
Stoichiometric Thiol Redox Proteomics for Quantifying Cellular Responses to Perturbations


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Post-Translational Modifications (PTMs) of Protein Cysteine Thiols
1.2. Maintenance of the Cellular Redox State
1.3. General Approaches for Measuring Redox PTMs
2. Stoichiometric Quantification of Thiol Redox Modifications Using Mass Spectrometry
2.1. PTM Stoichiometry
2.2. Applying Stoichiometry to Redox Proteomics
2.3. Methods That Measure Stoichiometry in Thiol Redox Proteomics
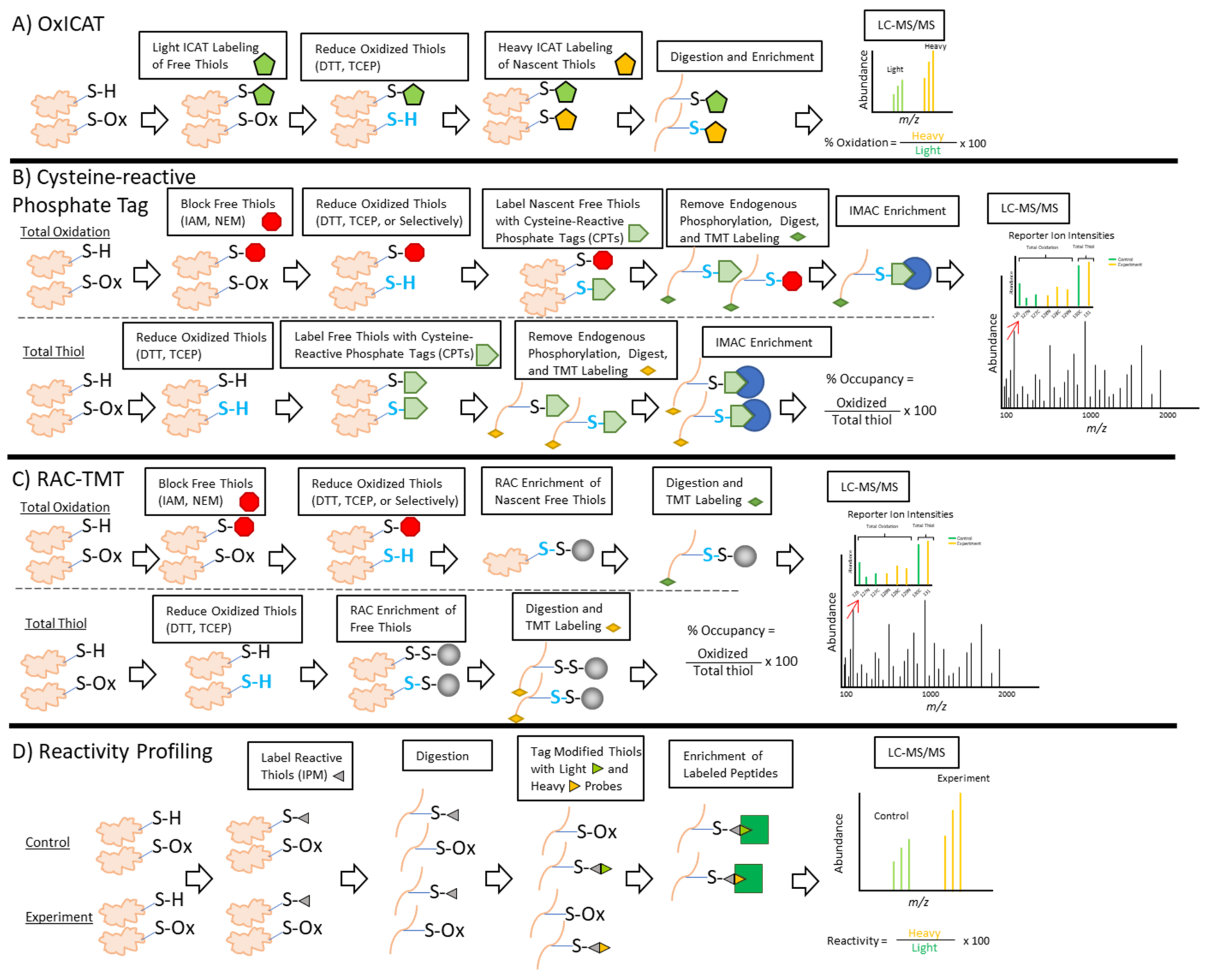
2.3.1. OxICAT
2.3.2. Cysteine-Reactive Phosphate Tags
2.3.3. RAC
2.3.4. Thiol Reactivity Profiling
2.4. Caveats of Current Quantitative Approaches
3. Applications of Thiol Redox Proteomics
3.1. Integrative Studies
3.2. Structural Insight
3.3. Potential Health and Clinical Applications
4. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Miseta, A.; Csutora, P. Relationship Between the Occurrence of Cysteine in Proteins and the Complexity of Organisms. Mol. Biol. Evol. 2000, 17, 1232–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leichert, L.I.; Dick, T.P. Incidence and physiological relevance of protein thiol switches. Biol. Chem. 2015, 396, 389–399. [Google Scholar] [CrossRef]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free. Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Winterbourn, C.C.; Hampton, M.B. Thiol chemistry and specificity in redox signaling. Free. Radic. Biol. Med. 2008, 45, 549–561. [Google Scholar] [CrossRef]
- Klomsiri, C.; Karplus, P.A.; Poole, L.B. Cysteine-Based Redox Switches in Enzymes. Antioxid. Redox Signal. 2011, 14, 1065–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bak, D.W.; Bechtel, T.J.; Falco, A.J.; Weerapana, E. Cysteine reactivity across the subcellular universe. Curr. Opin. Chem. Biol. 2019, 48, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Bachi, A.; Dalle-Donne, I.; Scaloni, A. Redox Proteomics: Chemical Principles, Methodological Approaches and Biological/Biomedical Promises. Chem. Rev. 2013, 113, 596–698. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.-M.; Jones, D.P. Thiol/disulfide redox states in signaling and sensing. Crit. Rev. Biochem. Mol. Biol. 2012, 48, 173–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernando, V.; Zheng, X.; Walia, Y.; Sharma, V.; Letson, J.; Furuta, S. S-Nitrosylation: An Emerging Paradigm of Redox Signaling. Antioxidants 2019, 8, 404. [Google Scholar] [CrossRef] [Green Version]
- Devarie-Baez, N.O.; Lopez, E.I.S.; Furdui, C.M. Biological chemistry and functionality of protein sulfenic acids and related thiol modifications. Free. Radic. Res. 2016, 50, 172–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grek, C.L.; Zhang, J.; Manevich, Y.; Townsend, D.M.; Tew, K.D. Causes and Consequences of Cysteine S-Glutathionylation. J. Biol. Chem. 2013, 288, 26497–26504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasamatsu, S.; Nishimura, A.; Morita, M.; Matsunaga, T.; Hamid, H.A.; Akaike, T. Redox Signaling Regulated by Cysteine Persulfide and Protein Polysulfidation. Molecules 2016, 21, 1721. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Zhi, X.; Wang, X.; Meng, D. Protein palmitoylation and its pathophysiological relevance. J. Cell. Physiol. 2021, 236, 3220–3233. [Google Scholar] [CrossRef]
- Musaogullari, A.; Chai, Y.-C. Redox Regulation by Protein S-Glutathionylation: From Molecular Mechanisms to Implications in Health and Disease. Int. J. Mol. Sci. 2020, 21, 8113. [Google Scholar] [CrossRef] [PubMed]
- Van Bergen, L.A.H.; Roos, G.; De Proft, F. From Thiol to Sulfonic Acid: Modeling the Oxidation Pathway of Protein Thiols by Hydrogen Peroxide. J. Phys. Chem. A 2014, 118, 6078–6084. [Google Scholar] [CrossRef]
- Chauvin, J.-P.R.; Pratt, D.A. On the Reactions of Thiols, Sulfenic Acids, and Sulfinic Acids with Hydrogen Peroxide. Angew. Chem. Int. Ed. 2017, 56, 6255–6259. [Google Scholar] [CrossRef]
- Beedle, A.E.M.; Lynham, S.; Garcia-Manyes, A.E.M.B.S. Protein S-sulfenylation is a fleeting molecular switch that regulates non-enzymatic oxidative folding. Nat. Commun. 2016, 7, 12490. [Google Scholar] [CrossRef] [Green Version]
- Akter, S.; Fu, L.; Jung, Y.; Conte, M.L.; Lawson, J.R.; Lowther, W.T.; Sun, R.; Liu, K.; Yang, J.; Carroll, K.S. Chemical proteomics reveals new targets of cysteine sulfinic acid reductase. Nat. Chem. Biol. 2018, 14, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.S.H.; Higgins, M.; Hams, E.; Szpyt, J.; Runtsch, M.C.; King, M.S.; McGouran, J.F.; Fischer, R.; Kessler, B.M.; McGettrick, A.F.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar]
