1. Introduction
In order to adapt to a changing environment, cells continuously translate extracellular cues into appropriate cellular responses through cascades of protein-protein interactions and post-translational modifications known as signal transduction. A recently discovered form of signal transduction, termed redox signaling, uses hydrogen peroxide (H
2O
2) as a second messenger, and proceeds through the reversible oxidation of specific cysteine thiols in proteins (for a review, see ref. [
1]). To function as a reliable second messenger, H
2O
2 should be able to discriminate which cysteines it needs to oxidize specifically in order to trigger the proper signaling cascade. Although numerous H
2O
2-regulated proteins and processes have been discovered, it is unclear how exactly redox signaling achieves the required reactivity and specificity, which are fundamental requirements for coherent cellular signaling.
H
2O
2 is considered the major reactive oxygen species (ROS) for signaling because of its relative stability compared to other cellular reactive oxygen species (ROS, i.e., O
2•− and
•OH) [
2]. However, this relative stability also means that H
2O
2 reacts poorly with most cysteine thiols, with rate constants ranging from 20 to 200 M
−1s
−1 [
3,
4,
5]. Additionally, dedicated H
2O
2 scavengers like peroxiredoxins (PRDXs) are estimated to eliminate >99% of cellular H
2O
2 [
6], because their catalytic cysteines react with many orders of magnitude faster with H
2O
2 than other thiols in cysteine side chains in proteins, including those found to be redox regulated. Peroxiredoxins are highly abundant and ubiquitous proteins, with isoforms localized to cytoplasm, mitochondria, endoplasmic reticulum (ER) and other cellular compartments [
7]. The poor reactivity of thiols with H
2O
2 combined with the effective elimination of H
2O
2 by peroxiredoxins seems to challenge the idea that reactivity and selectivity in redox signaling can be achieved by a simple molecule like H
2O
2.
Peroxiredoxins do not only scavenge H
2O
2; in fact, oxidized 2-Cys peroxiredoxins have also been shown to act as peroxidases and facilitate H
2O
2-dependent protein oxidation via disulfide exchange reactions. For example, in
Saccharomyces cerevisiae, Tsa1 and Orp1 peroxidases relay towards the Yap1 transcription factor [
8] and a similar mechanism was identified for Sty1 in in
Schizosaccharomyces pombe [
9]. In mammalian cells, the ASK1 kinase and STAT3 transcription factor are oxidized by PRDX1 and PRDX2, respectively [
10,
11], and ER-localized PRDX4 is known to induce disulfide formation through the oxidation of protein disulfide isomerase (PDI) [
12].
Others have shown a more widespread role for peroxiredoxins in H
2O
2-induced thiol oxidation [
13,
14]. In this so-called peroxiredoxin-based relay model, the extremely reactive peroxidatic cysteine of peroxiredoxins first reacts with H
2O
2 and subsequently the oxidized peroxiredoxin catalyzes the oxidation of low reactivity thiols in redox-regulated proteins (see
Figure 1A). This mechanism could explain how so many intrinsically unreactive protein thiols can be found to be reversibly oxidized in response to H
2O
2-dependent redox signaling, despite the presence of a highly abundant and reactive H
2O
2 scavenging system.
Although the peroxiredoxin-based relay model may explain how the reactivity of H2O2 with protein thiols is overcome, it does not as yet explain how the selectivity in H2O2-dependent redox signaling is achieved. In order to produce relevant biological signals, selective substrate targeting is required to achieve proper signaling outputs. In redox signaling, this would mean that in the presence of numerous potential substrates, specific subsets of redox-regulated proteins should be oxidized dependent on, for instance, the subcellular localization or the local concentration of H2O2. Mammalian cells express five 2-Cys peroxiredoxin isoforms, each with their own localization, oxidation kinetics and structural differences around their catalytic sites. We therefore hypothesized that reactivity and selectivity in redox signaling could also be provided by the different 2-Cys peroxiredoxins.
