
17β-Estradiol Abrogates Oxidative Stress and Neuroinflammation after Cortical Stab Wound Injury


 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Animal Model and Drug Administration
2.3. Protein Extraction for Biochemical Analysis
2.4. Immunoblotting
2.5. Brain Tissue Preparation and Sectioning
2.6. Immunofluorescence Analysis
2.7. Immunohistochemistry
2.8. Lesion Volume Assessment
2.9. Antibodies
2.10. Reactive Oxygen Species (ROS) and Glutathione Assay
2.11. Glutathione S-Transferase (GST) and Glutathione Reductase (GR) Assay
2.12. Statistical Analysis
3. Results
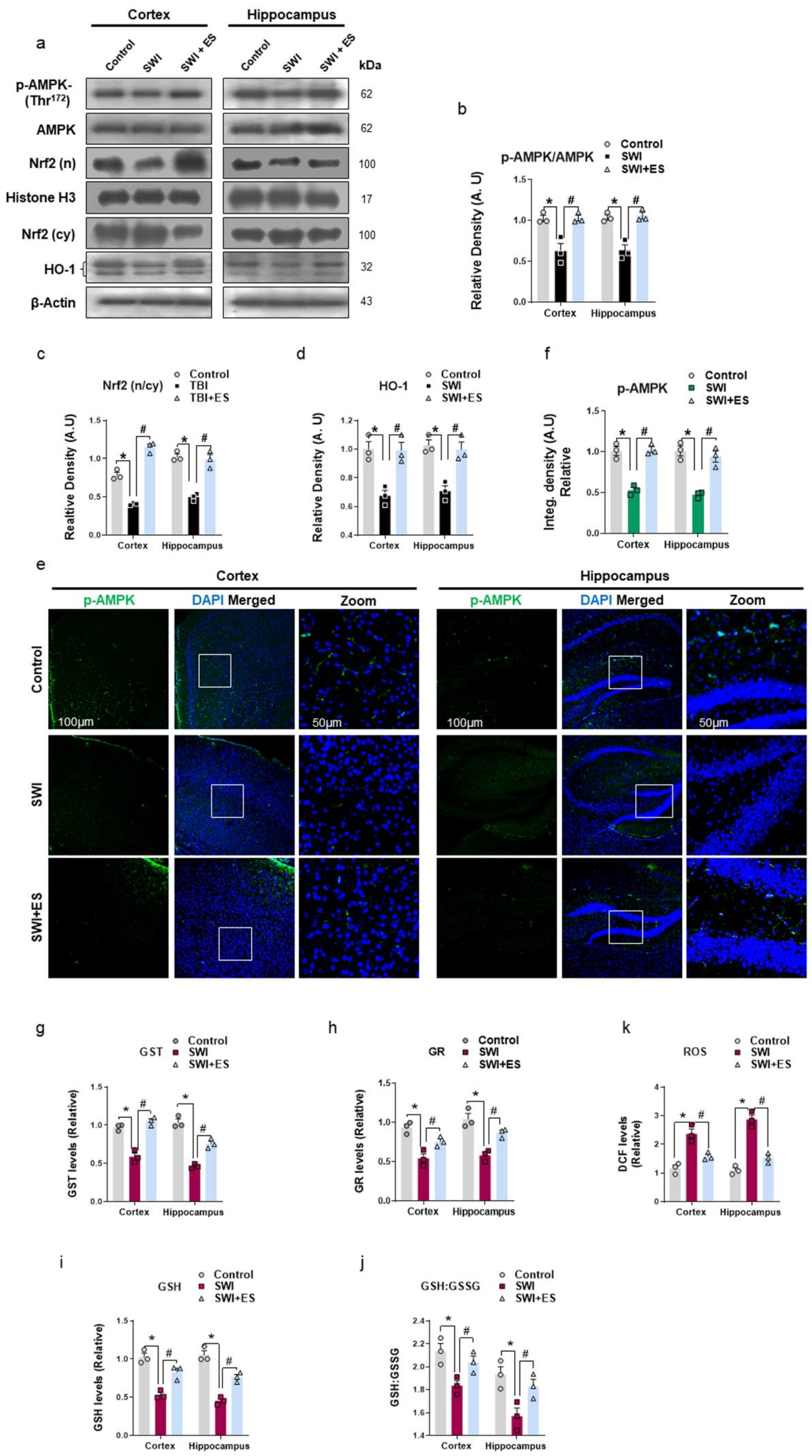
3.1. 17β-Estradiol Alleviates Energy Dyshomeostasis and Oxidative Stress after Brain Injury
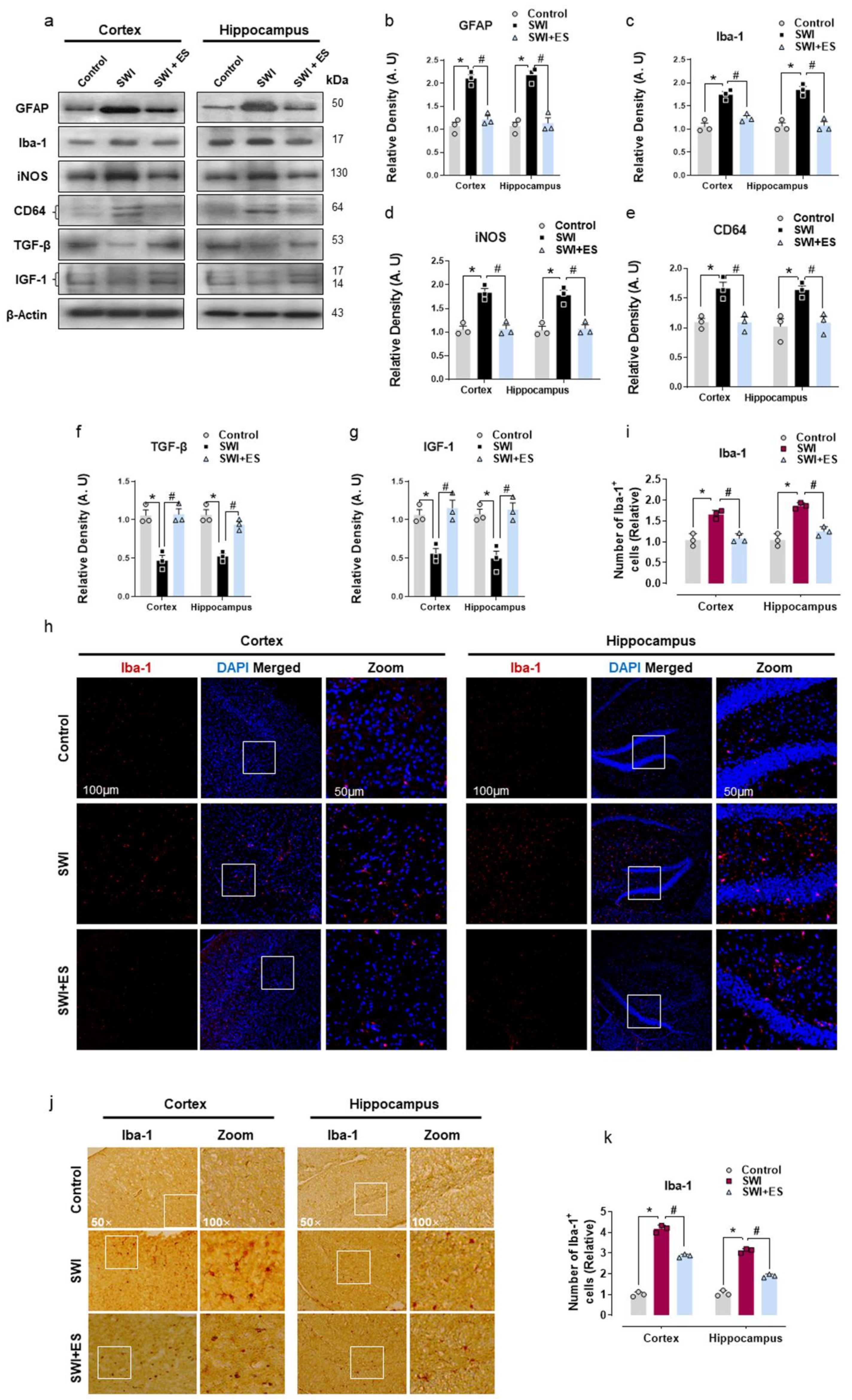
3.2. 17β-Estradiol Inhibits Gliosis and Inflammatory Response after Brain Injury
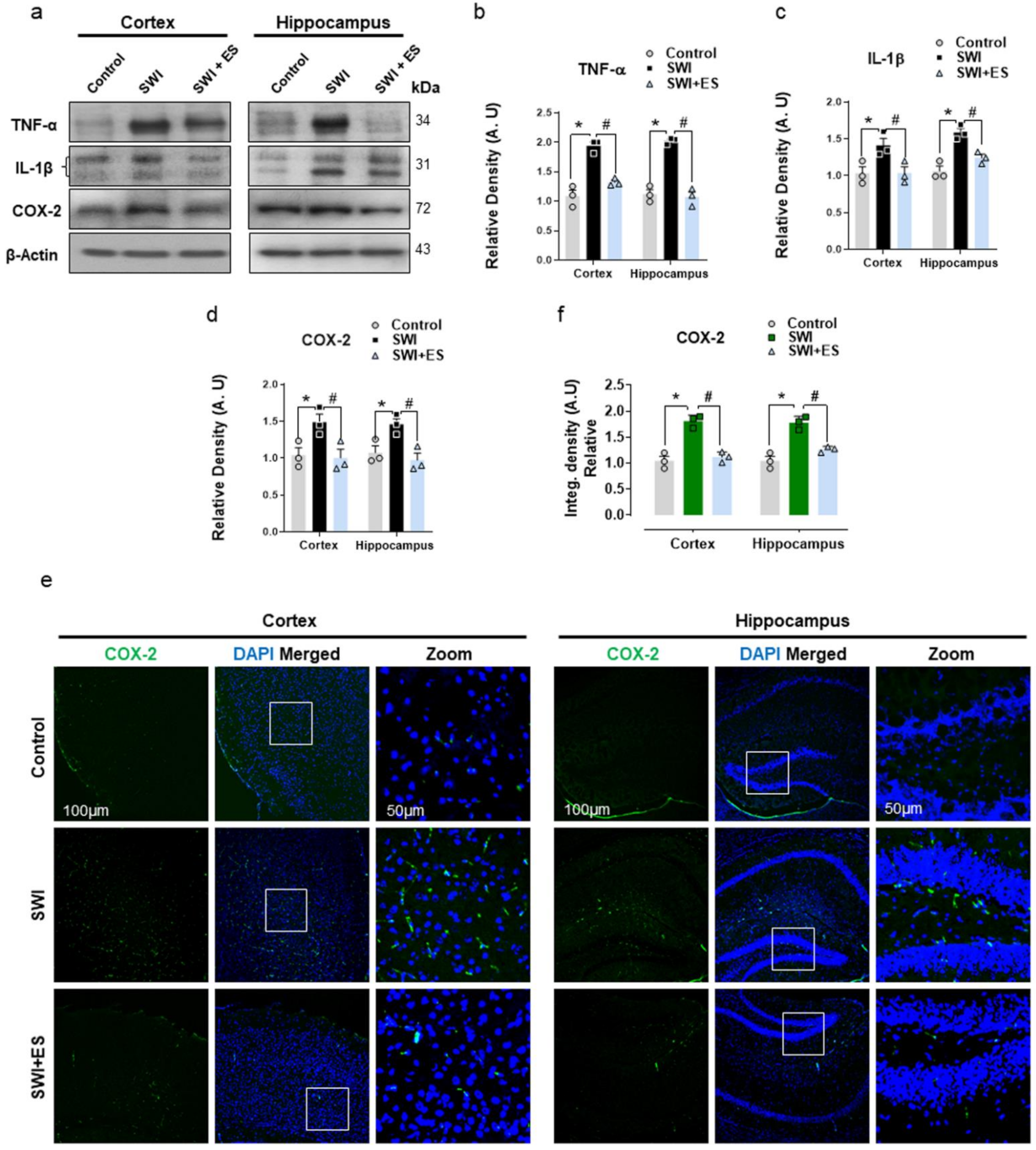
3.3. 17β-Estradiol Restrains Inflammatory Factors in the Injured Mouse Brain
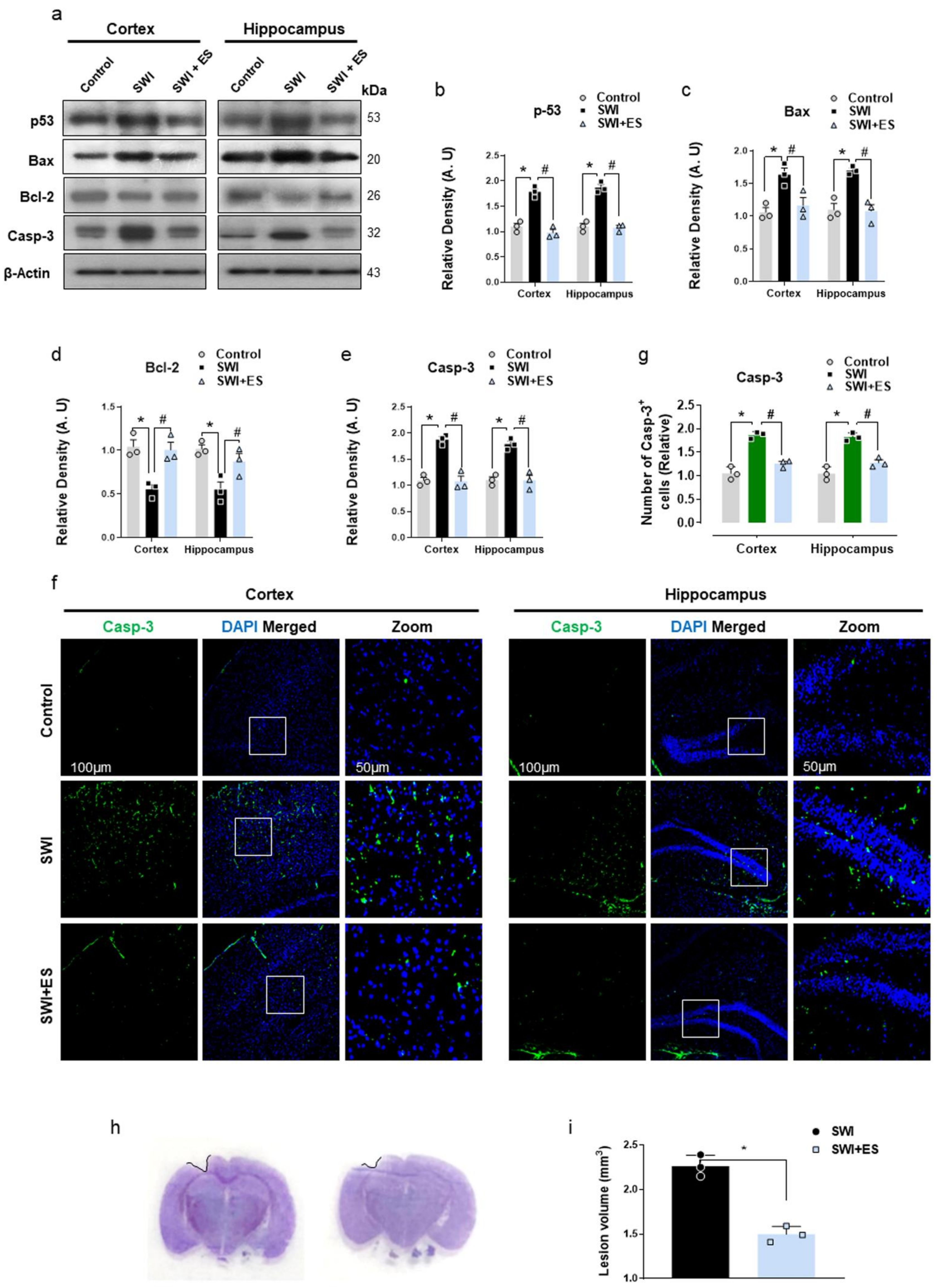
3.4. 17β-Estradiol Rescued Neuronal Apoptosis and Cell Death after Brain Injury
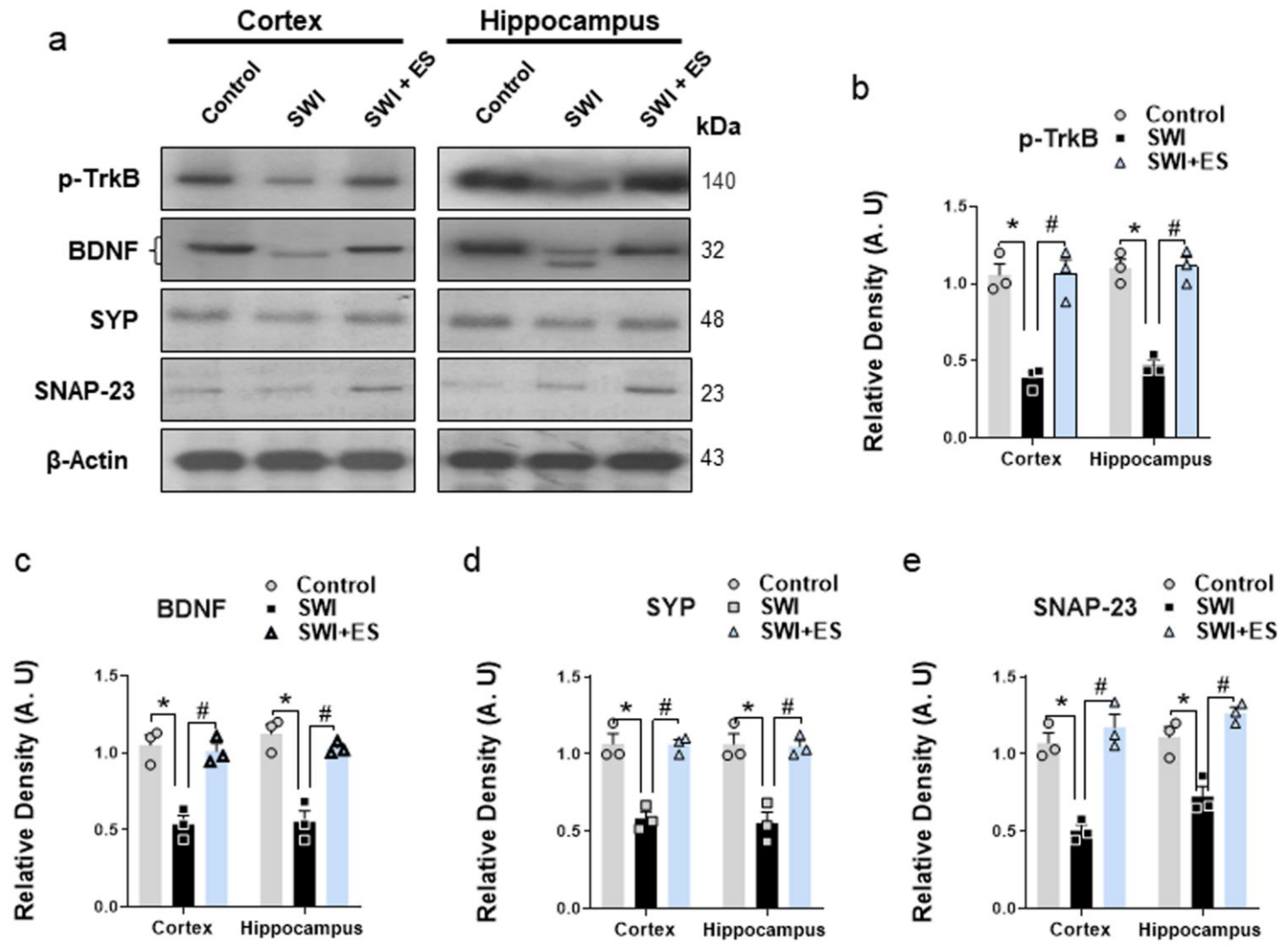
3.5. 17β-Estradiol Enhanced the Neurotrophic Effect and the Expression of Synaptic Proteins after Brain Injury
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | 5’AMP-activated protein kinase |
| Bax | Bcl-2-associated X protein |
| Bcl-2 | B-cell lymphoma 2 |