- Bambouskova, M.; Gorvel, L.; Lampropoulou, V.; Sergushichev, A.; Loginicheva, E.; Johnson, K.; Korenfeld, D.; Mathyer, M.E.; Kim, H.; Huang, L.-H.; et al. Electrophilic properties of itaconate and derivatives regulate the IκBζ-ATF3 inflammatory axis. Nat. Cell Biol. 2018, 556, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, R.A.; Montgomery, D.C.; Meier, J.L. Epigenetic regulation by endogenous metabolite pharmacology. Curr. Opin. Chem. Biol. 2019, 51, 30–39. [Google Scholar] [CrossRef]
- Harmel, R.; Fiedler, D. Features and regulation of non-enzymatic post-translational modifications. Nat. Chem. Biol. 2018, 14, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Hoch, D.G.; Abegg, D.; Adibekian, A. Cysteine-reactive probes and their use in chemical proteomics. Chem. Commun. 2018, 54, 4501–4512. [Google Scholar] [CrossRef]
- Su, D.; Shukla, A.K.; Chen, B.; Kim, J.-S.; Nakayasu, E.; Qu, Y.; Aryal, U.; Weitz, K.; Clauss, T.R.; Monroe, M.E.; et al. Quantitative site-specific reactivity profiling of S-nitrosylation in mouse skeletal muscle using cysteinyl peptide enrichment coupled with mass spectrometry. Free. Radic. Biol. Med. 2013, 57, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Lia, A.; Dowle, A.; Taylor, C.; Santino, A.; Roversi, P. Partial catalytic Cys oxidation of human GAPDH to Cys-sulfonic acid. Wellcome Open Res. 2020, 5, 114. [Google Scholar] [CrossRef] [PubMed]
- Barinova, K.; Serebryakova, M.; Muronetz, V.; Schmalhausen, E. S-glutathionylation of glyceraldehyde-3-phosphate dehydrogenase induces formation of C150-C154 intrasubunit disulfide bond in the active site of the enzyme. Biochim. Biophys. Acta BBA Gen. Subj. 2017, 1861, 3167–3177. [Google Scholar] [CrossRef]
- Zaffagnini, M.; Marchand, C.H.; Malferrari, M.; Murail, S.; Bonacchi, S.; Genovese, D.; Montalti, M.; Venturoli, G.; Falini, G.; Baaden, M.; et al. Glutathionylation primes soluble glyceraldehyde-3-phosphate dehydrogenase for late collapse into insoluble aggregates. Proc. Natl. Acad. Sci. USA 2019, 116, 26057–26065. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Itakura, M.; Kubo, T.; Kaneshige, A.; Harada, N.; Izawa, T.; Azuma, Y.-T.; Kuwamura, M.; Yamaji, R.; Takeuchi, T. Glyceraldehyde-3-phosphate Dehydrogenase (GAPDH) Aggregation Causes Mitochondrial Dysfunction during Oxidative Stress-induced Cell Death. J. Biol. Chem. 2017, 292, 4727–4742. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, J.L.; Tanner, J.J. High-resolution structure of humanD-glyceraldehyde-3-phosphate dehydrogenase. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 290–301. [Google Scholar] [CrossRef]
- Heppner, D.E.; Hristova, M.; Dustin, C.M.; Danyal, K.; Habibovic, A.; van der Vliet, A. The NADPH Oxidases DUOX1 and NOX2 Play Distinct Roles in Redox Regulation of Epidermal Growth Factor Receptor Signaling. J. Biol. Chem. 2016, 291, 23282–23293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, S.; Kukimoto-Niino, M.; Parker, L.; Handa, N.; Terada, T.; Fujimoto, T.; Terazawa, Y.; Wakiyama, M.; Sato, M.; Sano, S.; et al. Structural basis for the altered drug sensitivities of non-small cell lung cancer-associated mutants of human epidermal growth factor receptor. Oncogene 2013, 32, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, T.; Yasuda, H.; Hamamoto, J.; Nukaga, S.; Masuzawa, K.; Kawada, I.; Naoki, K.; Niimi, T.; Mimasu, S.; Sakagami, H.; et al. Pharmacological and Structural Characterizations of Naquotinib, a Novel Third-Generation EGFR Tyrosine Kinase Inhibitor, in EGFR-Mutated Non-Small Cell Lung Cancer. Mol. Cancer Ther. 2018, 17, 740–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulsen, E.C.; Truong, T.H.; Garcia, F.J.; Homann, A.; Gupta, V.; Leonard, S.E.; Carroll, K.S. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 2011, 8, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Truong, T.H.; Ung, P.M.-U.; Palde, P.B.; Paulsen, C.E.; Schlessinger, A.; Carroll, K.S. Molecular Basis for Redox Activation of Epidermal Growth Factor Receptor Kinase. Cell Chem. Biol. 2016, 23, 837–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Little, A.C.; Hristova, M.; Van Lith, L.; Schiffers, C.; Dustin, C.M.; Habibovic, A.; Danyal, K.; Heppner, D.E.; Lin, M.-C.J.; Van Der Velden, J.; et al. Dysregulated Redox Regulation Contributes to Nuclear EGFR Localization and Pathogenicity in Lung Cancer. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, P.A.; Kuzmic, P.; Solowiej, J.; Bergqvist, S.; Bolanos, B.; Almaden, C.; Nagata, A.; Ryan, K.; Feng, J.; Dalvie, D.; et al. Covalent EGFR inhibitor analysis reveals importance of reversible interactions to potency and mechanisms of drug resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Heppner, D.E.; van der Vliet, A. Redox-dependent regulation of epidermal growth factor receptor signaling. Redox Biol. 2016, 8, 24–27. [Google Scholar] [CrossRef] [Green Version]
