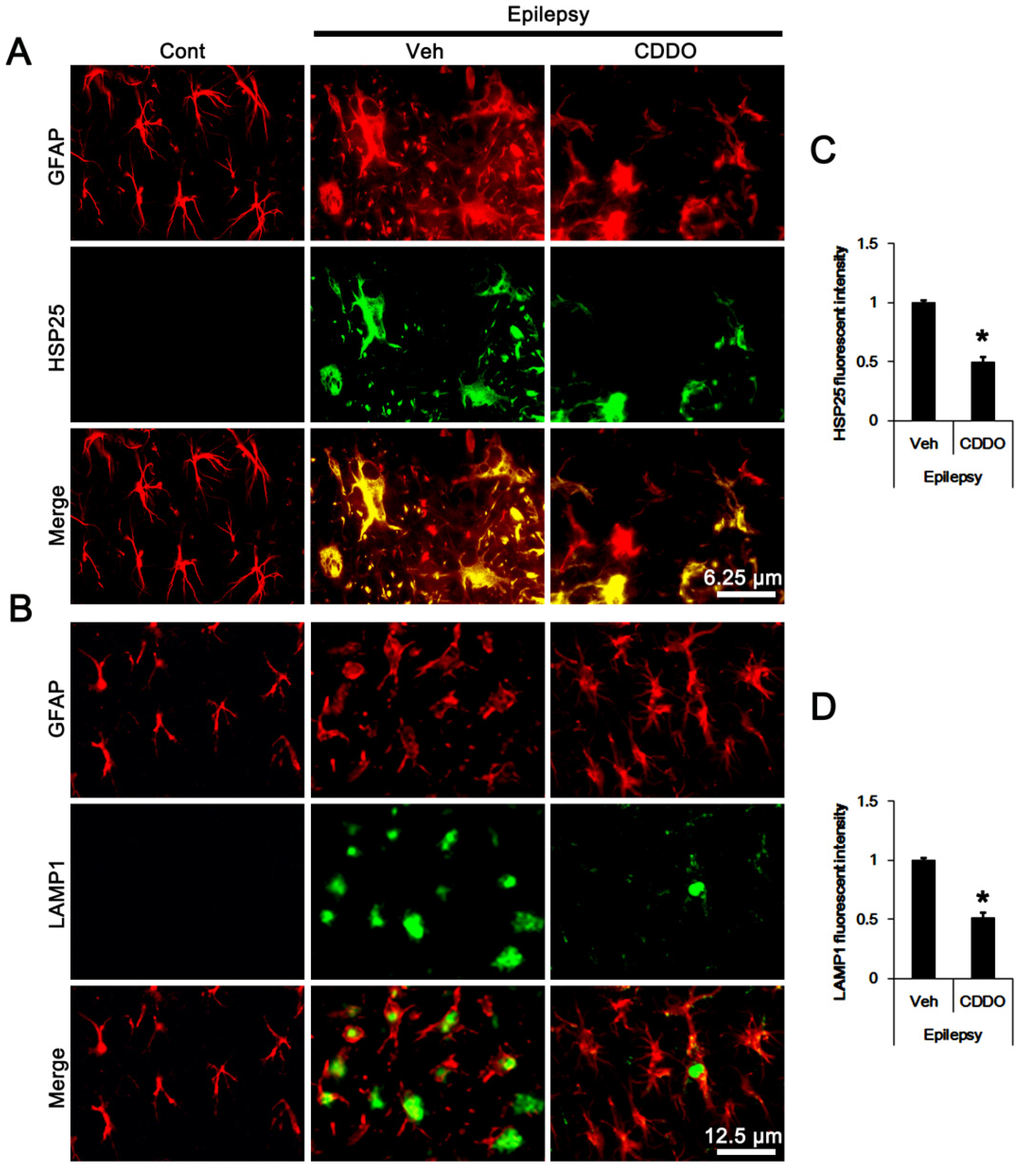
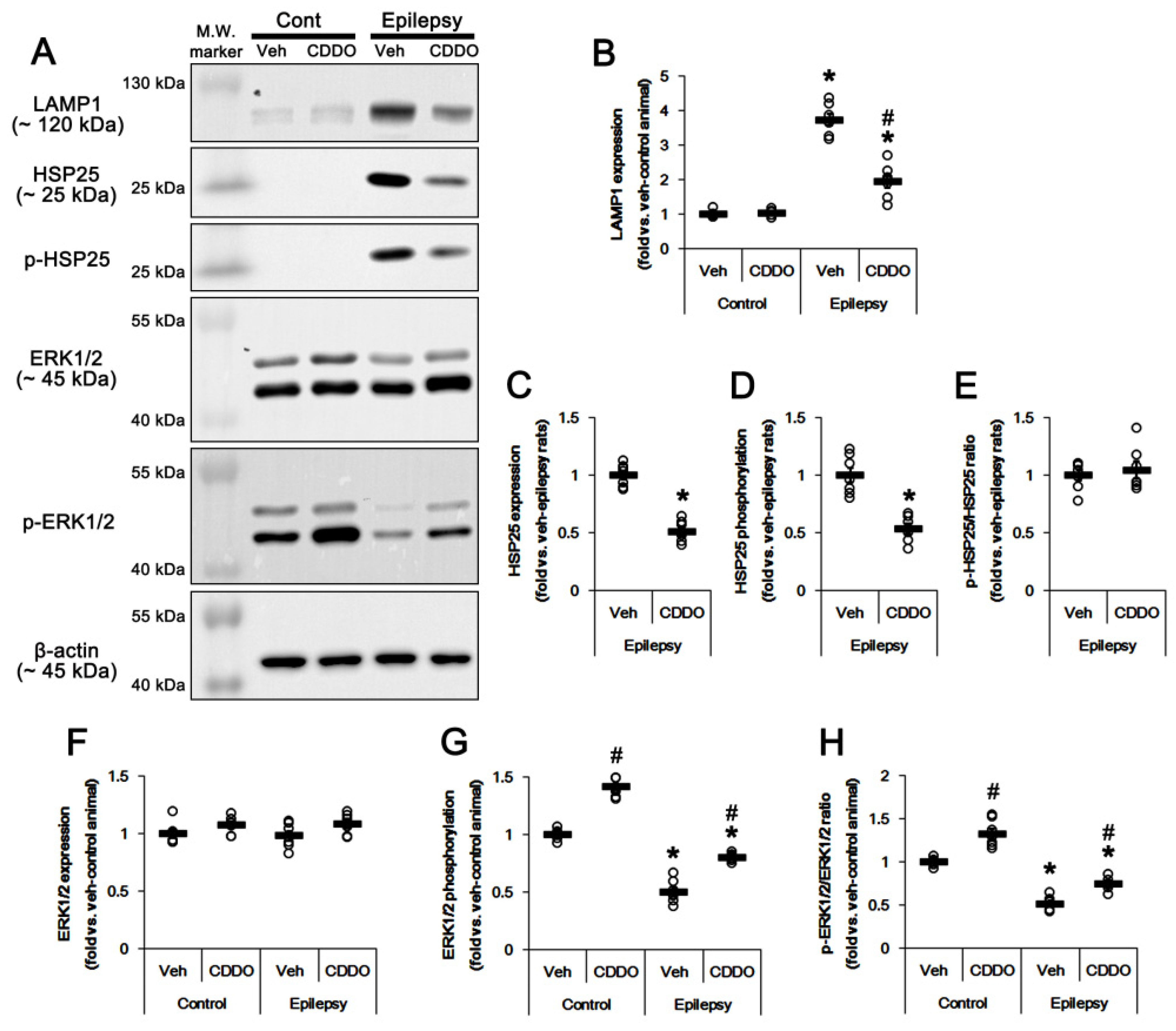
3.2. CDDO-Me Regulates ERK1/2-Mediated HSP25 Induction in CA1 Astrocytes
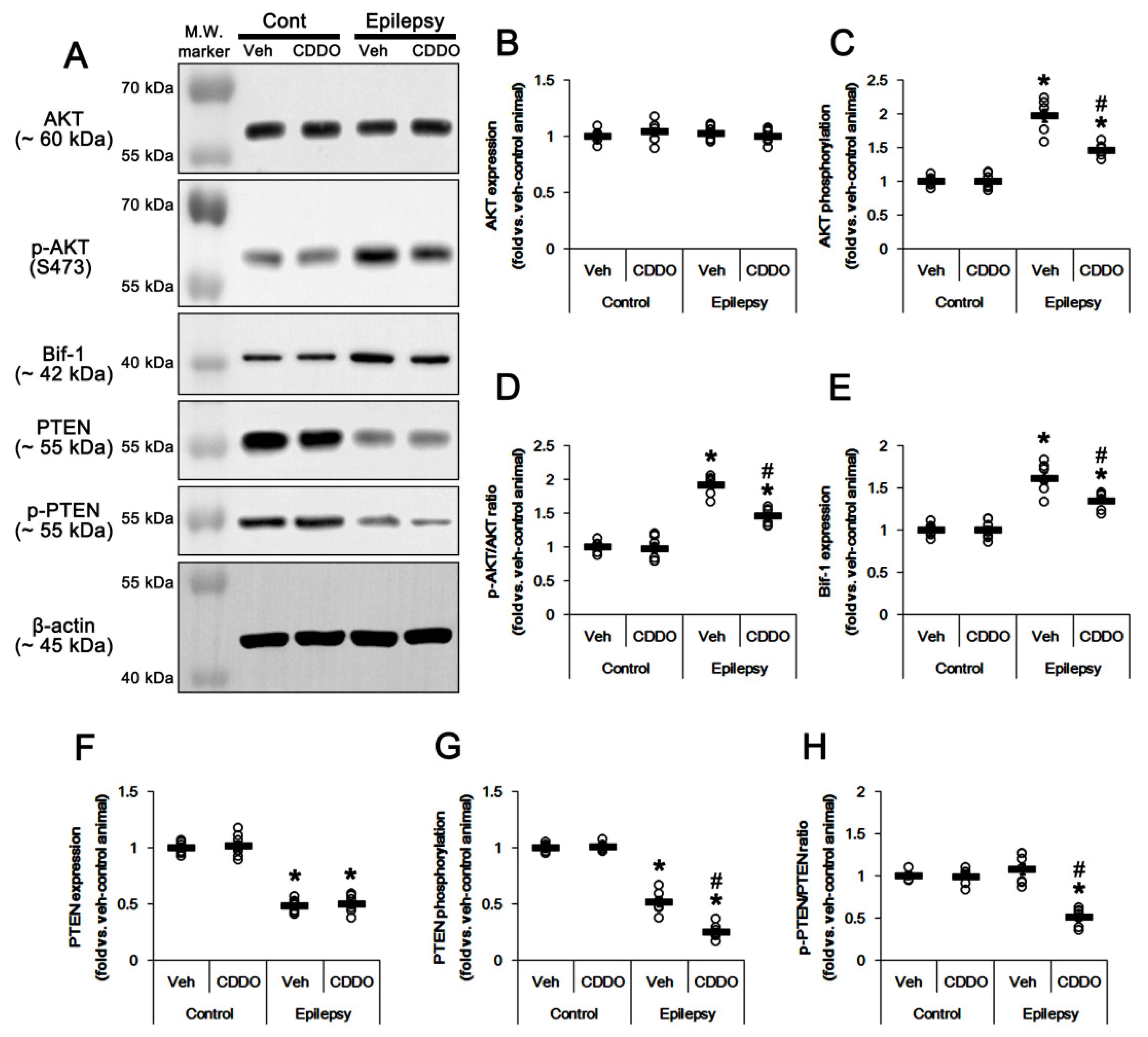
3.3. CDDO-Me Leads to PTEN-Mediated AKT Inhibition in the Epileptic Hippocampus
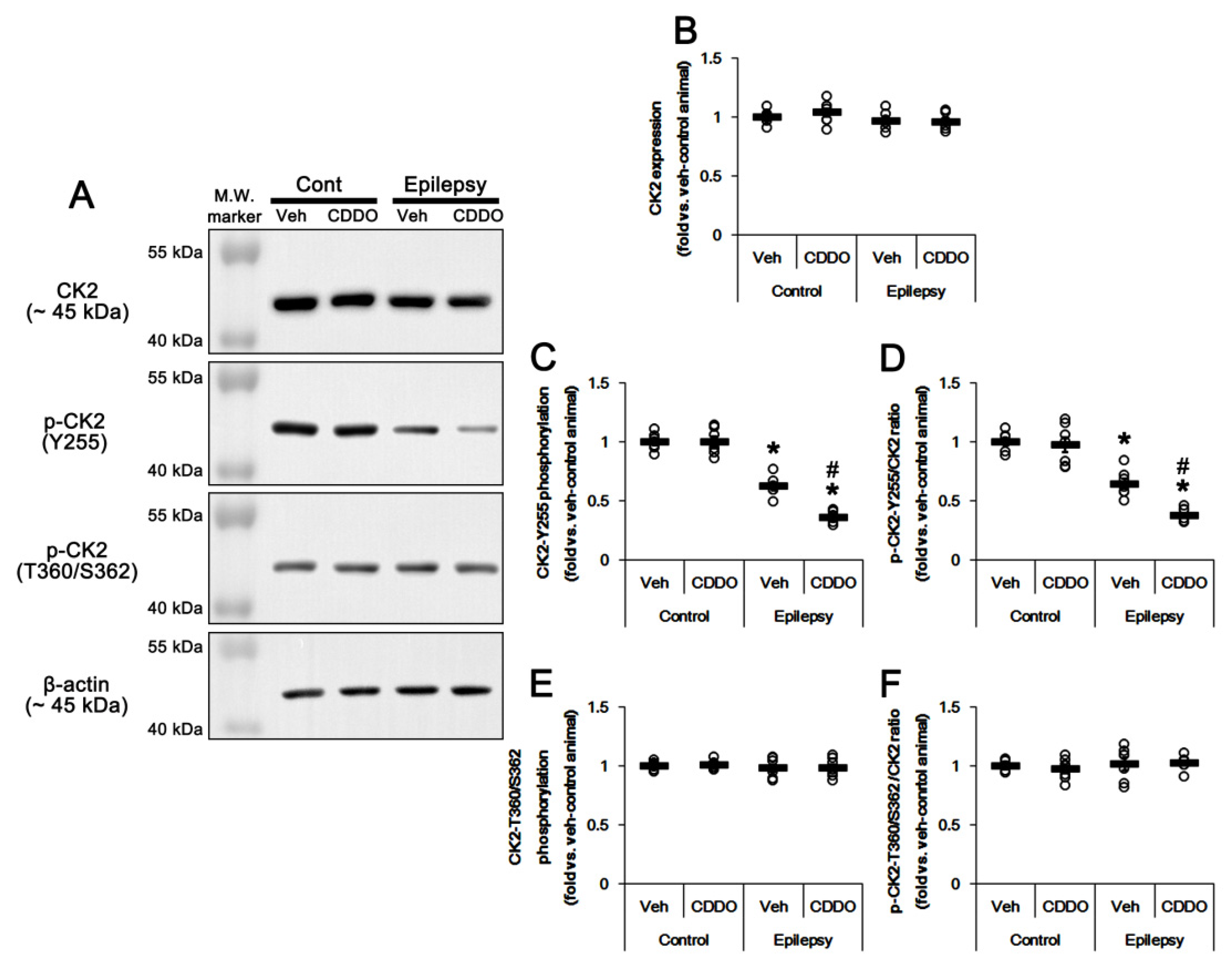
3.4. CDDO-Me Inhibits CK2 Phosphorylation in the Epileptic Hippocampus
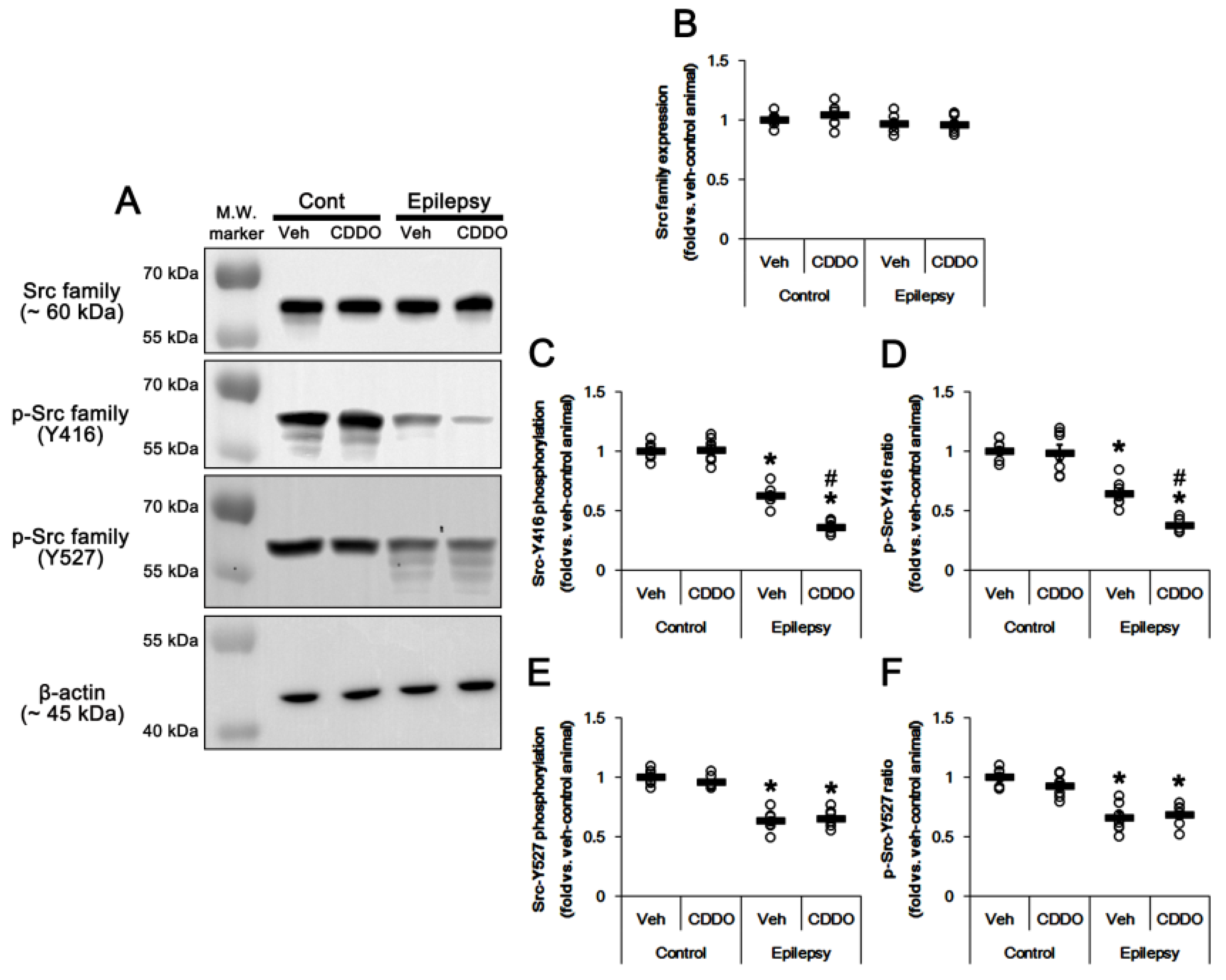
3.5. CDDO-Me Inhibits Src Family Phosphorylation at Y416 Site in the Epileptic Hippocampus
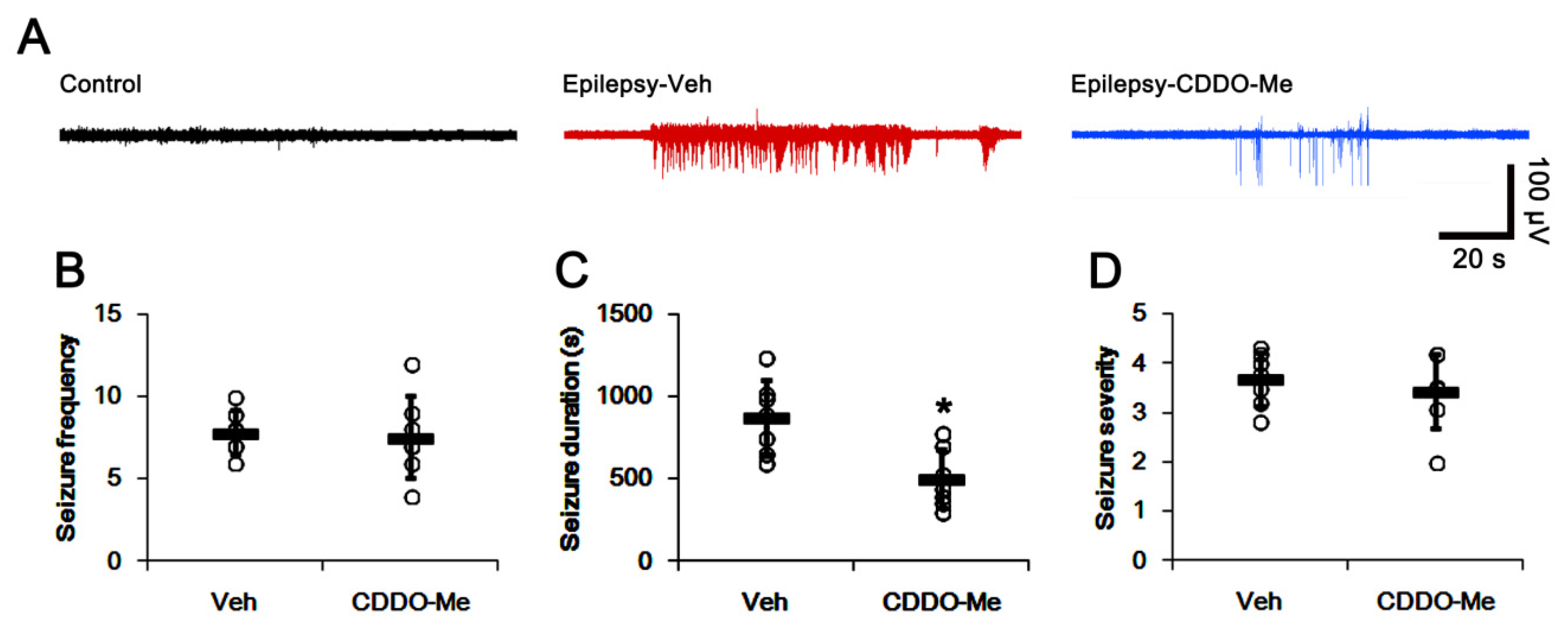
3.6. CDDO-Me Decreases Seizure Duration, But Not Seizure Frequency or Its Intensity
4. Discussion
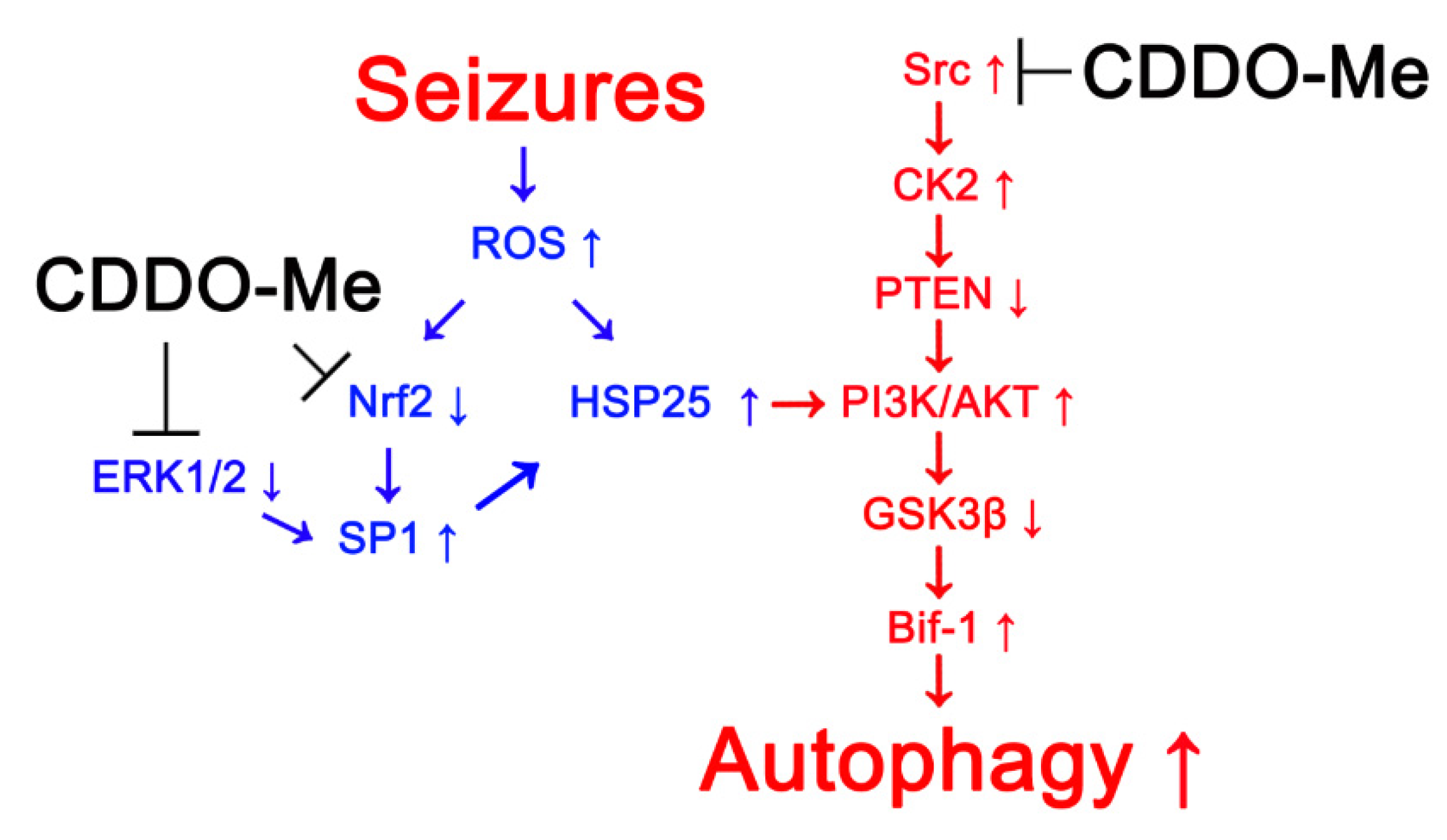
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Dayan, P.S.; Lillis, K.; Bennett, J.; Conners, G.; Bailey, P.; Callahan, J.; Akman, C.; Feldstein, N.; Kriger, J.; Hauser, W.A.; et al. Prevalence of and risk factors for intracranial abnormalities in unprovoked seizures. Pediatrics 2015, 136, e351–e360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadr, S.S.; Javanbakht, J.; Javidan, A.N.; Ghaffarpour, M.; Khamse, S.; Naghshband, Z. Descriptive epidemiology: Prevalence, incidence, sociodemographic factors, socioeconomic domains, and quality of life of epilepsy: An update and systematic review. Arch. Med. Sci. 2018, 14, 717–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathern, G.W.; Babb, T.L.; Pretorius, J.K.; Melendez, M.; Lévesque, M.F. The pathophysiologic relationships between lesion pathology, intracranial ictal EEG onsets, and hippocampal neuron losses in temporal lobe epilepsy. Epilepsy Res. 1995, 21, 133–147. [Google Scholar] [CrossRef]
- Wittner, L.; Maglóczky, Z.; Borhegyi, Z.; Halász, P.; Tóth, S.; Eross, L.; Szabó, Z.; Freund, T.F. Preservation of perisomatic inhibitory input of granule cells in the epileptic human dentate gyrus. Neuroscience 2001, 108, 587–600. [Google Scholar] [CrossRef]
- Briellmann, R.S.; Kalnins, R.M.; Berkovic, S.F.; Jackson, G.D. Hippocampal pathology in refractory temporal lobe epilepsy: T2-weighted signal change reflects dentate gliosis. Neurology 2002, 58, 265–271. [Google Scholar] [CrossRef]
- Vessal, M.; Dugani, C.B.; Solomon, D.A.; McIntyre Burnham, W.; Ivy, G.O. Might astrocytes play a role in maintaining the seizure-prone state? Brain Res. 2005, 1044, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.C.; Kim, D.S.; Kwak, S.E.; Kim, J.E.; Won, M.H.; Kim, D.W.; Choi, S.Y.; Kwon, O.S. Epileptogenic roles of astroglial death and regeneration in the dentate gyrus of experimental temporal lobe epilepsy. Glia 2006, 54, 258–271. [Google Scholar] [CrossRef]
- Anderson, C.M.; Swanson, R.A. Astrocyte glutamate transport: Review of properties, regulation, and physiological functions. Glia 2000, 32, 1–14. [Google Scholar] [CrossRef]
- Ransom, B.; Behar, T.; Nedergaard, M. New roles for astrocytes (stars at last). Trends Neurosci. 2003, 26, 520–522. [Google Scholar] [CrossRef]