| CD64 | Fc γ receptor I |
| COX-2 | Cyclooxygenase-2 |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DG | Dentate gyrus |
| ES | 17β-estradiol |
| GFAP | Glial fibrillary acidic protein |
| GST | Glutathione S-transferase |
| GR | Glutathione reductase |
| Iba-1 | Ionized calcium-binding adaptor molecule 1 |
| IHC | Immunohistochemistry |
| iNOS | Inducible nitric oxide synthase |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| ROS | Reactive oxygen species |
| SNAP-23 | Synaptosomal-associated protein 23 |
| SWI | Stab wound injury |
| SYP | Synaptophysin |
| TGF-β | Transforming growth factor beta |
References
- Hackenberg, K.; Unterberg, A. Traumatic brain injury. Der Nervenarzt 2016, 87, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Hardy, J.; Zetterberg, H. The neuropathology and neurobiology of traumatic brain injury. Neuron 2012, 76, 886–899. [Google Scholar] [CrossRef] [Green Version]
- Abe, H.; Shimoji, K.; Nagamine, Y.; Fujiwara, S.; Izumi, S.I. Corrigendum to “Predictors of Recovery from Traumatic Brain Injury-Induced Prolonged Consciousness Disorder”. Neural Plast. 2020, 2020, 7169025. [Google Scholar] [CrossRef] [PubMed]
- Eastman, C.L.; D’Ambrosio, R.; Ganesh, T. Modulating neuroinflammation and oxidative stress to prevent epilepsy and improve outcomes after traumatic brain injury. Neuropharmacology 2020, 172, 107907. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, C.; Crupi, R.; Calabrese, V.; Graziano, A.; Milone, P.; Pennisi, G.; Radak, Z.; Calabrese, E.J.; Cuzzocrea, S. Traumatic brain injury: Oxidative stress and neuroprotection. Antioxid. Redox Signal. 2013, 19, 836–853. [Google Scholar] [CrossRef]
- Briones, T.L.; Woods, J.; Rogozinska, M. Decreased neuroinflammation and increased brain energy homeostasis following environmental enrichment after mild traumatic brain injury is associated with improvement in cognitive function. Acta Neuropathol. Commun. 2013, 1, 57. [Google Scholar] [CrossRef] [Green Version]
- Rehman, S.U.; Ikram, M.; Ullah, N.; Alam, S.I.; Park, H.Y.; Badshah, H.; Choe, K.; Kim, M.O. Neurological enhancement effects of melatonin against brain injury-induced oxidative stress, neuroinflammation, and neurodegeneration via AMPK/CREB signaling. Cells 2019, 8, 760. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Rodriguez, A.; Egea-Guerrero, J.J.; Murillo-Cabezas, F.; Carrillo-Vico, A. Oxidative stress in traumatic brain injury. Curr. Med. Chem. 2014, 21, 1201–1211. [Google Scholar] [CrossRef]
- Ikram, M.; Park, T.J.; Ali, T.; Kim, M.O. Antioxidant and neuroprotective effects of caffeine against Alzheimer’s and Parkinson’s disease: Insight into the role of Nrf-2 and A2AR signaling. Antioxidants 2020, 9, 902. [Google Scholar] [CrossRef]