- Kemp, M.; Go, Y.-M.; Jones, D.P. Nonequilibrium thermodynamics of thiol/disulfide redox systems: A perspective on redox systems biology. Free Radic. Biol. Med. 2008, 44, 921–937. [Google Scholar] [CrossRef] [Green Version]
- Held, J.M. Redox Systems Biology: Harnessing the Sentinels of the Cysteine Redoxome. Antioxid. Redox Signal. 2020, 32, 659–676. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxidative Med. Cell. Longev. 2016, 2016, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.-H.; Li, L.; Parisien, M.; Wu, J.; Bederman, I.; Gao, Z.; Krokowski, D.; Chirieleison, S.M.; Abbott, D.W.; Wang, B.; et al. Discovery of a Redox Thiol Switch: Implications for Cellular Energy Metabolism. Mol. Cell. Proteom. 2020, 19, 852–870. [Google Scholar] [CrossRef] [Green Version]
- Janssen-Heininger, Y.M.; Mossman, B.T.; Heintz, N.H.; Forman, H.J.; Kalyanaraman, B.; Finkel, T.; Stamler, J.S.; Rhee, S.G.; van der Vliet, A. Redox-based regulation of signal transduction: Principles, pitfalls, and promises. Free. Radic. Biol. Med. 2008, 45, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, Y.-M.; Jones, D.P. The Redox Proteome. J. Biol. Chem. 2013, 288, 26512–26520. [Google Scholar] [CrossRef] [Green Version]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Secchi, C.; Carta, M.; Crescio, C.; Spano, A.; Arras, M.; Caocci, G.; Galimi, F.; La Nasa, G.; Pippia, P.; Turrini, F.; et al. T cell tyrosine phosphorylation response to transient redox stress. Cell. Signal 2015, 27, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Secchi, C.; Orecchioni, M.; Carta, M.; Galimi, F.; Turrini, F.; Pantaleo, A. Signaling Response to Transient Redox Stress in Human Isolated T Cells: Molecular Sensor Role of Syk Kinase and Functional Involvement of IL2 Receptor and L-Selectine. Sensors 2020, 20, 466. [Google Scholar] [CrossRef] [Green Version]
- Pires, P.W.; Earley, S. Redox regulation of transient receptor potential channels in the endothelium. Microcirculation 2017, 24, e12329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozai, D.; Ogawa, N.; Mori, Y. Redox Regulation of Transient Receptor Potential Channels. Antioxid. Redox Signal. 2014, 21, 971–986. [Google Scholar] [CrossRef] [Green Version]
- Benhar, M. Oxidants, Antioxidants and Thiol Redox Switches in the Control of Regulated Cell Death Pathways. Antioxidants 2020, 9, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sart, S.; Song, L.; Li, Y. Controlling Redox Status for Stem Cell Survival, Expansion, and Differentiation. Oxidative Med. Cell. Longev. 2015, 2015, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höhn, A.; König, J.; Grune, T. Protein oxidation in aging and the removal of oxidized proteins. J. Proteom. 2013, 92, 132–159. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, I.; Mullen, L.; Bekeschus, S.; Hanschmann, E.-M. Redox Regulation of Inflammatory Processes Is Enzymatically Controlled. Oxidative Med. Cell. Longev. 2017, 2017, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Van Der Reest, J.; Lilla, S.; Zheng, L.; Zanivan, S.; Gottlieb, E. Proteome-wide analysis of cysteine oxidation reveals metabolic sensitivity to redox stress. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Baba, S.P.; Bhatnagar, A. Role of thiols in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 133–139. [Google Scholar] [CrossRef]
- Ulrich, K.; Jakob, U. The role of thiols in antioxidant systems. Free Radic. Biol. Med. 2019, 140, 14–27. [Google Scholar] [CrossRef]
- Jakubczyk, K.; Kałduńska, J.; Dec, K.; Kawczuga, D.; Janda, K. Antioxidant properties of small-molecule non-enzymatic compounds. Pol. Merkur. Lekarski 2020, 48, 128–132. [Google Scholar] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2015, 15, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef] [Green Version]
- Mashamaite, L.N.; Rohwer, J.M.; Pillay, C.S. The glutaredoxin mono- and di-thiol mechanisms for deglutathionylation are functionally equivalent: Implications for redox systems biology. Biosci. Rep. 2015, 35, 00173. [Google Scholar] [CrossRef] [PubMed]