- Penfield, W. Neuroglia and microglia—The interstitial tissue of the central nervous system. In Special Cytology, the Form and Function of the Cell in Health and Disease; Cowdry, E.V., Ed.; Hoeber: New York, NY, USA, 1928; pp. 1033–1068. [Google Scholar]
- Revuelta, M.; Castaño, A.; Machado, A.; Cano, J.; Venero, J.L. Kainate-induced zinc translocation from presynaptic terminals causes neuronal and astroglial cell death and mRNA loss of BDNF receptors in the hippocampal formation and amygdala. J. Neurosci. Res. 2005, 82, 184–195. [Google Scholar] [CrossRef]
- Ryu, H.J.; Kim, J.E.; Yeo, S.I.; Kang, T.C. p65/RelA-Ser529 NF-κB subunit phosphorylation induces autophagic astroglial death (Clasmatodendrosis) following status epilepticus. Cell. Mol. Neurobiol. 2011, 31, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.J.; Kim, J.E.; Yeo, S.I.; Kim, D.W.; Kwon, O.S.; Choi, S.Y.; Kang, T.C. F-actin depolymerization accelerates clasmatodendrosis via activation of lysosome-derived autophagic astroglial death. Brain Res. Bull. 2011, 85, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Kim, Y.J.; Kim, J.Y.; Kang, T.C. PARP1 activation/expression modulates regional-specific neuronal and glial responses to seizure in a hemodynamic-independent manner. Cell Death Dis. 2014, 5, e1362. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.R.; Hyun, H.W.; Min, S.J.; Kim, J.E. The Differential DRP1 Phosphorylation and Mitochondrial Dynamics in the Regional Specific Astroglial Death Induced by Status Epilepticus. Front. Cell. Neurosci. 2016, 10, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.S.; Kim, J.E.; Kwak, S.E.; Choi, K.C.; Kim, D.W.; Kwon, O.S.; Choi, S.Y.; Kang, T.C. Spatiotemporal characteristics of astroglial death in the rat hippocampo-entorhinal complex following pilocarpine-induced status epilepticus. J. Comp. Neurol. 2008, 511, 581–598. [Google Scholar] [CrossRef]
- Kim, J.E.; Ryu, H.J.; Yeo, S.I.; Seo, C.H.; Lee, B.C.; Choi, I.G.; Kim, D.S.; Kang, T.C. Differential expressions of aquaporin subtypes in astroglia in the hippocampus of chronic epileptic rats. Neuroscience 2009, 163, 781–789. [Google Scholar] [CrossRef]
- Sakai, K.; Fukuda, T.; Iwadate, K. Beading of the astrocytic processes (clasmatodendrosis) following head trauma is associated with protein degradation pathways. Brain Inj. 2013, 27, 1692–1697. [Google Scholar] [CrossRef]
- Bouchat, J.; Gilloteaux, J.; Suain, V.; Van Vlaender, D.; Brion, J.P.; Nicaise, C. Ultrastructural Analysis of Thalamus Damages in a Mouse Model of Osmotic-Induced Demyelination. Neurotox. Res. 2019, 36, 144–162. [Google Scholar] [CrossRef] [Green Version]
- Bursch, W.; Ellinger, A.; Kienzl, H.; Török, L.; Pandey, S.; Sikorska, M.; Walker, R.; Hermann, R.S. Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: The role of autophagy. Carcinogenesis 1996, 17, 1595–1607. [Google Scholar] [CrossRef] [Green Version]
- Tsujimoto, Y.; Shimizu, S. Another way to die: Autophagic programmed cell death. Cell Death Differ. 2005, 12, 1528–1534. [Google Scholar] [CrossRef] [Green Version]