According to this line of reasoning, peroxiredoxins would be expected to participate in temporary covalent complexes with isoform-specific subsets of target proteins, mediated by disulfides that form between their catalytic cysteine and a cysteine in these target proteins (see
Figure 1A). To test this hypothesis, we used a systematic mass-spectrometry-based approach to identify cysteine-dependent interactors of the five human 2-Cys peroxiredoxins. Indeed, our results suggest that all five human 2-Cys peroxiredoxins are capable of forming disulfide-dependent heterodimers with a large set of proteins, and that each peroxiredoxin isoform displays a preference for a subset of disulfide-dependent binding partners. We explore what isoform-specific properties underlie these observations and we provide evidence that peroxiredoxin-based redox relays can proceed via two distinct molecular mechanisms. These findings support the idea that peroxiredoxins could play a role in providing not only reactivity but also selectivity in the transduction of peroxide signals to generate complex cellular signaling responses.
2. Materials and Methods
2.1. Cell Lines and Culture
HEK293T cells (Manassas, VA, USA, ATCC)were cultured in bicarbonate-buffered DMEM (Basel, Switzerland, Lonza, BE12-604Q), supplemented with 10% FBS (Alkmaar, Netherlands, Bodinco BDC-40506-C05), 2 mM L-glutamine (Basel, Switzerland, Lonza, BE17-605E) and 100 U/mL penicillin-streptomycin (Basel, Switzerland, Lonza, DE17-602E) and kept at 37 °C and under a 6% CO2 atmosphere. Transfections of HEK293T cells were carried out using PEI (St. Louis, MO, USA, Sigma-Aldrich, P3640) or FugeneHD reagent (Madison, WI, USA, Promega, E2311) following the manufacturer’s instructions. After two days, cells were harvested for further analysis.
2.2. Plasmids and Reagents
Human PRDX1-5 with att recombination sites were cloned from cDNA using the following primers. PRDX1 (NCBI RefSeq NM_002574)):_Fwd 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTAATGTCTTCAGGAAATGCTAAAATTGGGC-3, Rev 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCCTACTTCTGCTTGGAGAAATATTCTTTGCT-3′. PRDX2 (NCBI RefSeq NM_005809): Fwd 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTAATGGCCTCCGGTAACGC-3′, Rev 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTCTAATTGTGTTTGGAGAAATATTCCTTGCTGT-3′. PRDX3 (NCBI RefSeq NM_006793): _Fwd 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTAATGGCGGCTGCTGTAGG-3′, Rev 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTCTACTGATTTACCTTCTGAAAGTACTCTTTGGAAG-3′. PRDX4 (NCBI RefSeq NM_006406):_Fwd 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTAATGGAGGCGCTGCCG-3′, Rev 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTTTTCAGTTTATCGAAATACTTCAGCTTTCCAG-3′. PRDX5 (NCBI RefSeq NM_0129094): Fwd 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTAATGGGACTAGCTGGCGTG-3′, Rev 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTTCAGAGCTGTGAGATGATATTGGGTG-3′. Using Gateway technology (Invitrogen, now owned by Waltham, MA, USA, Thermo Scientific) entry clones were generated. The peroxidatic and resolving cysteine mutants of PRDX1-5 were created by site-directed mutagenesis PCR using the following primers: PRDX1_C52S_F-5′-CTTTGTGTCCCCCACGGAG-3′, PRDX1_C52S._R-5′-CTCCGTGGGGGACACAAAG-3′, PRDX1_C173S_F-5′-GGGAAGTGTCCCCAGCTGG-3′, PRDX1_C173S._R-5′-CCAGCTGGGGACACTTCCC-3′, PRDX2_C51S_F-5′-TCACTTTTGTGTCTCCCACCGAGATCATCGCG-3′, PRDX2_C51S._R-5′-CGCGATGATCTCGGTGGGAGACACAAAAGTGA-3′, PRDX2_C172S_F-5′-CATGGGGAAGTTTCTCCCGCTGGCT-3′, PRDX2_C172S._R-5′-AGCCAGCGGGAGAAACTTCCCCATG-3′, PRDX3_C108S_F-5′-TCACCTTTGTGTCTCCTACAGAAATTGTTGCT-3′, PRDX3_C108S._R-5′-AGCAACAATTTCTGTAGGAGACACAAAGGTGA-3′, PRDX3_C229S_F-5′-ACACATGGAGAAGTCTCTCCAGCGAACTGGACA-3′, PRDX3_C229S._R-5′-TGTCCAGTTCGCTGGAGAGACTTCTCCATGTGT-3′, PRDX4_C124S_F-5′-ATTTCACATTTGTGTCTCCAACTGAAATTATCGCTTTTGG-3′, PRDX4_C124S._R-5′-CCAAAAGCGATAATTTCAGTTGGAGACACAAATGTGAAAT-3′, PRDX4_C245S_F-5′-GGAGAAGTCTCCCCTGCTGGCTGGAA-3′, PRDX4_C245S._R-5′-TTCCAGCCAGCAGGGGAGACTTCTCC-3′, PRDX5_C47S_F-5′-TTCACCCCTGGATCTTCCAAGACACACCTG-3′, PRDX5_C47S._R-5′-CAGGTGTGTCTTGGAAGATCCAGGGGTGAA-3′, PRDX5_C151S_F-5′-CAGGCCTCACCTCCAGCCTGGCA-3′, PRDX5_C151S._R-5′-TGCCAGGCTGGAGGTGAGGCCTG-3′. Gateway technology (Waltham, MA, USA, Thermo Scientific) was used to create N-terminally tagged FLAG-HIS-PRDX1-3 and 5, and C-terminally tagged PRDX4-FLAG-HIS constructs (backbone pcDNA3). Furthermore, 30% H2O2 (St. Louis, MO, USA, Sigma-Aldrich 31642) was freshly diluted to a stock of 10 or 100 mM in H2O for every experiment. Unless stated otherwise, H2O2 (St. Louis, MO, USA, Sigma-Aldrich) treatments were 25 μM (PRDX2) and 100 μM (other isoforms) for 2 min.
2.3. Co-Immunoprecipitation Experiments and Western Blotting
After treatment with H2O2, cells were scraped in 100 mM N-ethylmaleimide (NEM, St. Louis, MO, USA, Sigma-Aldrich E3876) in PBS for 5 min at 37 °C to trap free thiols in their in vivo redox state during sample preparation and collected by centrifugation at 1200 rpm for 3 min. Cells were lysed using a buffer containing 50 mM Tris-HCl pH 7.5, 1% TX100, 1.5 mM MgCl2, 5 mM EDTA, 100 mM NaCl, NaF, Leupeptin and Aprotinin (all from St. Louis, MO, USA, Sigma-Aldrich). Furthermore, 100 mM iodoacetamide (St. Louis, MO, USA, Sigma-Aldrich) was added to the lysis buffer to prevent post-lysis cysteine oxidation and to inactivate disulfide reducing enzymes. After centrifugation at 14,000 rpm for 10 min, 5% of the supernatant was kept as a control (denoted ‘input’ in figures) and the remaining supernatant was used for immunoprecipitation with anti-Flag-M2 affinity gel (St. Louis, MO, USA, Sigma-Aldrich A222). After a 2 h incubation, immunoprecipitates were washed 4 times with lysis buffer containing 1 M NaCl and samples were boiled for 5 min in sample buffer with or without the reducing agent β-mercaptoethanol. Samples were separated on a 10% polyacrylamide gel and transferred to immobilon-FL membranes (Burlington, MA, USA, Merck) before staining.
2.4. Antibodies
The following antibodies were used in this study: anti-Flag antibody and anti-Flag-M2 beads (St. Louis, MO, USA Sigma-Aldrich F3165 and A222, respectively), monoclonal anti-HA antibody (12CA5) was prepared using hybridoma cell lines, anti-tubulin (Burlington, MA, USA, Merck Millipore CP06), anti-peroxiredoxin-SO3 (Cambridge, MA, USA, Abcam ab16830), anti-peroxiredoxin 1 (Cambridge, MA, USA, Abcam ab15571), anti-peroxiredoxin 2 (Cambridge, MA, USA, Abcam ab15572), anti-peroxiredoxin 3 (Cambridge, MA, USA, Abcam ab73349), anti-peroxiredoxin 4 (Cambridge, MA, USA, Abcam ab59542) and anti-peroxiredoxin 5 (Cambridge, MA, USA, Abcam ab16944). Detection of 2 fluorescent secondary antibodies was done simultaneously using the LI-COR Biosciences Odyssey Infrared Imaging System or the Amersham Typhoon NIR Plus Biomolecular Imager (Chicago IL, USA, GE Healthcare), detection of HRP-coupled secondary antibodies was performed using the FUJIFILM Luminescent Image Analyzer LAS-3000.