- Begas, P.; Liedgens, L.; Moseler, A.; Meyer, A.J.; DePonte, M. Glutaredoxin catalysis requires two distinct glutathione interaction sites. Nat. Commun. 2017, 8, 14835. [Google Scholar] [CrossRef] [PubMed]
- Matsui, R.; Ferran, B.; Oh, A.; Croteau, D.; Shao, D.; Han, J.; Pimentel, D.R.; Bachschmid, M.M. Redox RegulationviaGlutaredoxin-1 and ProteinS-Glutathionylation. Antioxid. Redox Signal. 2020, 32, 677–700. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Parrott, A.M.; Fu, C.; Liu, T.; Marino, S.M.; Gladyshev, V.N.; Jain, M.R.; Baykal, A.T.; Li, Q.; Oka, S.; et al. Thioredoxin 1-Mediated Post-Translational Modifications: Reduction, Transnitrosylation, Denitrosylation, and Related Proteomics Methodologies. Antioxid. Redox Signal. 2011, 15, 2565–2604. [Google Scholar] [CrossRef] [Green Version]
- Conte, M.L.; Carroll, K.S. The Redox Biochemistry of Protein Sulfenylation and Sulfinylation. J. Biol. Chem. 2013, 288, 26480–26488. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Collet, J.-F.; Messens, J. Structure, Function, and Mechanism of Thioredoxin Proteins. Antioxid. Redox Signal. 2010, 13, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.; Bae, S.H.; Toledano, M.B.; Rhee, S.G. Role of sulfiredoxin as a regulator of peroxiredoxin function and regulation of its expression. Free Radic. Biol. Med. 2012, 53, 447–456. [Google Scholar] [CrossRef]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, M.; Jiang, H.; Wu, L.; Chawsheen, H.A.; Wei, Q. The sulfiredoxin-peroxiredoxin (Srx-Prx) axis in cell signal transduction and cancer development. Cancer Lett. 2015, 366, 150–159. [Google Scholar] [CrossRef] [Green Version]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, C. Redox signalling via the cellular thiolstat. Biochem. Soc. Trans. 2011, 39, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.; Battaglia, E.; Burkholz, T.; Peng, D.; Bagrel, D.; Montenarh, M. Control of Oxidative Posttranslational Cysteine Modifications: From Intricate Chemistry to Widespread Biological and Medical Applications. Chem. Res. Toxicol. 2011, 25, 588–604. [Google Scholar] [CrossRef]
- Jones, D.P.; Sies, H. The Redox Code. Antioxid. Redox Signal. 2015, 23, 734–746. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P. Disruption of mitochondrial redox circuitry in oxidative stress. Chem. Interact. 2006, 163, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P.; Go, Y.-M.; Anderson, C.L.; Ziegler, T.R.; Kinkade, J.J.M.; Kirlin, W.G. Cysteine/cystine couple is a newly recognized node in the circuitry for biologic redox signaling and control. FASEB J. 2004, 18, 1246–1248. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P.; Go, Y.-M. Redox compartmentalization and cellular stress. Diabetes Obes. Metab. 2010, 12, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, Y.-M.; Jones, D.P. Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta BBA Gen. Subj. 2008, 1780, 1273–1290. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.M.; Go, Y.-M.; Jones, D.P. Nuclear and Mitochondrial Compartmentation of Oxidative Stress and Redox Signaling. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Lismont, C.; Nordgren, M.; Van Veldhoven, P.P.; Fransen, M. Redox interplay between mitochondria and peroxisomes. Front. Cell Dev. Biol. 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Jiang, J.; Lei, Y.; Zhou, S.; Wei, Y.; Huang, C. Targeting Metabolic-Redox Circuits for Cancer Therapy. Trends Biochem. Sci. 2019, 44, 401–414. [Google Scholar] [CrossRef]
- Antunes, F.; Brito, P.M. Quantitative biology of hydrogen peroxide signaling. Redox Biol. 2017, 13, 1–7. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Pillay, C.S.; Eagling, B.D.; Driscoll, S.R.; Rohwer, J.M. Quantitative measures for redox signaling. Free Radic. Biol. Med. 2016, 96, 290–303. [Google Scholar] [CrossRef]
- Posen, Y.; Kalchenko, V.; Seger, R.; Brandis, A.; Scherz, A.; Salomon, Y. Manipulation of redox signaling in mammalian cells enabled by controlled photogeneration of reactive oxygen species. J. Cell Sci. 2005, 118, 1957–1969. [Google Scholar] [CrossRef] [Green Version]
- Duan, J.; Kodali, V.K.; Gaffrey, M.J.; Guo, J.; Chu, R.K.; Camp, D.G.; Smith, R.D.; Thrall, B.D.; Qian, W.-J. Quantitative Profiling of Protein S-Glutathionylation Reveals Redox-Dependent Regulation of Macrophage Function during Nanoparticle-Induced Oxidative Stress. ACS Nano 2016, 10, 524–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couvertier, S.M.; Zhou, Y.; Weerapana, E. Chemical-proteomic strategies to investigate cysteine posttranslational modifications. Biochim. Biophys. Acta BBA Proteins Proteom. 2014, 1844, 2315–2330. [Google Scholar] [CrossRef] [PubMed]