- Muhammad, T.; Ikram, M.; Ullah, R.; Rehman, S.U.; Kim, M.O. Hesperetin, a citrus flavonoid, attenuates LPS-induced neuroinflammation, apoptosis and memory impairments by modulating TLR4/NF-κB signaling. Nutrients 2019, 11, 648. [Google Scholar] [CrossRef] [Green Version]
- Saeed, K.; Shah, S.A.; Ullah, R.; Alam, S.I.; Park, J.S.; Saleem, S.; Jo, M.H.; Kim, M.W.; Hahm, J.R.; Kim, M.O. Quinovic acid impedes cholesterol dyshomeostasis, oxidative stress, and neurodegeneration in an amyloid-beta-induced mouse model. Oxidative Med. Cell. Longev. 2020, 2020, 9523758. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Okamoto, S.I.; Cui, J.; Watanabe, Y.; Furuta, K.; Suzuki, M.; Tohyama, K.; Lipton, S.A. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophilic [correction of electrophillic] phase II inducers. Proc. Natl. Acad. Sci. USA 2006, 103, 768–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, D.S.A.; Oliver, P.L. ROS generation in microglia: Understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018, 285, 3576–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Towler, M.C.; Hardie, D.G. AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 2007, 100, 328–341. [Google Scholar] [CrossRef]