- Gozuacik, D.; Kimchi, A. Autophagy and cell death. Curr. Top. Dev. Biol. 2007, 78, 217–245. [Google Scholar]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Hyun, H.W.; Min, S.J.; Kang, T.C. Sustained HSP25 Expression Induces Clasmatodendrosis via ER Stress in the Rat Hippocampus. Front. Cell. Neurosci. 2017, 11, 47. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Ko, A.R.; Hyun, H.W.; Min, S.J.; Kang, T.C. P2RX7-MAPK1/2-SP1 axis inhibits MTOR independent HSPB1-mediated astroglial autophagy. Cell Death Dis. 2018, 9, 546. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000, 275, 16023–16029. [Google Scholar] [CrossRef] [Green Version]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, A.; Ohta, T.; Yamamoto, M. Unique function of the Nrf2-Keap1 pathway in the inducible expression of antioxidant and detoxifying enzymes. Methods Enzymol. 2004, 378, 273–286. [Google Scholar] [PubMed]
- Kang, T.C. Nuclear factor-erythroid 2-related factor 2 (Nrf2) and mitochondrial dynamics/mitophagy in neurological diseases. Antioxidants 2020, 9, 617. [Google Scholar] [CrossRef]
- Gao, P.; Li, L.; Ji, L.; Wei, Y.; Li, H.; Shang, G.; Zhao, Z.; Chen, Q.; Jiang, T.; Zhang, N. Nrf2 ameliorates diabetic nephropathy progression by transcriptional repression of TGFβ1 through interactions with c-Jun and SP1. Biochim. Biophys. Acta. 2014, 1839, 1110–1120. [Google Scholar] [CrossRef]
- Takagi, T.; Kitashoji, A.; Iwawaki, T.; Tsuruma, K.; Shimazawa, M.; Yoshimura, S.; Iwama, T.; Hara, H. Temporal activation of Nrf2 in the penumbra and Nrf2 activator-mediated neuroprotection in ischemia-reperfusion injury. Free Radic. Biol. Med. 2014, 72, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Takagi, T.; Kitashoji, A.; Yamauchi, K.; Shimazawa, M.; Hara, H. Nrf2 activator ameliorates hemorrhagic transformation in focal cerebral ischemia under warfarin anticoagulation. Neurobiol. Dis. 2016, 89, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Park, H.; Choi, S.H.; Kong, M.J.; Kang, T.C. CDDO-Me selectively attenuates CA1 neuronal death induced by status epilepticus via facilitating mitochondrial fission independent of LONP1. Cells 2019, 8, 833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.J.; Park, H.; Choi, S.H.; Kong, M.J.; Kim, J.E.; Kang, T.C. CDDO-Me attenuates vasogenic edema and astroglial death by regulating NF-κB p65 phosphorylations and Nrf2 expression following status epilepticus. Int. J. Mol. Sci. 2019, 20, 4862. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Lee, D.S.; Park, H.; Jeong, M.J.; Kang, T.C. Src/CK2/PTEN-mediated GluN2B and CREB dephosphorylations regulate the responsiveness to AMPA receptor antagonist in chronic epilepsy rat. Int. J. Mol. Sci. 2020, 21, 9633. [Google Scholar] [CrossRef]