2.5. Mass Spectrometry Sample Preparation
For the identification of cysteine-dependent interactors the lysate of 4 × 20 cm dishes were used for each pulldown on 75 μL of Flag-M2 beads similar to the immunoprecipitation experiments described above. All immunoprecipitations were performed using three biological replicates. After washing, beads were resuspended with 8 M urea in 1 M ammonium bicarbonate (ABC), reduced and alkylated in 10 mM TCEP and 40 mM chloroacetamide (CAA) for 30 min at RT. After fourfold dilution with 1 M ABC, proteins were digested overnight on-bead with 250 ng Trypsin/LysC (Madison, WI, USA, Promega V5071) per sample at 37 °C. Samples were cleaned up with in-house-made C18 stagetips.
2.6. Mass Spectrometry
Mass spectrometry was performed as previously described [
15]. Peptides were separated on a 30-cm pico-tip column (75 μm ID, New Objective) and were packed in-house with 3 μm aquapur gold C-18 material (Dr. Maisch) using a 140-min gradient (7–80% ACN 0.1% FA), delivered by an easy-nLC 1000 (LC 120, Waltham, MA, USA, Thermo Scientific), and electro-sprayed directly into an Orbitrap Fusion Tribrid Mass Spectrometer (LC 120, Waltham, MA, USA, Thermo Scientific). Raw files were analyzed with MaxQuant software version 1.5.2.8 with oxidation of methionine, alkylation with N-ethylmaleimide and carbamidomethylation set as variable modifications. The human protein database of UniProt was searched with both the peptide as well as the protein false discovery rate set to 1%. The mass spectrometry proteomics data were uploaded into the ProteomeXchange Consortium via the PRIDE [
16] partner repository with the dataset identifier PXD024114. Downstream analysis was done using R version 4.0.2.
2.7. Data Filtering and proDA Modeling
The code used was uploaded to GitHub at
https://github.com/loesoe/peroxiredoxin (accessed on 2 July 2020). In short, LFQ data from the MaxQuant proteinGroups file and corresponding protein information was used. Proteins were filtered for reverse hits and standard contaminants. Next, we selected proteins that were identified with three or more unique peptides and were measured in at least one sample in two or more replicates. Data was log
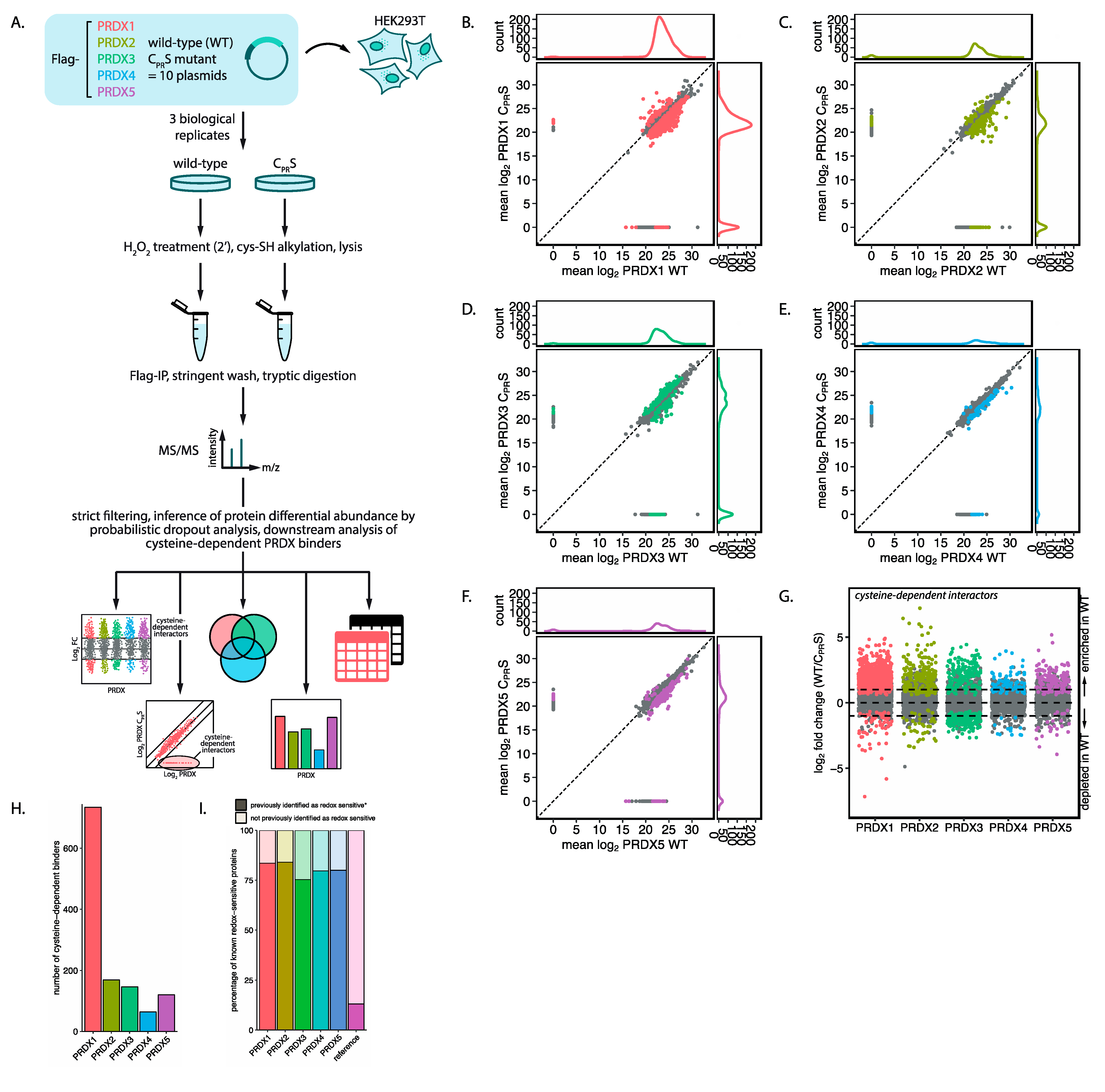