- Hill, J.L.; Kobori, N.; Zhao, J.; Rozas, N.S.; Hylin, M.J.; Moore, A.N.; Dash, P.K. Traumatic brain injury decreases AMP-activated protein kinase activity and pharmacological enhancement of its activity improves cognitive outcome. J. Neurochem. 2016, 139, 106–119. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.M.; Wong, M.D.; Samii, A.; Hovda, D.A. Evidence for energy failure following irreversible traumatic brain injury. Ann. N. Y. Acad. Sci. 1999, 893, 337–340. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ashford, M.L. AMPK: Regulating energy balance at the cellular and whole body levels. Physiology 2014, 29, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, T.; Ali, T.; Ikram, M.; Khan, A.; Alam, S.I.; Kim, M.O. Melatonin rescue oxidative stress-mediated neuroinflammation/neurodegeneration and memory impairment in scopolamine-induced amnesia mice model. J. Neuroimmune Pharmacol. 2019, 14, 278–294. [Google Scholar] [CrossRef]
- Karve, I.P.; Taylor, J.M.; Crack, P.J. The contribution of astrocytes and microglia to traumatic brain injury. Br. J. Pharm. 2016, 173, 692–702. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.C.; Ma, L.S.; Chu, Z.H.; Xu, H.; Wu, W.Q.; Liu, F. Regulation of microglial activation in stroke. Acta Pharm. Sin. 2017, 38, 445–458. [Google Scholar] [CrossRef]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.W.; Chen, J.H.; Lin, H.Y.; Liu, Y.S.; Tsai, C.F.; Chang, P.C.; Lu, D.Y.; Lin, C. Regulatory effects of neuroinflammatory responses through brain-derived neurotrophic factor signaling in microglial cells. Mol. Neurobiol. 