
Reprograming of Tumor-Associated Macrophages in Breast Tumor-Bearing Mice under Chemotherapy by Targeting Heme Oxygenase-1

 ,
,  , , ,
, , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Bioinformatic Analysis
2.2. Kaplan–Meier (Relapse-Free Survival) Analysis
2.3. Syngeneic Murine Breast Tumor Model
2.4. Macrophages Depletion
2.5. Cell Culture
2.6. Macrophage Polarization
2.7. Generation of Tumor Cell Debris
2.8. Isolation of Single Cells from Mouse Tumors
2.9. Flow Cytometric Analysis
2.10. Cell Sorting
2.11. Immunofluorescence Analysis
2.12. Phagocytosis Assay
2.13. Quantitative PCR
2.14. Statistical Analysis
3. Results
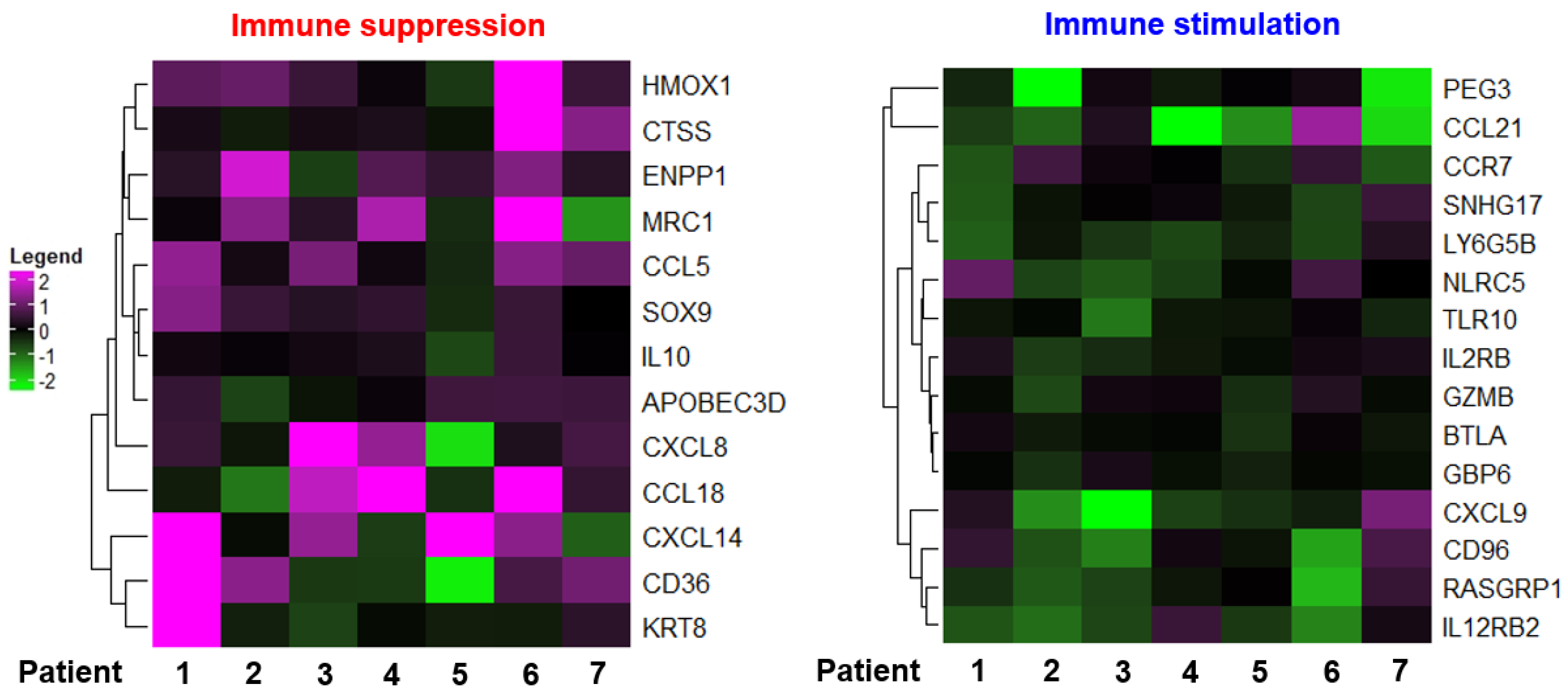
3.1. Chemotherapy Induces TAM Polarization toward the M2 Phenotype
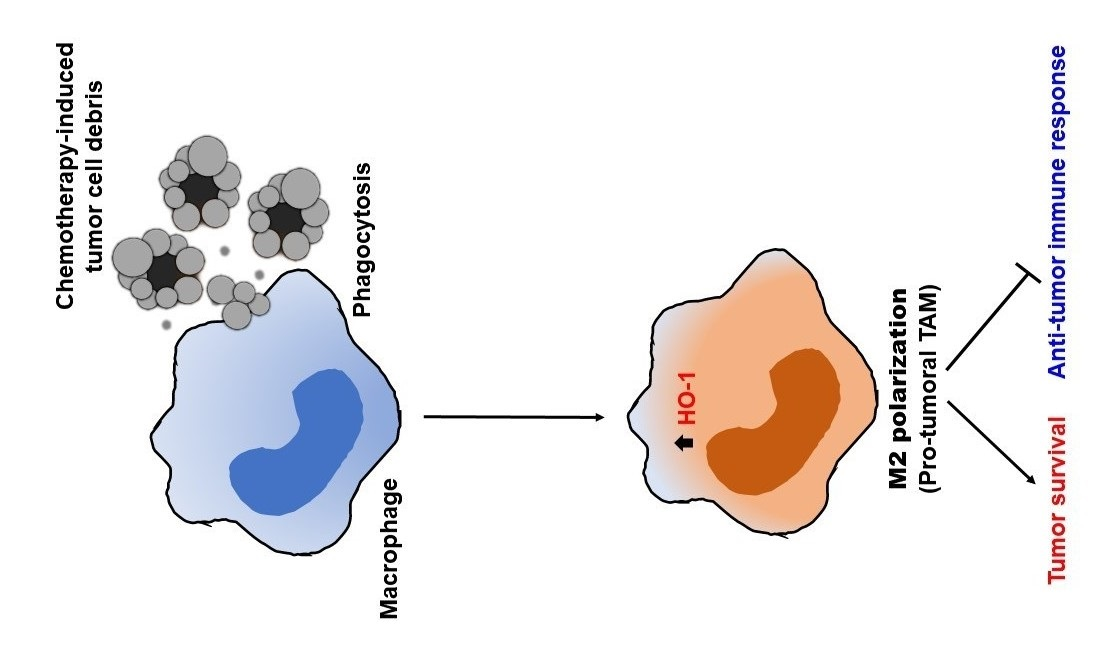
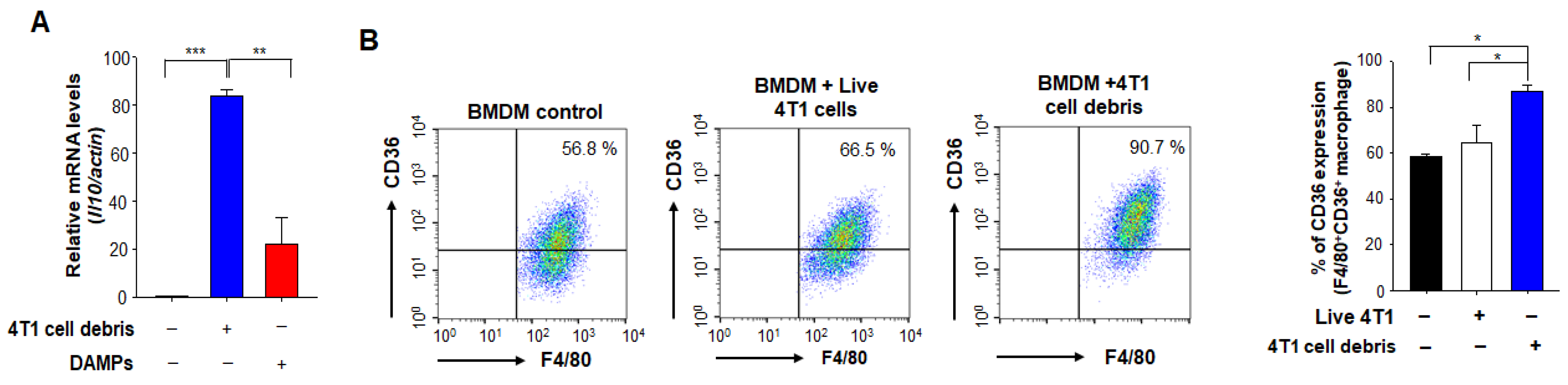
3.2. Phagocytic Engulfment of Tumor Cell Debris Re-Educates Macrophages toward M2-Like Polarization
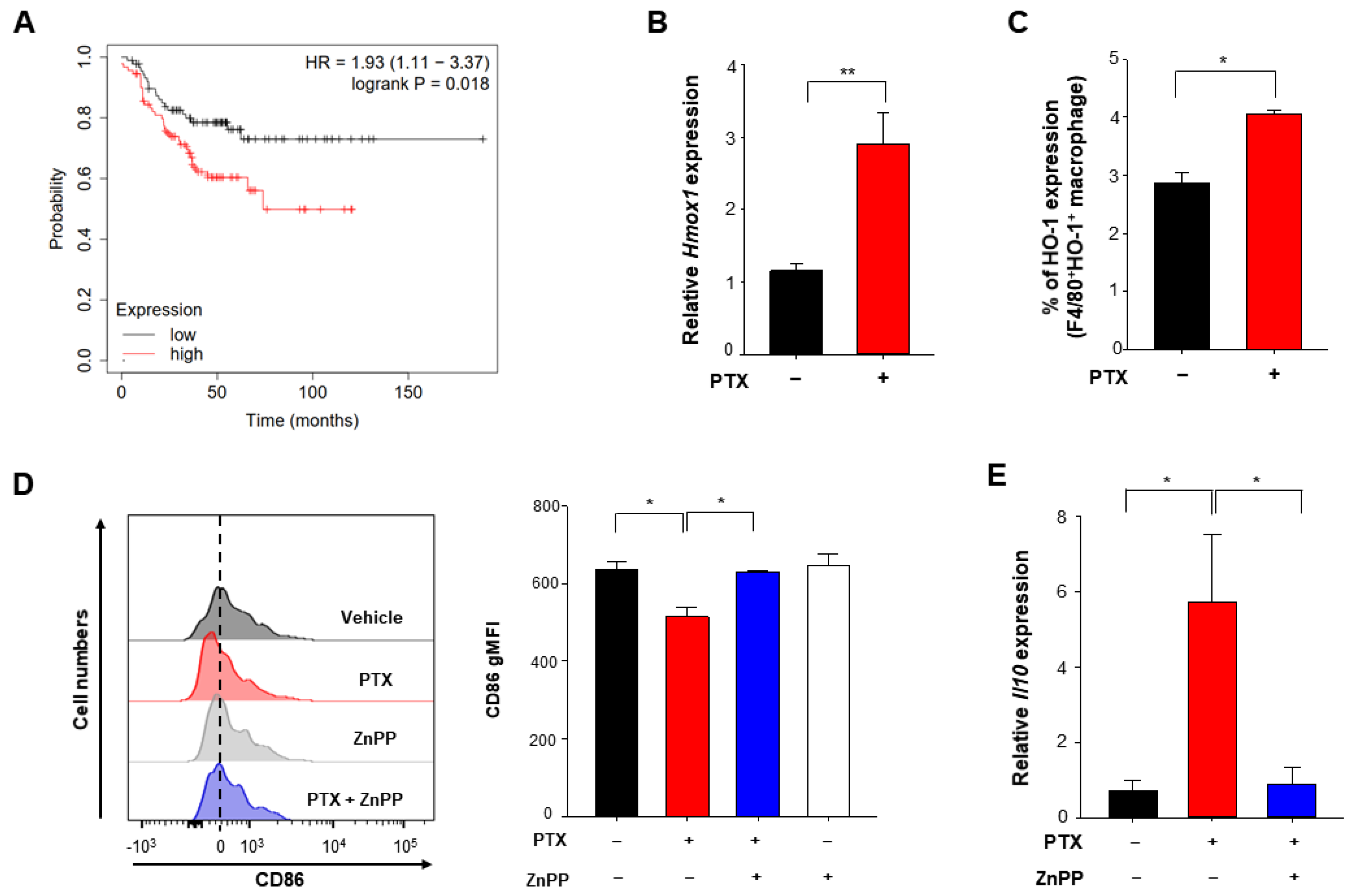
3.3. Chemotherapy Enhances Expression of HO-1 Which Is Associated with Poor Prognosis
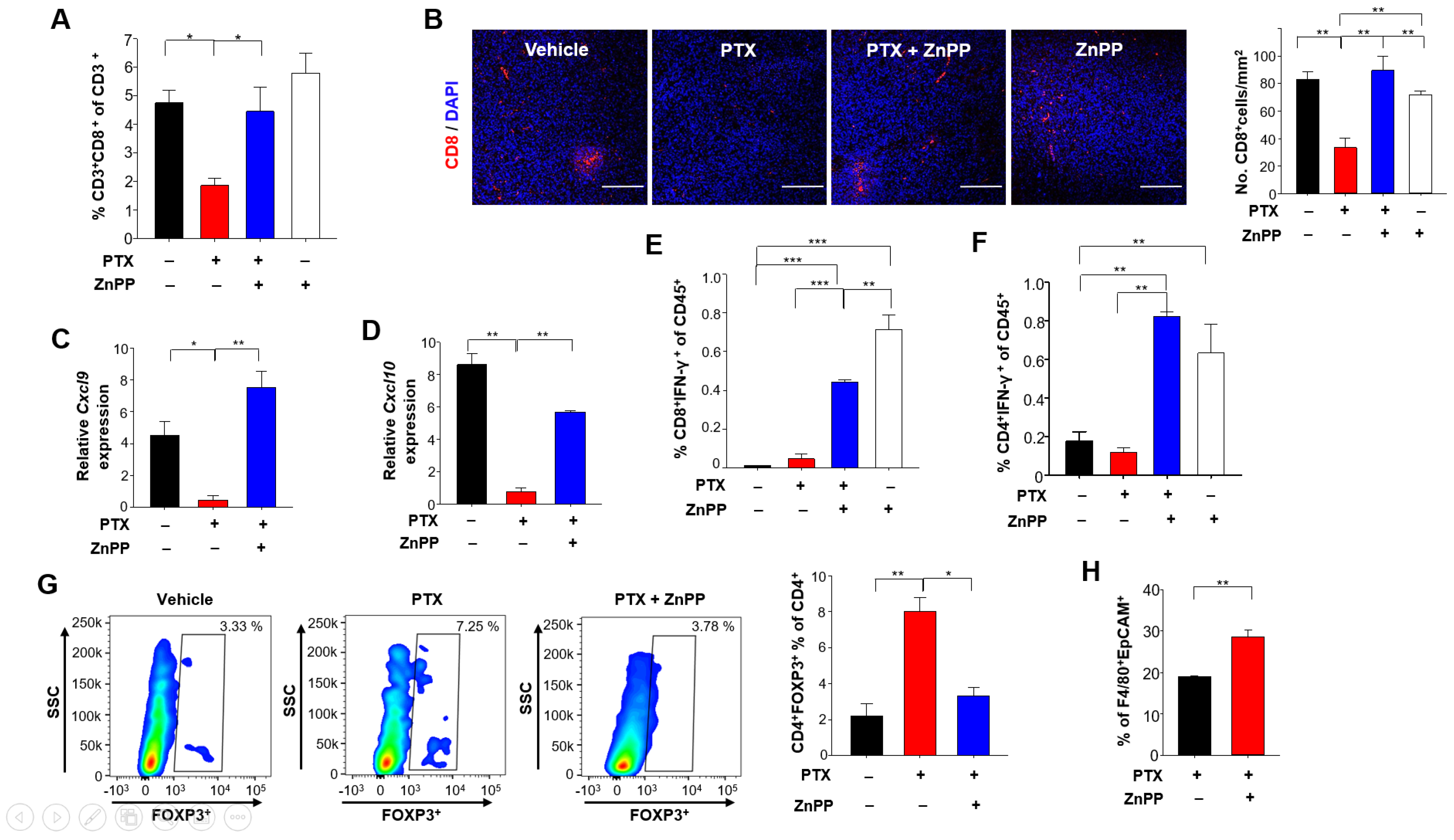
3.4. HO-1 Inhibition Potentiates Anti-Tumor T Cell Function in Response to PTX Treatment
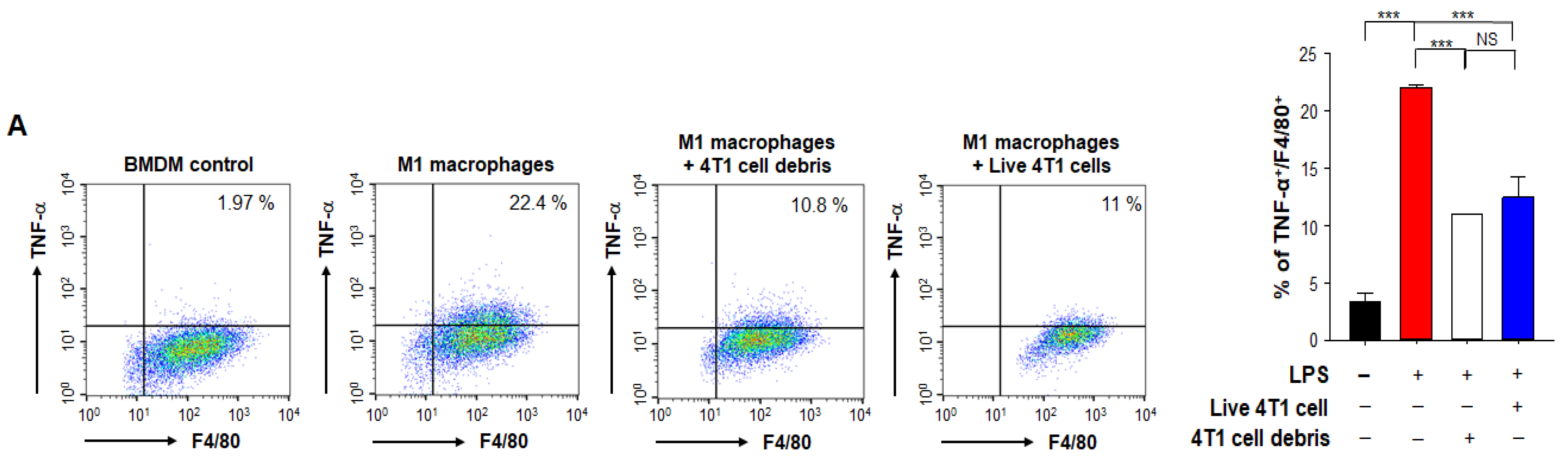
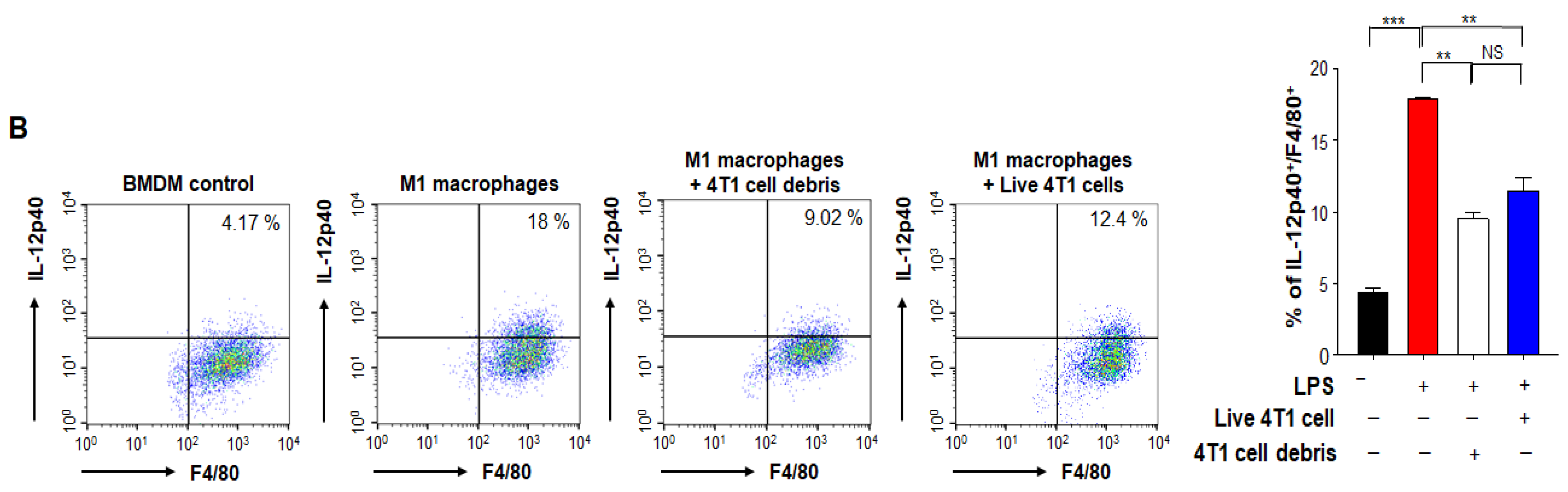
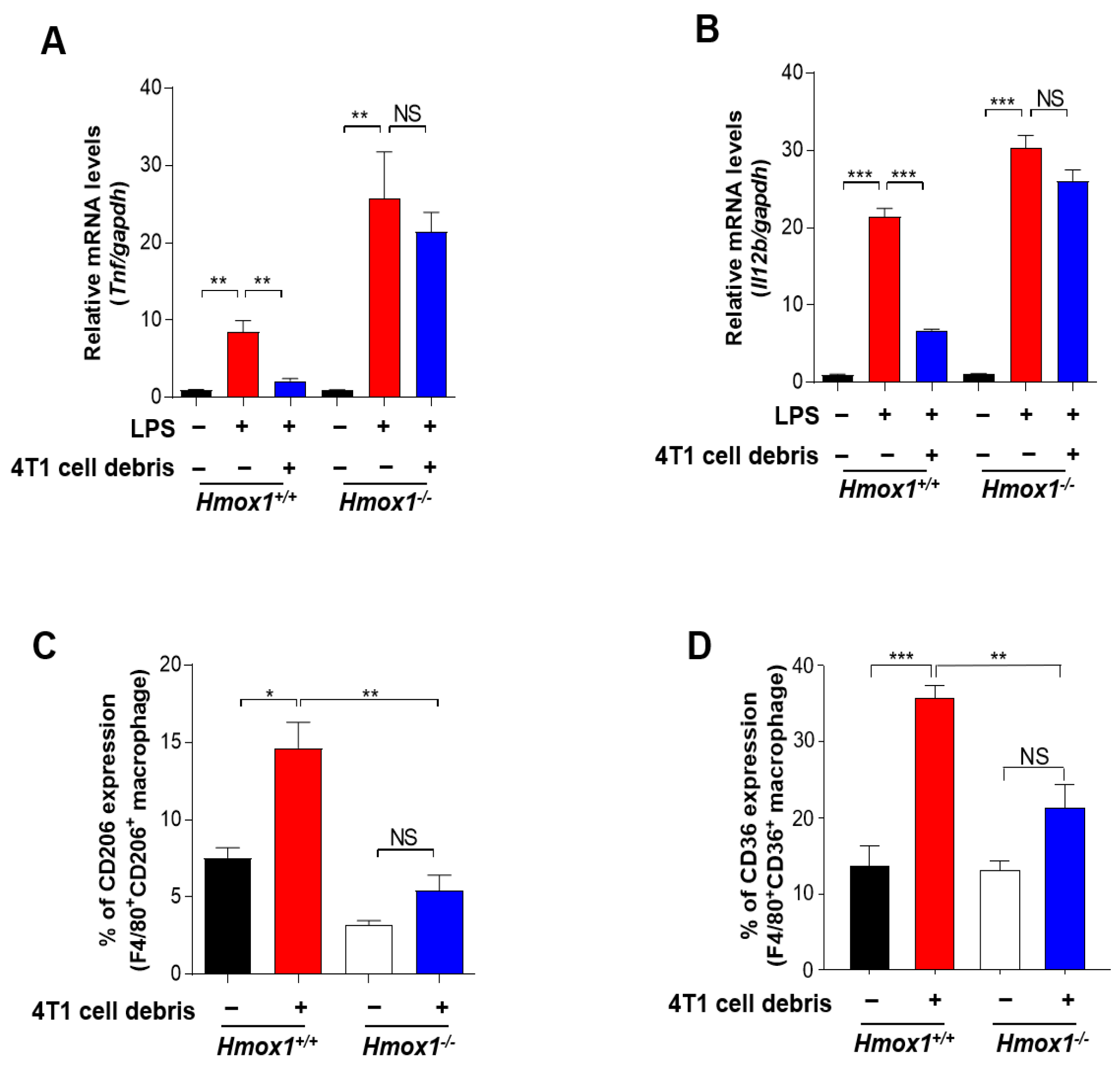
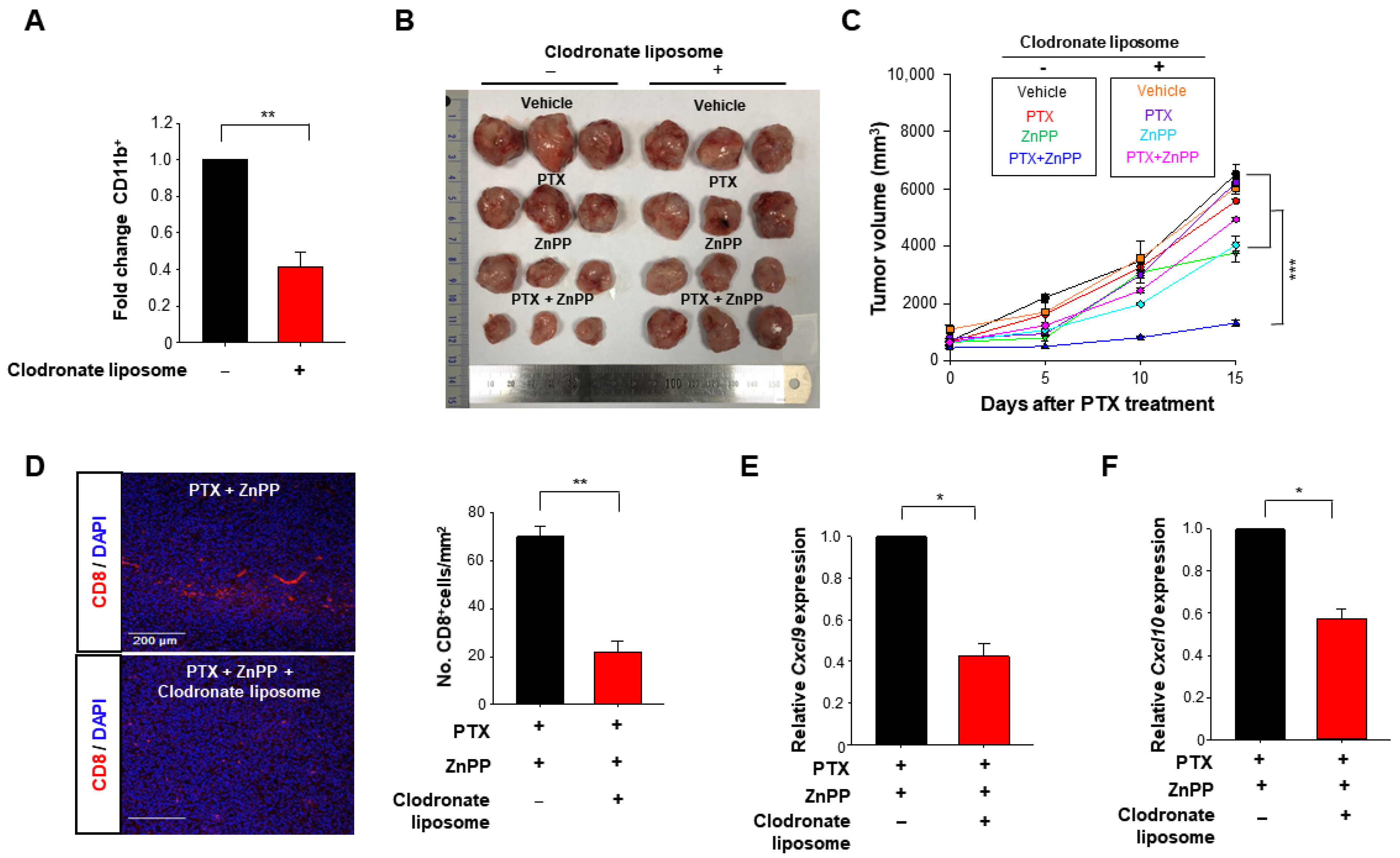
3.5. HO-1 Inactivation-induced M1 TAMs Are Crucial for the Enhanced Response to PTX Therapy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spill, F.; Reynolds, D.S.; Kamm, R.D.; Zaman, M.H. Impact of the physical microenvironment on tumor progression and metastasis. Curr. Opin. Biotechnol. 