- Kim, J.E.; Choi, H.C.; Song, H.K.; Kang, T.C. Perampanel affects up-stream regulatory signaling pathways of GluA1 phosphorylation in normal and epileptic rats. Front. Cell. Neurosci. 2019, 13, 80. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Park, H.; Lee, J.E.; Kim, T.H.; Kang, T.C. PTEN is required for the anti-epileptic effects of AMPA receptor antagonists in chronic epileptic rats. Int. J. Mol. Sci. 2020, 21, 5643. [Google Scholar] [CrossRef]
- Park, K.K.; Liu, K.; Hu, Y.; Kanter, J.L.; He, Z. PTEN/mTOR and axon regeneration. Exp. Neurol. 2010, 223, 45–50. [Google Scholar] [CrossRef]
- Tang, C.; Gu, Y.; Wang, H.; Wu, H.; Wang, Y.; Meng, Y.; Han, Z.; Gu, Y.; Ma, W.; Jiang, Z.; et al. Targeting of microRNA-21-5p protects against seizure damage in a kainic acid-induced status epilepticus model via PTEN-mTOR. Epilepsy Res. 2018, 144, 34–42. [Google Scholar] [CrossRef]
- Liu, H. LncRNA CASC2 inhibits astrocytic activation and adenosine metabolism by regulating PTEN in pentylenetetrazol-induced epilepsy model. J. Chem. Neuroanat. 2020, 17, 101749. [Google Scholar]
- Ross, A.H.; Gericke, A. Phosphorylation keeps PTEN phosphatase closed for business. Proc. Natl. Acad. Sci. USA 2009, 106, 1297–1298. [Google Scholar] [CrossRef] [Green Version]
- Torres, J.; Pulido, R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J. Biol. Chem. 2001, 276, 993–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fragoso, R.; Barata, J.T. Kinases, tails and more: Regulation of PTEN function by phosphorylation. Methods 2015, 77–78, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Donella-Deana, A.; Cesaro, L.; Sarno, S.; Ruzzene, M.; Brunati, A.M.; Marin, O.; Vilk, G.; Doherty-Kirby, A.; Lajoie, G.; Litchfield, D.W.; et al. Tyrosine phosphorylation of protein kinase CK2 by Src-related tyrosine kinases correlates with increased catalytic activity. Biochem. J. 2003, 372, 841–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, H.; Wang, J.; Nika, H.; Hawke, D.; Keezer, S.; Ge, Q.; Fang, B.; Fang, X.; Fang, D.; Litchfield, D.W.; et al. EGF-induced ERK activation promotes CK2-mediated disassociation of alpha-Catenin from beta-Catenin and transactivation of beta-Catenin. Mol. Cell. 2009, 36, 547–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyneken, U.; Smalla, K.H.; Marengo, J.J.; Soto, D.; de la Cerda, A.; Tischmeyer, W.; Grimm, R.; Boeckers, T.M.; Wolf, G.; Orrego, F.; et al. Kainate-induced seizures alter protein composition and N-methyl-D-aspartate receptor function of rat forebrain postsynaptic densities. Neuroscience 2001, 102, 65–74. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Src kinase regulation by phosphorylation and dephosphorylation. Biochem. Biophys. Res. Commun. 2005, 331, 1–14. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure and regulation. Biochem. Biophys. Res. Commun. 2004, 324, 1155–1164. [Google Scholar] [CrossRef]
- Ling, X.; Konopleva, M.; Zeng, Z.; Ruvolo, V.; Stephens, L.C.; Schober, W.; McQueen, T.; Dietrich, M.; Madden, T.L.; Andreeff, M. The novel triterpenoid C-28 methyl ester of 2-cyano-3, 12-dioxoolen-1, 9-dien-28-oic acid inhibits metastatic murine breast tumor growth through inactivation of STAT3 signaling. Cancer Res. 2007, 67, 4210–4218. [Google Scholar] [CrossRef] [Green Version]