- Marouga, R.; David, S.; Hawkins, E. The development of the DIGE system: 2D fluorescence difference gel analysis technology. Anal. Bioanal. Chem. 2005, 382, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Charles, R.; Jayawardhana, T.; Eaton, P. Gel-based methods in redox proteomics. Biochim. Biophys. Acta BBA Gen. Subj. 2014, 1840, 830–837. [Google Scholar] [CrossRef]
- Fratelli, M.; Demol, H.; Puype, M.; Casagrande, S.; Eberini, I.; Salmona, M.; Bonetto, V.; Mengozzi, M.; Duffieux, F.; Miclet, E.; et al. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 3505–3510. [Google Scholar] [CrossRef] [Green Version]
- McDonagh, B. Detection of ROS Induced Proteomic Signatures by Mass Spectrometry. Front. Physiol. 2017, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Alcock, L.J.; Perkins, M.V.; Chalker, J.M. Chemical methods for mapping cysteine oxidation. Chem. Soc. Rev. 2017, 47, 231–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulsen, C.E.; Carroll, K.S. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef]
- Abo, M.; Weerapana, E. Chemical Probes for Redox Signaling and Oxidative Stress. Antioxid. Redox Signal. 2019, 30, 1369–1386. [Google Scholar] [CrossRef]
- Pan, J.; Carroll, K.S. Chemical biology approaches to study protein cysteine sulfenylation. Biopolymers 2014, 101, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Gupta, V.; Tallman, K.A.; Porter, N.A.; Carroll, K.S.; Liebler, D.C. Global, in situ, site-specific analysis of protein S-sulfenylation. Nat. Protoc. 2015, 10, 1022–1037. [Google Scholar] [CrossRef]
- Doulias, P.-T.; Raju, K.; Greene, J.L.; Tenopoulou, M.; Ischiropoulos, H. Mass spectrometry-based identification of S-nitrosocysteine in vivo using organic mercury assisted enrichment. Methods 2013, 62, 165–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, J.; Gaffrey, M.J.; Qian, W.-J. Quantitative proteomic characterization of redox-dependent post-translational modifications on protein cysteines. Mol. BioSyst. 2017, 13, 816–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrester, M.T.; Foster, M.W.; Benhar, M.; Stamler, J.S. Detection of protein S-nitrosylation with the biotin-switch technique. Free Radic. Biol. Med. 2009, 46, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Jaffrey, S.R.; Erdjument-Bromage, H.; Ferris, C.D.; Tempst, P.; Snyder, S.H. Protein S-nitrosylation: A physiological signal for neuronal nitric oxide. Nat. Cell Biol. 2001, 3, 193–197. [Google Scholar] [CrossRef]
- Urban, P.L. Quantitative mass spectrometry: An overview. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, C.I.; Van Eyk, J.E. A Twist on Quantification. Circ. Res. 2012, 111, 1253–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prus, G.; Hoegl, A.; Weinert, B.T.; Choudhary, C. Analysis and Interpretation of Protein Post-Translational Modification Site Stoichiometry. Trends Biochem. Sci. 2019, 44, 943–960. [Google Scholar] [CrossRef]
- Chiappetta, G.; Ndiaye, S.; Igbaria, A.; Kumar, C.; Vinh, J.; Toledano, M.B. Proteome Screens for Cys Residues Oxidation. Methods Enzymol. 2010, 473, 199–216. [Google Scholar] [CrossRef]
- Thamsen, M.; Jakob, U. The redoxome. Curr. Opin. Chem. Biol. 2011, 15, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed Protein Quantitation in Saccharomyces cerevisiae Using Amine-reactive Isobaric Tagging Reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, A.; Schäfer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, A.T.; Hamon, C. Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.Y.; O’Brien, J.; Paulo, J.A.; Gygi, S.P. Improved Method for Determining Absolute Phosphorylation Stoichiometry Using Bayesian Statistics and Isobaric Labeling. J. Proteome Res. 2017, 16, 4217–4226. [Google Scholar] [CrossRef]
- Duan, J.; Zhang, T.; Gaffrey, M.J.; Weitz, K.K.; Moore, R.J.; Li, X.; Xian, M.; Thrall, B.D.; Qian, W.-J. Stochiometric quantification of the thiol redox proteome of macrophages reveals subcellular compartmentalization and susceptibility to oxidative perturbations. Redox Biol. 2020, 36, 101649. [Google Scholar] [CrossRef]