2016, 40, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, D.; Cang, H.; Guo, B. Crosstalk between cancer and immune cells: Role of tumor-associated macrophages in the tumor microenvironment. Cancer Med. 2019, 8, 4709–4721. [Google Scholar] [CrossRef]
- Nishida-Aoki, N.; Gujral1, T.S. Emerging approaches to study cell–cell interactions in tumor microenvironment. Oncotarget 2019, 10, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Luo, Z.; Li, X.; Han, X.; Shi, S.; Zhang, T. Tumor-associated macrophages: Role in tumorigenesis and immunotherapy implications. J. Cancer 2021, 12, 54–64. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef]
- Puthenveetil, A.; Dubey, S. Metabolic reprograming of tumor-associated macrophages. Ann. Transl. Med. 2020, 8, 1030. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Jan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Wang, S.; Liu, R.; Yu, Q.; Dong, L.; Bi, Y.; Liu, G. Metabolic reprogramming of macrophages during infections and cancer. Cancer Lett. 2019, 452, 14–22. [Google Scholar] [CrossRef]
- Malyshev, I.; Malyshev, Y. Current concept and update of the macrophage plasticity concept: Intracellular mechanisms of reprogramming and M3 macrophage (Switch) phenotype. BioMed Res. Int. 2015, 2015, 341308. [Google Scholar] [CrossRef] [Green Version]
- Genard, G.; Lucas, S.; Michiels, C. Reprogramming of tumor-associated macrophages with anticancer therapies: Radiotherapy versus chemo- and immunotherapies. Front. Immunol. 2017, 8, 828. [Google Scholar] [CrossRef] [Green Version]
- Schultze, J.L. Reprogramming of macrophages-new opportunities for therapeutic targeting. Curr. Opin. Pharmacol. 2016, 26, 10–15. [Google Scholar] [CrossRef]