2018, 55, 7487–7499. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.T.; Rao, V.L.; Sailor, K.A.; Bowen, K.K.; Dempsey, R.J. Protective effects of glial cell line-derived neurotrophic factor on hippocampal neurons after traumatic brain injury in rats. J. Neurosurg. 2001, 95, 674–679. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Z.; Li, J.; Wu, H.; Peng, Y.; Fan, L.; Chen, J.; Gu, C.; Yan, F.; Wang, L.; et al. The polarization states of microglia in TBI: A new paradigm for pharmacological intervention. Neural Plast. 2017, 2017, 5405104. [Google Scholar] [CrossRef] [PubMed]
- Chio, C.C.; Lin, M.T.; Chang, C.P. Microglial activation as a compelling target for treating acute traumatic brain injury. Curr. Med. Chem. 2015, 22, 759–770. [Google Scholar] [CrossRef]
- Crupi, R.; Cordaro, M.; Cuzzocrea, S.; Impellizzeri, D. Management of traumatic brain injury: From present to future. Antioxidants 2020, 9, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubal, D.B.; Zhu, H.; Yu, J.; Rau, S.W.; Shughrue, P.J.; Merchenthaler, I.; Kindy, M.S.; Wise, P.M. Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc. Natl. Acad. Sci. USA 2001, 98, 1952–1957. [Google Scholar] [CrossRef] [PubMed]
- Chiueh, C.; Lee, S.; Andoh, T.; Murphy, D. Induction of antioxidative and antiapoptotic thioredoxin supports neuroprotective hypothesis of estrogen. Endocrine 2003, 21, 27–31. [Google Scholar] [CrossRef]