- Paulech, J.; Liddy, K.A.; Engholm-Keller, K.; White, M.Y.; Cordwell, S.J. Global Analysis of Myocardial Peptides Containing Cysteines with Irreversible Sulfinic and Sulfonic Acid Post-Translational Modifications. Mol. Cell. Proteom. 2015, 14, 609–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leichert, L.I.; Gehrke, F.; Gudiseva, H.V.; Blackwell, T.; Ilbert, M.; Walker, A.K.; Strahler, J.R.; Andrews, P.C.; Jakob, U. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 8197–8202. [Google Scholar] [CrossRef] [Green Version]
- Brandes, N.; Reichmann, D.; Tienson, H.; Leichert, L.I.; Jakob, U. Using Quantitative Redox Proteomics to Dissect the Yeast Redoxome. J. Biol. Chem. 2011, 286, 41893–41903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topf, U.; Suppanz, I.; Samluk, L.; Wrobel, L.; Böser, A.; Sakowska, P.; Knapp, B.; Pietrzyk, M.K.; Chacinska, A.; Warscheid, B. Quantitative proteomics identifies redox switches for global translation modulation by mitochondrially produced reactive oxygen species. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Menger, K.E.; James, A.M.; Cochemé, H.M.; Harbour, M.E.; Chouchani, E.T.; Ding, S.; Fearnley, I.M.; Partridge, L.; Murphy, M.P. Fasting, but Not Aging, Dramatically Alters the Redox Status of Cysteine Residues on Proteins in Drosophila melanogaster. Cell Rep. 2015, 11, 1856–1865. [Google Scholar] [CrossRef] [Green Version]
- Xie, K.; Bunse, C.; Marcus, K.; Leichert, L.I. Quantifying changes in the bacterial thiol redox proteome during host-pathogen interaction. Redox Biol. 2019, 21, 101087. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Carroll, K.S.; Liebler, D.C. The Expanding Landscape of the Thiol Redox Proteome. Mol. Cell. Proteom. 2016, 15, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Luo, Z.; Wu, X.; Zhu, J.; Yu, K.; Jin, Y.; Zhang, Z.; Zhao, S.; Zhou, L. Proteomic Analyses of Cysteine Redox in High-Fat-Fed and Fasted Mouse Livers: Implications for Liver Metabolic Homeostasis. J. Proteome Res. 2017, 17, 129–140. [Google Scholar] [CrossRef]
- Kramer, P.A.; Duan, J.; Gaffrey, M.J.; Shukla, A.K.; Wang, L.; Bammler, T.K.; Qian, W.-J.; Marcinek, D.J. Fatiguing contractions increase protein S-glutathionylation occupancy in mouse skeletal muscle. Redox Biol. 2018, 17, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, D.B.; Bernhardt, O.M.; Hogrebe, A.; Martinez-Val, A.; Verbeke, L.; Gandhi, T.; Kelstrup, C.D.; Reiter, L.; Olsen, J.V. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.-K.; Zhang, G.; Lawlor, K.; Nazarian, A.; Philip, J.; Tempst, P.; Dephoure, N.; Neubert, T.A. Deep Coverage of Global Protein Expression and Phosphorylation in Breast Tumor Cell Lines Using TMT 10-plex Isobaric Labeling. J. Proteome Res. 2017, 16, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Petersen, M.H.; Ibañez-Vea, M.; Lassen, P.S.; Larsen, M.R.; Palmisano, G. Simultaneous Enrichment of Cysteine-containing Peptides and Phosphopeptides Using a Cysteine-specific Phosphonate Adaptable Tag (CysPAT) in Combination with titanium dioxide (TiO2) Chromatography. Mol. Cell. Proteom. 2016, 15, 3282–3296. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; Long, J.; Mills, E.L.; Szpyt, J.; et al. A Quantitative Tissue-Specific Landscape of Protein Redox Regulation during Aging. Cell 2020, 180, 968–983. [Google Scholar] [CrossRef]
- Guo, J.; Gaffrey, M.J.; Su, D.; Liu, T.; Camp, D.G.; Smith, R.D.; Qian, W.-J. Resin-assisted enrichment of thiols as a general strategy for proteomic profiling of cysteine-based reversible modifications. Nat. Protoc. 2014, 9, 64–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Nguyen, A.Y.; Dai, Z.; Su, D.; Gaffrey, M.J.; Moore, R.J.; Jacobs, J.M.; Monroe, M.E.; Smith, R.D.; Koppenaal, D.W.; et al. Proteome-wide Light/Dark Modulation of Thiol Oxidation in Cyanobacteria Revealed by Quantitative Site-specific Redox Proteomics. Mol. Cell. Proteom. 2014, 13, 3270–3285. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Li, Z.; Liu, K.; Tian, C.; He, J.; He, J.; He, F.; Xu, P.; Yang, J. A quantitative thiol reactivity profiling platform to analyze redox and electrophile reactive cysteine proteomes. Nat. Protoc. 2020, 15, 2891–2919. [Google Scholar] [CrossRef] [PubMed]
- Weerapana, E.; Speers, E.A.; Cravatt, B.F. Tandem orthogonal proteolysis-activity-based protein profiling (TOP-ABPP)—A general method for mapping sites of probe modification in proteomes. Nat. Protoc. 2007, 2, 1414–1425. [Google Scholar] [CrossRef]