- Gok Yavuz, B.; Gunaydin, G.; Gedik, M.E.; Kosemehmetoglu, K.; Karakoc, D.; Ozgur, F.; Guc, D. Cancer associated fibroblasts sculpt tumour microenvironment by recruiting monocytes and inducing immunosuppressive PD-1+ TAMs. Sci. Rep. 2019, 28, 3172. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 macrophages: Phenotypical vs. functional differentiation. Front. Immunol. 2014, 17, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Guo, N.; Zhou, Y.; Chen, J.; Wei, Q.; Han, M. The role of tumor-associated macrophages (TAMs) in tumor progression and relevant advance in targeted therapy. Acta Pharm. Sin. 2020, 10, 2156–2170. [Google Scholar] [CrossRef]
- Zhang, F.; Parayath, N.N.; Ene, C.I.; Stephan, S.B.; Koehne, A.L.; Coon, M.E.; Holland, E.C.; Stephan, M.T. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat. Commun. 2019, 10, 3974. [Google Scholar] [CrossRef]
- Hörhold, F.; Eisel, D.; Oswald, M.; Kolte, A.; Röll, D.; Osen, W.; Eichmüller, S.B.; König, R. Reprogramming of macrophages employing gene regulatory and metabolic network models. PLoS Comput. Biol. 2020, 16, e1007657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarode, P.; Schaefer, M.B.; Grimminger, F.; Seeger, W.; Savai, R. Macrophage and tumor cell cross-talk is fundamental for lung tumor progression: We need to talk. Front. Oncol. 2020, 10, 324. [Google Scholar] [CrossRef] [Green Version]