- Duan, Z.; Ames, R.Y.; Ryan, M.; Hornicek, F.J.; Mankin, H.; Seiden, M.V. CDDO-Me, a synthetic triterpenoid, inhibits expression of IL-6 and Stat3 phosphorylation in multi-drug resistant ovarian cancer cells. Cancer Chemother. Pharmacol. 2009, 63, 681–689. [Google Scholar] [CrossRef] [Green Version]
- Tomimoto, H.; Akiguchi, I.; Wakita, H.; Suenaga, T.; Nakamura, S.; Kimura, J. Regressive changes of astroglia in white matter lesions in cerebrovascular disease and Alzheimer’s disease patients. Acta Neuropathol. 1997, 94, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Akinyemi, R.O.; Hase, Y.; Firbank, M.J.; Ndung’u, M.N.; Foster, V.; Craggs, L.J.; Washida, K.; Okamoto, Y.; Thomas, A.J.; et al. Frontal white matter hyperintensities, clasmatodendrosis and gliovascular abnormalities in ageing and post-stroke dementia. Version 2. Brain 2016, 139, 242–258. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Mohri, I.; Hirata, I.; Kuwada, A.; Kimura-Ohba, S.; Kagitani-Shimono, K.; Fushimi, H.; Inoue, T.; Shiomi, M.; Kakuta, Y.; et al. Clasmatodendrosis is associated with dendritic spines and does not represent autophagic astrocyte death in influenza-associated encephalopathy. Brain Dev. 2019, 41, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Early, A.N.; Gorman, A.A.; Van Eldik, L.J.; Bachstetter, A.D.; Morganti, J.M. Effects of advanced age upon astrocyte-specific responses to acute traumatic brain injury in mice. J. Neuroinflammation 2020, 17, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Gong, Q.; Feng, X.; Zhang, D.; Quan, L. Astrocytic clasmatodendrosis in the cerebral cortex of methamphetamine abusers. Forensic. Sci. Res. 2017, 2, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Misu, T.; Höftberger, R.; Fujihara, K.; Wimmer, I.; Takai, Y.; Nishiyama, S.; Nakashima, I.; Konno, H.; Bradl, M.; Garzuly, F.; et al. Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol. 2013, 125, 815–827. [Google Scholar] [CrossRef] [Green Version]
- Canchi, S.; Sarntinoranont, M.; Hong, Y.; Flint, J.J.; Subhash, G.; King, M.A. Simulated blast overpressure induces specific astrocyte injury in an ex vivo brain slice model. PLoS ONE 2017, 12, e0175396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerbai, F.; Lana, D.; Nosi, D.; Petkova-Kirova, P.; Zecchi, S.; Brothers, H.M.; Wenk, G.L.; Giovannini, M.G. The neuron-astrocyte-microglia triad in normal brain ageing and in a model of neuroinflammation in the rat hippocampus. PLoS ONE 2012, 7, e45250. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Ko, P.W.; Lee, H.W.; Jeong, J.Y.; Lee, M.G.; Kim, J.H.; Lee, W.H.; Yu, R.; Oh, W.J.; Suk, K. Astrocyte-derived lipocalin-2 mediates hippocampal damage and cognitive deficits in experimental models of vascular dementia. Glia 2017, 65, 1471–1490. [Google Scholar] [CrossRef]