2-transformed and normalized using quantile normalization to simultaneously correct for overall protein content and immunoprecipitation (IP) efficiency. ProDA model fitting was performed using the number of proteins in the data as the number of subsamples. To test for differential protein abundance, a proDa model was fit to compare WT against mutant for each peroxiredoxin.
2.8. Threshold Cutoff Calculation
To determine the cutoff for out data, we fitted a proDA model for wild-type and mutant peroxiredoxin, without considering the isoform. Next, we repeated this 100 times, but instead with randomized labels. The difference at which 5% of the randomized data was included was determined as the cutoff.
2.9. Known Redox-Sensitive Proteins
A reference proteome containing 75,071 human entries from Uniprot tagged with the keyword reference proteome was downloaded from
https://www.uniprot.org/help/reference_proteome (accessed on 2 August 2020). Proteins that were previously identified in a screen for redox-sensitive proteins in human cell lines (HEK293T and HCT116) were taken from [
17].
2.10. Upset Plot and Venn Diagrams
To visualize the isoform-specific set of intersections we used the UpsetR package [
18]. Venn diagrams were created using the Venn version 1.9 package and colored manually.
2.11. Localization Analysis
We analyzed the localization of isoform-specific interactors using the neighborhood/compartment predictions data for A431 cells from
https://www.subcellbarcode.org (accessed on 2 January 2020) [
19]. Peroxiredoxin interactors with the highest fold change (>10-fold) were matched with their neighborhood data and their fold enrichment were calculated compared to the cell line data. Main localization of peroxiredoxin isoforms from this tool are as follows: PRDX1 PRDX2 and PRDX5, cytosol; PRDX3, mitochondria; PRDX4, unclassified.
2.12. Sequence Similarity
PRDX Sequences were loaded as a FASTA file. Pairwise alignment was calculated using EMBOSS needle (
https://www.ebi.ac.uk/Tools/psa/emboss_needl, accessed on 2 July 2016), which uses the Needleman–Wunsch global alignment algorithm to find the optimum global alignment. Similarity (the percentage of matches between the two aligned sequences) was plotted for each peroxiredoxin isoform pair.