- Larionova, I.; Cherdyntseva, N.; Liu, T.; Patysheva, M.; Rakina, R.; Kzhyshkowska, J. Interaction of tumor-associated macrophages and cancer chemotherapy. Oncoimmunology 2019, 8, e1596004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning cold into hot: Firing up the tumor microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Sulciner, M.L.; Serhan, C.N.; Gilligan, M.M.; Mudge, D.K.; Chang, J.; Gartung, A.; Lehner, K.A.; Bielenberg, D.R.; Schmidt, B.; Dalli, J.; et al. Resolvins suppress tumor growth and enhance cancer therapy. J. Exp. Med. 2018, 215, 115–140. [Google Scholar] [CrossRef]
- Chang, J.; Bhasin, S.S.; Bielenberg, D.R.; Sukhatme, V.P.; Bhasin, M.; Huang, S.; Kieran, M.W.; Panigrahy, D. Chemotherapy-generated cell debris stimulates colon carcinoma tumor growth via osteopontin. FASEB J. 2019, 33, 114–125. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.J.; Gu, D.N.; Dai, J.J.; Huang, Q.; Tian, L. Dark side of cytotoxic therapy: Chemoradiation-induced cell death and tumor repopulation. Trends Cancer. 2020, 6, 419–431. [Google Scholar] [CrossRef]
- Jozkowicz, A.; Was, H.; Dulak, J. Heme oxygenase-1 in tumors: Is it a false friend? Antioxid. Redox Signal. 2007, 9, 2099–2117. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Saeidi, S.; Xiancai, Z.; Gwak, S.-Y.; Muna, I.K.; Park, S.-A.; Kim, S.H.; Na, H.-K.; Joe, Y.; Chung, H.T.; et al. Breast cancer cell debris diminishes therapeutic efficacy through heme oxygenase-1-mediated inactivation of M1-like tumor-associated. Neoplasia 2020, 22, 606–616. [Google Scholar] [CrossRef]
- Ma, X. TNF- and IL-12: A balancing act in macrophage functioning. Microbes Infect. 2001, 3, 121–129. [Google Scholar] [CrossRef]