- Hulse, R.E.; Winterfield, J.; Kunkler, P.E.; Kraig, R.P. Astrocytic clasmatodendrosis in hippocampal organ culture. Glia 2001, 33, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Tsai, K.L.; Wang, S.M.; Chen, C.C.; Fong, T.H.; Wu, M.L. Mechanism of oxidative stress-induced intracellular acidosis in rat cerebellar astrocytes and C6 glioma cells. J. Physiol. 1997, 502, 161–174. [Google Scholar] [CrossRef]
- Muranyi, M.; Ding, C.; He, Q.; Lin, Y.; Li, P.A. Streptozotocin-induced diabetes causes astrocyte death after ischemia and reperfusion injury. Diabetes 2006, 55, 349–355. [Google Scholar] [CrossRef] [Green Version]
- Lewerenz, J.; Dargusch, R.; Maher, P. Lactacidosis modulates glutathione metabolism and oxidative glutamate toxicity. J. Neurochem. 2010, 113, 502–514. [Google Scholar] [CrossRef]
- Steele, M.L.; Fuller, S.; Patel, M.; Kersaitis, C.; Ooi, L.; Münch, G. Effect of Nrf2 activators on release of glutathione, cysteinylglycine and homocysteine by human U373 astroglial cells. Redox Biol. 2013, 1, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Parcellier, A.; Schmitt, E.; Gurbuxani, S.; Seigneurin-Berny, D.; Pance, A.; Chantôme, A.; Plenchette, S.; Khochbin, S.; Solary, E.; Garrido, C. HSP27 is a ubiquitin-binding protein involved in I-kappaBalpha proteasomal degradation. Mol. Cell. Biol. 2003, 23, 5790–5802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Cho, H.N.; Jeoung, D.I.; Soh, J.W.; Cho, C.K.; Bae, S.; Chung, H.Y.; Lee, S.J.; Lee, Y.S. HSP25 overexpression attenuates oxidative stress-induced apoptosis: Roles of ERK1/2 signaling and manganese superoxide dismutase. Free Radic. Biol. Med. 2004, 36, 429–444. [Google Scholar] [CrossRef]
- Didelot, C.; Schmitt, E.; Brunet, M.; Maingret, L.; Parcellier, A.; Garrido, C. Heat shock proteins: Endogenous modulators of apoptotic cell death. Handb. Exp. Pharmacol. 2006, 172, 71–98. [Google Scholar]
- Arrigo, A.P. The cellular “networking” of mammalian Hsp27 and its functions in the control of protein folding, redox state and apoptosis. Adv. Exp. Med. Biol. 2007, 594, 14–26. [Google Scholar] [PubMed]
- Che, Y.; Piao, C.S.; Han, P.L.; Lee, J.K. Delayed induction of alpha B-crystallin in activated glia cells of hippocampus in kainic acid-treated mouse brain. J. Neurosci. Res. 2001, 65, 425–431. [Google Scholar] [CrossRef]
- Kirschstein, T.; Mikkat, S.; Mikkat, U.; Bender, R.; Kreutzer, M.; Schulz, R.; Köhling, R.; Glocker, M.O. The 27-kDa heat shock protein (HSP27) is a reliable hippocampal marker of full development of pilocarpine-induced status epilepticus. Epilepsy Res. 2012, 98, 35–43. [Google Scholar] [CrossRef]
- Cuesta, R.; Laroia, G.; Schneider, R.J. Chaperone hsp27 inhibits translation during heat shock by binding eIF4G and facilitating dissociation of cap-initiation complexes. Genes Dev. 2000, 14, 1460–1470. [Google Scholar] [PubMed]
- Jänen, S.B.; Chaachouay, H.; Richter-Landsberg, C. Autophagy is activated by proteasomal inhibition and involved in aggresome clearance in cultured astrocytes. Glia 2010, 58, 1766–1774. [Google Scholar] [CrossRef]