- Weerapana, E.; Wang, C.; Simon, G.M.; Richter, F.; Khare, S.D.; Dillon, M.B.D.; Bachovchin, D.A.; Mowen, A.K.; Baker, D.; Cravatt, B.F. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nat. Cell Biol. 2010, 468, 790–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, C.; Sun, R.; Liu, K.; Fu, L.; Liu, X.; Zhou, W.; Yang, Y.; Yang, J. Multiplexed Thiol Reactivity Profiling for Target Discovery of Electrophilic Natural Products. Cell Chem. Biol. 2017, 24, 1416–1427. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Liu, K.; Sun, M.; Tian, C.; Sun, R.; Betanzos, C.M.; Tallman, K.A.; Porter, N.A.; Yang, Y.; Guo, D.; et al. Systematic and Quantitative Assessment of Hydrogen Peroxide Reactivity with Cysteines Across Human Proteomes. Mol. Cell. Proteom. 2017, 16, 1815–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, R.; Shi, F.; Liu, K.; Fu, L.; Tian, C.; Yang, Y.; Tallman, K.A.; Porter, N.A.; Yang, J. A Chemoproteomic Platform to Assess Bioactivation Potential of Drugs. Chem. Res. Toxicol. 2017, 30, 1797–1803. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Liu, K.; Ferreira, R.B.; Carroll, K.S.; Yang, J. Proteome-Wide Analysis of Cysteine S-Sulfenylation Using a Benzothiazine-Based Probe. Curr. Protoc. Protein Sci. 2018, 95, 76. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Carroll, K.S. Activity-Based Sensing for Site-Specific Proteomic Analysis of Cysteine Oxidation. Acc. Chem. Res. 2020, 53, 20–31. [Google Scholar] [CrossRef]
- Winther, J.R.; Thorpe, C. Quantification of thiols and disulfides. Biochim. Biophys. Acta BBA Gen. Subj. 2014, 1840, 838–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, T.; Inaba, K.; Kadokura, H. Methods to identify the substrates of thiol-disulfide oxidoreductases. Protein Sci. 2018, 28, 30–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ow, S.Y.; Salim, M.; Noirel, J.; Evans, C.; Rehman, I.; Wright, P.C. iTRAQ Underestimation in Simple and Complex Mixtures: “The Good, the Bad and the Ugly”. J. Proteome Res. 2009, 8, 5347–5355. [Google Scholar] [CrossRef]
- Bantscheff, M.; Boesche, M.; Eberhard, D.; Matthieson, T.; Sweetman, G.; Kuster, B. Robust and Sensitive iTRAQ Quantification on an LTQ Orbitrap Mass Spectrometer. Mol. Cell. Proteom. 2008, 7, 1702–1713. [Google Scholar] [CrossRef] [Green Version]
- Savitski, M.M.; Mathieson, T.; Zinn, N.; Sweetman, G.; Doce, C.; Becher, I.; Pachl, F.; Kuster, B.; Bantscheff, M. Measuring and Managing Ratio Compression for Accurate iTRAQ/TMT Quantification. J. Proteome Res. 2013, 12, 3586–3598. [Google Scholar] [CrossRef]
- Searle, B.C.; Yergey, A.L. An efficient solution for resolving iTRAQ and TMT channel cross-talk. J. Mass Spectrom. 2020, 55, e4354. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Val, A.; Garcia, F.; Ximénez-Embún, P.; Ibarz, N.; Zarzuela, E.; Ruppen, I.; Mohammed, S.; Munoz, J. On the Statistical Significance of Compressed Ratios in Isobaric Labeling: A Cross-Platform Comparison. J. Proteome Res. 2016, 15, 3029–3038. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.-M.; Roede, J.R.; Orr, M.; Liang, Y.; Jones, D.P. Integrated Redox Proteomics and Metabolomics of Mitochondria to Identify Mechanisms of Cd Toxicity. Toxicol. Sci. 2014, 139, 59–73. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.; Burchfield, J.G.; Yang, P.; Humphrey, S.J.; Yang, G.; Francis, D.; Yasmin, S.; Shin, S.-Y.; Norris, D.M.; Kearney, A.L.; et al. Global redox proteome and phosphoproteome analysis reveals redox switch in Akt. Nat. Commun. 2019, 10, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.-A.; Zhang, Q.; Wang, Y.; Ge, W.; Guo, D. Prediction of redox-sensitive cysteines using sequential distance and other sequence-based features. BMC Bioinform. 2016, 17, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chen, X.; Li, C.; Liu, Y.; Yang, F.; Wang, C. Sequence-Based Prediction of Cysteine Reactivity Using Machine Learning. Biochemistry 2018, 57, 451–460. [Google Scholar] [CrossRef]
- Mapes, N.J.; Rodriguez, C.; Chowriappa, P.; Dua, S. Residue Adjacency Matrix Based Feature Engineering for Predicting Cysteine Reactivity in Proteins. Comput. Struct. Biotechnol. J. 2019, 17, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Erdős, G.; Mészáros, B.; Reichmann, D.; Dosztányi, Z. Large-Scale Analysis of Redox-Sensitive Conditionally Disordered Protein Regions Reveals Their Widespread Nature and Key Roles in High-Level Eukaryotic Processes. Proteomics 2019, 19, e1800070. [Google Scholar] [CrossRef]