- Ubil, E.; Caskey, L.; Holtzhausen, A.; Hunter, D.; Story, C.; Earp, H.S. Tumor-secreted Pros1 inhibits macrophage M1 polarization to reduce antitumor immune response. J. Clin. Investig. 2018, 128, 2356–2369. [Google Scholar] [CrossRef] [Green Version]
- Pandolfi, F.; Altamura, S.; Frosali, S.; Conti, P. Key role of DAMP in inflammation, cancer, and tissue repair. Clin. Ther. 2016, 38, 1017–1028. [Google Scholar] [CrossRef] [Green Version]
- Alaluf, E.; Vokaer, B.; Detavernier, A.; Azouz, A.; Splittgerber, M.; Carrette, A.; Boon, L.; Libert, F.; Soares, M.; Le Moine, A.; et al. Heme oxygenase-1 orchestrates the immunosuppressive program of tumor-associated macrophages. JCI Insight 2020, 5, e133929. [Google Scholar]
- House, I.G.; Savas, P.; Lai, J.; Chen, A.X.-Y.; Oliver, A.J.; Teo, Z.L.; Todd, K.L.; Henderson, M.A.; Giuffrida, L.; Petley, E.V.; et al. Macrophage-derived CXCL9 and CXCL10 are required for antitumor immune responses following immune checkpoint blockade. Clin. Cancer Res. 2020, 26, 487–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, P.; Leggatt, G.; Waterhouse, N.; Frazer, I.H. Interferon-γ derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis. 2017, 8, e2836. [Google Scholar] [CrossRef] [Green Version]
- Deng, R.; Wang, S.-M.; Yin, T.; Ye, T.-H.; Shen, G.-B.; Li, L.; Zhao, J.-Y.; Sang, Y.-X.; Duan, X.-G.; Wei, Y.-Q. Inhibition of tumor growth and alteration of associated macrophage cell type by an HO-1 inhibitor in breast carcinoma-bearing mice. Oncol. Res. 2013, 20, 473–482. [Google Scholar] [CrossRef]
- Arnold, J.N.; Magiera, L.; Kraman, M.; Fearon, D.T. Tumoral immune suppression by macrophages expressing fibroblast activation protein- and heme oxygenase-1. Cancer Immunol. Res. 2014, 2, 121–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muri, J.; Kopf, M. Redox regulation of immunometabolism. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef]