- Haslbeck, M. sHsps and their role in the chaperone network. Cell. Mol. Life Sci. 2002, 59, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Goldbaum, O.; Riedel, M.; Stahnke, T.; Richter-Landsberg, C. The small heat shock protein HSP25 protects astrocytes against stress induced by proteasomal inhibition. Glia 2009, 57, 1566–1577. [Google Scholar] [CrossRef]
- Gaestel, M.; Gotthardt, R.; Müller, T. Structure and organisation of a murine gene encoding small heat-shock protein Hsp25. Gene 1993, 128, 279–283. [Google Scholar] [CrossRef]
- Chuang, J.Y.; Wang, S.A.; Yang, W.B.; Yang, H.C.; Hung, C.Y.; Su, T.P.; Chang, W.C.; Hung, J.J. Sp1 phosphorylation by cyclin-dependent kinase 1/cyclin B1 represses its DNA-binding activity during mitosis in cancer cells. Oncogene 2012, 31, 4946–4959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jutooru, I.; Chadalapaka, G.; Abdelrahim, M.; Basha, M.R.; Samudio, I.; Konopleva, M.; Andreeff, M.; Safe, S. Methyl 2-cyano-3,12-dioxooleana-1,9-dien-28-oate decreases specificity protein transcription factors and inhibits pancreatic tumor growth: Role of microRNA-27a. Mol. Pharmacol. 2010, 78, 226–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murashov, A.K.; Haq, I.U.; Hill, C.; Park, E.; Smith, M.; Wang, X.; Wang, X.; Goldberg, D.J.; Wolgemuth, D.J. Crosstalk between p38, Hsp25 and Akt in spinal motor neurons after sciatic nerve injury. Brain Res. Mol. Brain Res. 2001, 93, 199–208. [Google Scholar] [CrossRef]
- Yang, J.; Takahashi, Y.; Cheng, E.; Liu, J.; Terranova, P.F.; Zhao, B.; Thrasher, J.B.; Wang, H.G.; Li, B. GSK-3beta promotes cell survival by modulating Bif-1-dependent autophagy and cell death. J. Cell. Sci. 2010, 123, 861–870. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.S.; Kim, J.E. P2X7 receptor inhibits astroglial autophagy via regulating FAK- and PHLPP1/2-mediated AKT-S473 phosphorylation following kainic acid-induced seizures. Int. J. Mol. Sci. 2020, 21, 6476. [Google Scholar] [CrossRef]
- Wu, R.; Kausar, H.; Johnson, P.; Montoya-Durango, D.E.; Merchant, M.; Rane, M.J. Hsp27 regulates Akt activation and polymorphonuclear leukocyte apoptosis by scaffolding MK2 to Akt signal complex. J. Biol. Chem. 2007, 282, 21598–21608. [Google Scholar] [CrossRef] [Green Version]
- Havasi, A.; Li, Z.; Wang, Z.; Martin, J.L.; Botla, V.; Ruchalski, K.; Schwartz, J.H.; Borkan, S.C. Hsp27 inhibits Bax activation and apoptosis via a phosphatidylinositol 3-kinase-dependent mechanism. J. Biol. Chem. 2008, 283, 12305–12313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konishi, H.; Matsuzaki, H.; Tanaka, M.; Takemura, Y.; Kuroda, S.; Ono, Y.; Kikkawa, U. Activation of protein kinase B (Akt/RAC-protein kinase) by cellular stress and its association with heat shock protein Hsp27. FEBS Lett. 1997, 410, 493–498. [Google Scholar] [CrossRef] [Green Version]