2.13. AA Composition and Motifs
The sequences for PRDX isoform-specific interactors were retrieved from Uniprot (
https://www.uniprot.org/uploadlists, accessed on 2 January 2021). Sequences were loaded using the Biostrings package, sequences were shuffled 100 times as a background using the universalmotif package. Motifs were extracted of 9 amino acids centered around each cysteine. The amino acid composition was calculated for the isoform-specific sequences, the shuffled background and total protein using the alphabetFrequency function. Fold enrichment and Benjamini–Hochberg adjusted
p-values were calculated per peroxiredoxin isoform using the amino acid composition of all other isoforms as a control. Motifs were analyzed using motif-x (rmotifx package) using the shuffled sequences as a background [
20].
4. Discussion
Peroxiredoxin-catalyzed oxidation has been suggested to answer the question as to how thiols with low intrinsic reactivity can be oxidized by low levels of H
2O
2 despite the presence of abundant and highly reactive peroxidases. Here we show that all peroxiredoxin isoforms are capable of forming numerous cysteine-dependent heterodimers. Our in-depth mass-spectrometry and complementary bioinformatics approach provides, first of all, a resource of potential 2-Cys peroxiredoxin-catalyzed cysteine oxidation substrates. Many of the proteins that we identified as cysteine-dependent peroxiredoxin binders were indeed identified previously to contain redox sensitive cysteines [
17]. This overlap could point at a major role for peroxiredoxins in cysteine oxidation in other proteins. It is not clear what follows after peroxiredoxin-dependent cysteine oxidation, but one could think of three possible scenarios following intermolecular disulfide formation between peroxiredoxin and a target protein. 1) The intermolecular disulfide could be rapidly resolved by disulfide exchange to the resolving cysteine of the peroxiredoxin, forming the canonical C
P-S-S-C
R and leaving the target reduced. 2) The intermolecular disulfide could be resolved by disulfide exchange to another cysteine in the binding protein (or protein complex), forming an intra- or intermolecular disulfide in that protein and leaving peroxiredoxin reduced. 3) The intermolecular disulfide dependent complex of peroxiredoxin and its target could represent a novel type of post-translational modification on cysteine, that for instance alters the function of the target, that we would like to coin S-peroxiredoxinylation (S-PRDXylation). Others have shown that many of the PRDX1 and PRDX2-dependent binders were also identified in a TRX-trap pull down, confirming that proteins binding to peroxiredoxin in a cysteine-dependent manner indeed become oxidized [
13]. This observation probably does not exclude S-PRDXylation. Widespread S-PRDXylation could also be in accordance with the identification of many oxidation-sensitive cysteines in redox proteomics studies, as these would not distinguish S-PRDXylation from other intra- or intermolecular disulfides [
28]. Besides a role as a post-translational modification impacting the function of specific proteins, widespread peroxiredoxinylation could in principle also serve as a redox buffer.