- Boada-Romero, E.; Martinez, J.; Heckmann, B.L.; Green, D.R. The clearance of dead cells by efferocytosis. Nat. Rev. Mol. Cell Biol. 2020, 21, 398–414. [Google Scholar] [CrossRef]
- Limongi, D.; Baldelli, S.; Checconi, P.; Marcocci, M.E.; De Chiara, G.; Fraternale, A.; Magnani, M.; Ciriolo, M.R.; Palamara, A.T. GSH-C4 Acts as anti inflammatory frug in different models of canonical and cell autonomous inflammation through NF B inhibition. Front. Immunol. 2019, 6, 155. [Google Scholar]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Guerriero, J.L. Macrophages: The road less traveled, changing anticancer therapy. Trends Mol. Med. 2018, 24, 472–489. [Google Scholar] [CrossRef]
- Kumar, S.; Calianese, D.; Birge, R.B. Efferocytosis of dying cells differentially modulate immunological outcomes in tumor microen vironment. Immunol. Rev. 2017, 280, 149–164. [Google Scholar] [CrossRef]
- Georgoudaki, A.M.; Prokopec, K.E.; Boura, V.F.; Hellqvist, E.; Sohn, S.; Ostling, J.; Dahan, R.; Harris, R.A.; Rantalainen, M.; Bring, D.K.; et al. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. 2016, 15, 2000–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanford, J.C.; Young, C.; Hicks, D.; Owens, P.; Williams, A.; Vaught, D.B.; Morrison, M.M.; Lim, J.; Williams, M.; Brantley-Sieders, D.M.; et al. Efferocytosis produces a prometastatic landscape during postpartum mammary gland involution. J. Clin. Investig. 2014, 124, 4737–4752. [Google Scholar] [CrossRef] [PubMed]