- Son, T.W.; Yun, S.P.; Yong, M.S.; Seo, B.N.; Ryu, J.M.; Youn, H.Y.; Oh, Y.M.; Han, H.J. Netrin-1 protects hypoxia-induced mitochondrial apoptosis through HSP25 expression via DCC- and integrin α6β4-dependent AKT, GSK-3β, and HSF-1 in mesenchymal stem cells. Cell Death Dis. 2013, 4, e563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Marsh, D.J.; Kum, J.B.; Lunetta, K.L.; Bennett, M.J.; Gorlin, R.J.; Ahmed, S.F.; Bodurtha, J.; Crowe, C.; Curtis, M.A.; Dasouki, M.; et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum. Mol. Genet. 1999, 8, 1461–1472. [Google Scholar] [CrossRef]
- LaSarge, C.L.; Pun, R.Y.; Muntifering, M.B.; Danzer, S.C. Disrupted hippocampal network physiology following PTEN deletion from newborn dentate granule cells. Neurobiol. Dis. 2016, 96, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Santos, V.R.; Pun, R.Y.K.; Arafa, S.R.; LaSarge, C.L.; Rowley, S.; Khademi, S.; Bouley, T.; Holland, K.D.; Garcia-Cairasco, N.; Danzer, S.C. PTEN deletion increases hippocampal granule cell excitability in male and female mice. Neurobiol. Dis. 2017, 108, 339–351. [Google Scholar] [CrossRef]
- Zhu, J.M.; Li, K.X.; Cao, S.X.; Chen, X.J.; Shen, C.J.; Zhang, Y.; Geng, H.Y.; Chen, B.Q.; Lian, H.; Zhang, J.M.; et al. Increased NRG1-ErbB4 signaling in human symptomatic epilepsy. Sci. Rep. 2017, 7, 141. [Google Scholar] [CrossRef] [Green Version]
- Walz, W. Role of glial cells in the regulation of the brain ion microenvironment. Prog. Neurobiol. 1989, 33, 309–333. [Google Scholar] [CrossRef]
- D’Antuono, M.; Benini, R.; Biagini, G.; D’Arcangelo, G.; Barbarosie, M.; Tancredi, V.; Avoli, M. Limbic network interactions leading to hyperexcitability in a model of temporal lobe epilepsy. J. Neurophysiol. 2002, 87, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Brehme, H.; Kirschstein, T.; Schulz, R.; Köhling, R. In vivo treatment with the casein kinase 2 inhibitor 4,5,6,7-tetrabromotriazole augments the slow afterhyperpolarizing potential and prevents acute epileptiform activity. Epilepsia 2014, 55, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, D.K.; Yao, X.; Zador, Z.; Sick, T.J.; Verkman, A.S.; Manley, G.T. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia 2006, 53, 631–636. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-E.; Kang, T.-C. CDDO-Me Attenuates Astroglial Autophagy via Nrf2-, ERK1/2-SP1- and Src-CK2-PTEN-PI3K/AKT-Mediated Signaling Pathways in the Hippocampus of Chronic Epilepsy Rats. Antioxidants 2021, 10, 655. https://doi.org/10.3390/antiox10050655
Kim J-E, Kang T-C. CDDO-Me Attenuates Astroglial Autophagy via Nrf2-, ERK1/2-SP1- and Src-CK2-PTEN-PI3K/AKT-Mediated Signaling Pathways in the Hippocampus of Chronic Epilepsy Rats. Antioxidants. 2021; 10(5):655. https://doi.org/10.3390/antiox10050655
Chicago/Turabian StyleKim, Ji-Eun, and Tae-Cheon Kang. 2021. "CDDO-Me Attenuates Astroglial Autophagy via Nrf2-, ERK1/2-SP1- and Src-CK2-PTEN-PI3K/AKT-Mediated Signaling Pathways in the Hippocampus of Chronic Epilepsy Rats" Antioxidants 10, no. 5: 655. https://doi.org/10.3390/antiox10050655