- Trivedi, M.V.; Laurence, J.S.; Siahaan, T.J. The Role of Thiols and Disulfides on Protein Stability. Curr. Protein Pept. Sci. 2009, 10, 614–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wouters, M.A.; Fan, S.W.; Haworth, N.L. Disulfides as Redox Switches: From Molecular Mechanisms to Functional Significance. Antioxid. Redox Signal. 2010, 12, 53–91. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.W.; George, R.A.; Haworth, N.L.; Feng, L.L.; Liu, J.Y.; Wouters, M.A. Conformational changes in redox pairs of protein structures. Protein Sci. 2009, 18, 1745–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, J.; Hogg, P.J. Allosteric disulfides: Sophisticated molecular structures enabling flexible protein regulation. J. Biol. Chem. 2019, 294, 2949–5908. [Google Scholar] [CrossRef] [Green Version]
- Zeida, A.; Guardia, C.M.; Lichtig, P.; Perissinotti, L.L.; DeFelipe, L.A.; Turjanski, A.G.; Radi, R.; Trujillo, M.; Estrin, D.A. Thiol redox biochemistry: Insights from computer simulations. Biophys. Rev. 2014, 6, 27–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrams, A.J.; Farooq, A.; Wang, G. S-Nitrosylation of ApoE in Alzheimer’s Disease. Biochemistry 2011, 50, 3405–3407. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Kang, E.; Wang, Y.; Yang, C.; Yu, H.; Wang, Q.; Chen, Z.; Zhang, C.; Christian, K.M.; Song, H.; et al. Brain-specific Crmp2 deletion leads to neuronal development deficits and behavioural impairments in mice. Nat. Commun. 2016, 7, 11773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moutal, A.; White, K.A.; Chefdeville, A.; Laufmann, R.N.; Vitiello, P.F.; Feinstein, D.; Weimer, J.M.; Khanna, R. Dysregulation of CRMP2 Post-Translational Modifications Drive Its Pathological Functions. Mol. Neurobiol. 2019, 56, 6736–6755. [Google Scholar] [CrossRef] [PubMed]
- Gellert, M.; Venz, S.; Mitlöhner, J.; Cott, C.; Hanschmann, E.-M.; Lillig, C.H. Identification of a Dithiol-disulfide Switch in Collapsin Response Mediator Protein 2 (CRMP2) That Is Toggled in a Model of Neuronal Differentiation. J. Biol. Chem. 2013, 288, 35117–35125. [Google Scholar] [CrossRef] [Green Version]
- Möller, D.; Gellert, M.; Langel, W.; Lillig, C.H. Molecular dynamics simulations and in vitro analysis of the CRMP2 thiol switch. Mol. BioSyst. 2017, 13, 1744–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheehan, D.; McDonagh, B. The clinical potential of thiol redox proteomics. Expert Rev. Proteom. 2020, 17, 41–48. [Google Scholar] [CrossRef]
- Paramasivan, S.; Adav, S.S.; Ngan, S.C.; Dalan, R.; Leow, M.K.-S.; Ho, H.H.; Sze, S.K. Serum albumin cysteine trioxidation is a potential oxidative stress biomarker of type 2 diabetes mellitus. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zago, M.P.; Wiktorowicz, J.E.; Spratt, H.; Koo, S.-J.; Barrientos, N.; Burgos, A.N.; Burgos, J.N.; Iñiguez, F.; Botelli, V.; De La Fuente, R.L.; et al. Potential Utility of Protein Targets of Cysteine-S-Nitrosylation in Identifying Clinical Disease Status in Human Chagas Disease. Front. Microbiol. 2019, 9, 3320. [Google Scholar] [CrossRef]
- Zhao, K.; Zhao, G.M.; Wu, D.; Soong, Y.; Birk, A.V.; Schiller, P.W.; Szeto, H.H. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J. Biol. Chem. 2004, 279, 34682–34690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birk, A.V.; Chao, W.M.; Bracken, C.; Warren, J.D.; Szeto, H.H. Targeting mitochondrial cardiolipin and the cytochromec/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 2014, 171, 2017–2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, M.D.; Duan, J.; Samuelson, A.T.; Gaffrey, M.J.; Merrihew, G.E.; Egertson, J.D.; Wang, L.; Bammler, T.K.; Moore, R.J.; White, C.C.; et al. Improving mitochondrial function with SS-31 reverses age-related redox stress and improves exercise tolerance in aged mice. Free Radic. Biol. Med. 2019, 134, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Chiao, Y.A.; Zhang, H.; Sweetwyne, M.; Whitson, J.; Ting, Y.S.; Basisty, N.; Pino, L.K.; Quarles, E.; Nguyen, N.-H.; Campbell, M.D.; et al. Late-life restoration of mitochondrial function reverses cardiac dysfunction in old mice. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Day, N.J.; Gaffrey, M.J.; Qian, W.-J. Stoichiometric Thiol Redox Proteomics for Quantifying Cellular Responses to Perturbations. Antioxidants 2021, 10, 499. https://doi.org/10.3390/antiox10030499
Day NJ, Gaffrey MJ, Qian W-J. Stoichiometric Thiol Redox Proteomics for Quantifying Cellular Responses to Perturbations. Antioxidants. 2021; 10(3):499. https://doi.org/10.3390/antiox10030499
Chicago/Turabian StyleDay, Nicholas J., Matthew J. Gaffrey, and Wei-Jun Qian. 2021. "Stoichiometric Thiol Redox Proteomics for Quantifying Cellular Responses to Perturbations" Antioxidants 10, no. 3: 499. https://doi.org/10.3390/antiox10030499