- Lauber, K.; Ernst, A.; Orth, M.; Herrmann, M.; Belka, C. Dying cell clearance and its impact on the outcome of tumor radiotherapy. Front. Oncol. 2012, 2, 116. [Google Scholar] [CrossRef] [Green Version]
- Shree, T.; Olson, O.C.; Elie, B.T.; Kester, J.C.; Garfall, A.L.; Simpson, K.; Bell-McGuinn, K.M.; Zabor, E.C.; Brogi, E.; Joyce, J.A. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011, 25, 2465–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poh, A.R.; Ernst, M. Targeting macrophages in cancer: From bench to bedside. Front. Oncol. 2018, 8, 49. [Google Scholar] [CrossRef] [Green Version]
- El Andaloussi, A.; Lesniak, M.S. CD4+ CD25+ FoxP3+ T-cell infiltration and heme oxygenase-1 expression correlate with tumor grade in human gliomas. J. Oncol. 2007, 83, 145–152. [Google Scholar] [CrossRef]
- Choi, B.M.; Pae, H.O.; Jeong, Y.R.; Kim, Y.M.; Chung, H.T. Critical role of heme oxygenase-1 in Foxp3-mediated immune suppression. Biochem. Biophys. Res. Commun. 2005, 327, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Lien, K.; Georgsdottir, S.; Sivanathan, L.; Chan, K.; Emmenegger, U. Low-dose metronomic chemotherapy: A systematic literature analysis. Eur. J. Cancer 2013, 49, 3387–3395. [Google Scholar] [CrossRef] [PubMed]
- Krajnak, S.; Schnatz, C.; Almstedt, K.; Brenner, W.; Haertner, F.; Heimes, A.S.; Lebrecht, A.; Makris, G.M.; Schwab, R.; Hasenburg, A.; et al. Low-dose metronomic chemotherapy as an efficient treatment option in metastatic breast cancer-results of an exploratory case-control study. Breast Cancer Res. Treat. 2020, 182, 389–399. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Primer Sequences |