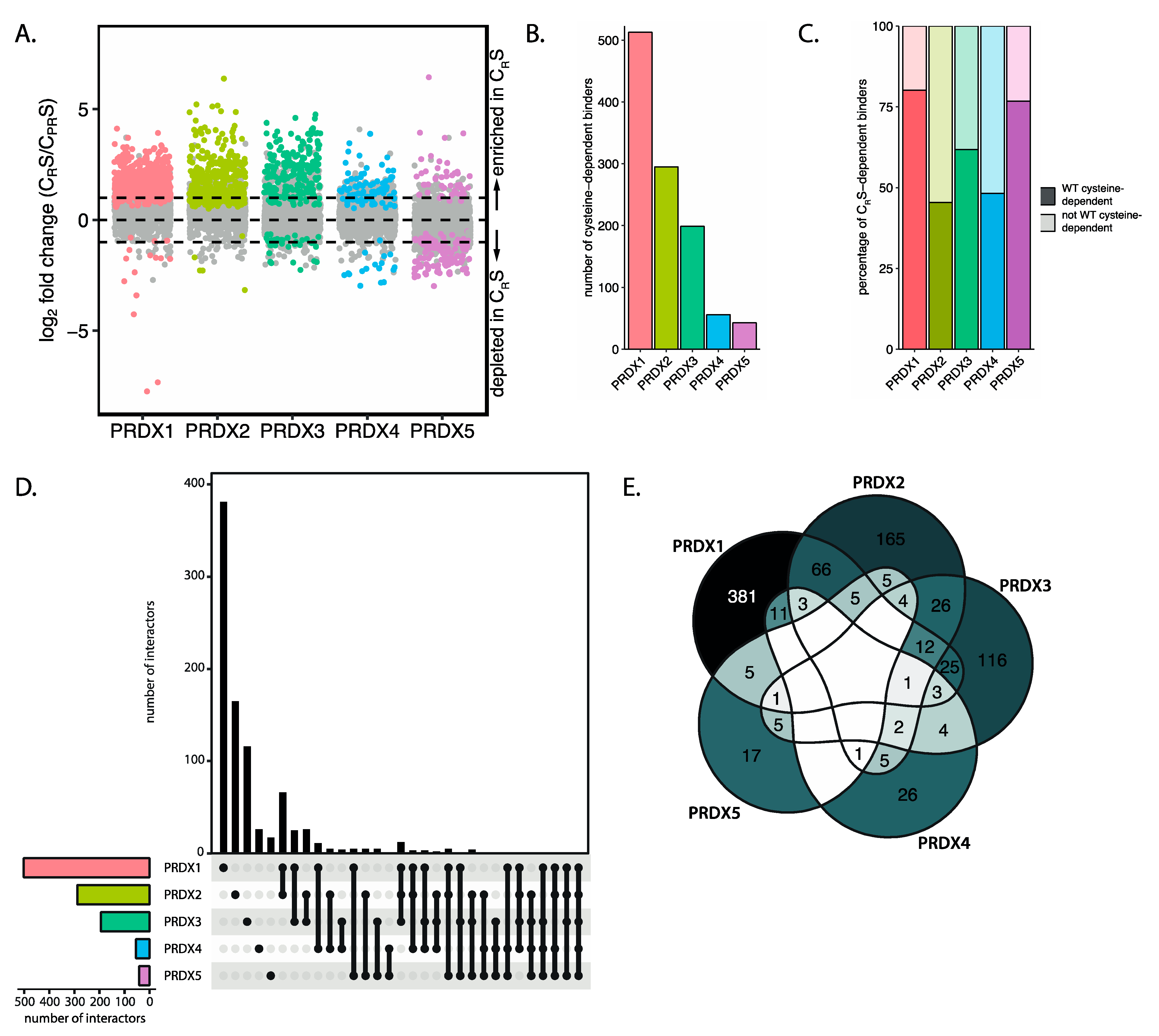
Our data furthermore provides evidence that the peroxiredoxin-dependent redox relay model could also explain how selectivity in redox signaling can be achieved. Selectivity stems from the observation that each peroxiredoxin isoform interacts with a largely specific subset of proteins. This could in part depend on isoform-specific subcellular localization, but the relatively low overlap in binders for PRDX1 and PRDX2, which share the same subcellular localization and a high sequence similarity, suggests that this is not the only determinant for binding of a protein to a specific peroxiredoxin isoform. Analysis of local structural differences surrounding the region around the cysteine of the binding protein could also contribute to selective binding of proteins to the different peroxiredoxins. A second layer of specificity is suggested by the observation that peroxiredoxin-mediated relays can proceed through two distinct molecular mechanisms, starting from either CP-SOH or CP-S-S-CR, and that peroxiredoxin isoforms and targets display varying preferences for these mechanisms. Each peroxiredoxin has different kinetics for CP-SOH and CP-S-S-CR formation and reduction, and these kinetics could dictate which cysteines in target proteins can be oxidized under specific conditions. For instance, at low levels of peroxide, PRDX2 would be the first to form CP-SOH, whereas only under conditions where TRX activity is limiting, oxidized peroxiredoxin in the CP-S-S-CR form would be sufficiently abundant to oxidize another set of targets. Interestingly, when we look at isoform-specific differences in the frequency of CP-SOH and CP-S-S-CR-mediated interactors, we find that there are large differences in the distribution of these relay mechanisms between isoforms.