|---|---|
| Hmox1 | Forward: GATAGAGCGCAACAAGCAGAA |
| Reverse: CAGTGAGGCCCATACCAGAAG | |
| Cxcl9 | Forward: GGAGTTCGAGGAACCCTAGTG |
| Reverse: GGGATTTGTAGTGGATCGTGC | |
| Cxcl10 | Forward: CCAAGTGCTGCCGTCATTTTC |
| Reverse: GGCTCGCAGGGATGATTTCAA | |
| Il10 | Forward: GCTCTTACTGACTGGCATGAG |
| Reverse: CGCAGCTCTAGGAGCATGTG | |
| Tnf | Forward: CCCTCACACTCAGATCATCTTCT |
| Reverse: GCTACGACGTGGGCTACAG | |
| Il12b | Forward: TGGTTTGCCATCGTTTTGCTG |
| Reverse: ACAGGTGAGGTTCACTGTTTG | |
| Actb | Forward: TGCTAGGAGCCAGAGCAGTA |
| Reverse: AGTGTGACGTTGACATCCGT | |
| Gapdh | Forward: GGGAAGCCCATCACCATCT |
| Reverse: CGGCCTCACCCCATTTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.H.; Kim, S.-J.; Park, J.; Joe, Y.; Lee, S.E.; Saeidi, S.; Zhong, X.; Kim, S.H.; Park, S.-A.; Na, H.-K.; et al. Reprograming of Tumor-Associated Macrophages in Breast Tumor-Bearing Mice under Chemotherapy by Targeting Heme Oxygenase-1. Antioxidants 2021, 10, 470. https://doi.org/10.3390/antiox10030470
Kim SH, Kim S-J, Park J, Joe Y, Lee SE, Saeidi S, Zhong X, Kim SH, Park S-A, Na H-K, et al. Reprograming of Tumor-Associated Macrophages in Breast Tumor-Bearing Mice under Chemotherapy by Targeting Heme Oxygenase-1. Antioxidants. 2021; 10(3):470. https://doi.org/10.3390/antiox10030470
Chicago/Turabian StyleKim, Seung Hyeon, Su-Jung Kim, Jeongmin Park, Yeonsoo Joe, So Eui Lee, Soma Saeidi, Xiancai Zhong, Seong Hoon Kim, Sin-Aye Park, Hye-Kyung Na, and et al. 2021. "Reprograming of Tumor-Associated Macrophages in Breast Tumor-Bearing Mice under Chemotherapy by Targeting Heme Oxygenase-1" Antioxidants 10, no. 3: 470. https://doi.org/10.3390/antiox10030470