A fair number of proteins seems to bind peroxiredoxins independent of its catalytic cysteines (
Figure 3L, grey dots), despite high-salt washing. The peroxiredoxin isoform dependent specificity irrespective of cysteine-dependency suggests that these interactions are probably not artefacts of the used method. This leaves the possibility that some of these proteins could function as adaptor proteins to facilitate peroxiredoxin-dependent relays to cysteine-dependent binding proteins. Although this would need to be explored, adaptor proteins have been shown to be involved in peroxidase-dependent redox relays. For instance, Orp1-dependent Yap1 oxidation is dependent on the presence of the adapter protein Ybp1, shielding oxidized Orp1 from reduction [
29,
30]. Similarly, the PRDX2-STAT2 redox relay depends on association with the membrane-associated scaffold protein ANXA2 [
31]. We indeed also identify ANXA2 as a PRDX2-specific and cysteine-dependent interactor in our screen (
Table S2). It is conceivable that many more peroxiredoxin-based redox relays may proceed via the formation of ternary complexes with scaffold proteins. This would not only increase the chances that a peroxiredoxin finds a target, but would also add another level of specificity, coming from the interaction of specific peroxiredoxin isoforms with specific scaffolds for the relay of oxidation to subsets of target proteins.
Taken together, our observations regarding widespread cysteine-dependent binding of proteins to the 2-Cys peroxiredoxins provides a model that could explain both the reactivity and selectivity of the extensive cysteine oxidation observed in response to low amounts of H2O2.
6. Limitations of this Study
The cut-offs used in the analysis of our mass-spectrometry screen are quite stringent, and whether proteins bind only in a cysteine-dependent manner or to only a certain peroxiredoxin isoform may not be as unambiguous. Furthermore, to keep the number of mass-spectrometry samples manageable the analysis was performed at a single timepoint following a single concentration of H2O2 treatment. It is not unthinkable that proteins found to interact specifically to one peroxiredoxin isoform in this study will in fact interact with others when analyzed at other timepoints or H2O2 concentrations. The use of the H2O2 treated CPRS mutants as a control rather than also including untreated samples for WT PRDX1–5 (for the same reason of keeping the number of mass-spectrometry samples manageable) may obscure whether proteins also bind peroxiredoxins under basal conditions, be it cysteine dependent or not. Future work will be needed to carefully validate each protein found as a cysteine-dependent peroxiredoxin interactor.
It is not unthinkable that differences in the level of overexpression of the Flag-tagged PRDX1–5 or their mutants may lead to variation in the number of proteins pulled down. In general, the C
PRS mutants of each PRDX1–5 isoform had very similar expression and IP efficiency as compared to their wildtype isoform counterparts (see for instance
Figure 1C, reducing IP and input), suggesting that whether an interactor is identified as a cysteine-dependent binder is not much affected by variable expression levels. The levels of overexpression of Flag-PRDX1–5 compared to each are somewhat variable, and for his reason MS/MS data was log
2-transformed followed by quantile normalization to simultaneously correct for overall protein content and IP efficiency in an attempt to lower the chance that differences in expression levels affect our analysis.
The analysis of the chemical environment of cysteines oxidized through peroxiredoxin dependent relays would greatly benefit from knowing which cysteine in a binding partner is being oxidized. Here we have analyzed all cysteines in the interactors which obviously dilutes any specific pattern. Combining peroxiredoxin-interactome screens as described here with redox proteomics or ways to keep the disulfide between peroxiredoxin and its targets intact and suitable for analysis by MS/MS in future studies could be a way to achieve this. It is difficult to unambiguously exclude that proteins no longer bind to the used peroxiredoxin mutants due to structural changes other than loss of the cysteine thiol. For the resolving cysteine mutants at least, a recent study shows that the cysteine to serine mutation has only a limited effect on the rate of oxidation of the peroxidatic cysteine in PRDX2 [
32]. Characterization of the functional consequences of specific peroxiredoxin-based interactions is outside the scope of this study. However, it would be interesting to investigate the mechanisms and fate of these complexes in more detail. Additionally, further work is needed to link the mechanisms of peroxiredoxin specificity to biological cues that determine downstream signaling.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