- Wise, P.M.; Dubal, D.B.; Wilson, M.E.; Rau, S.W.; Liu, Y. Estrogens: Trophic and protective factors in the adult brain. Front. Neuroendocr. 2001, 22, 33–66. [Google Scholar] [CrossRef]
- Barha, C.K.; Galea, L.A. Influence of different estrogens on neuroplasticity and cognition in the hippocampus. Biochim. Biophys. Acta 2010, 1800, 1056–1067. [Google Scholar] [CrossRef]
- Lu, H.; Ma, K.; Jin, L.; Zhu, H.; Cao, R. 17beta-estradiol rescues damages following traumatic brain injury from molecule to behavior in mice. J. Cell Physiol. 2018, 233, 1712–1722. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Saeed, K.; Jo, M.G.; Kim, M.O. 17-beta estradiol rescued immature rat brain against glutamate-induced oxidative stress and neurodegeneration via regulating Nrf2/HO-1 and MAP-kinase signaling pathway. Antioxidants 2021, 10, 892. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.I.; Jo, M.G.; Park, T.J.; Ullah, R.; Ahmad, S.; Rehman, S.U.; Kim, M.O. Quinpirole-mediated regulation of dopamine D2 receptors inhibits glial cell-induced neuroinflammation in cortex and striatum after brain injury. Biomedicines 2021, 9, 47. [Google Scholar] [CrossRef]
- Shah, S.A.; Yoon, G.H.; Chung, S.S.; Abid, M.N.; Kim, T.H.; Lee, H.Y.; Kim, M.O. Novel osmotin inhibits SREBP2 via the AdipoR1/AMPK/SIRT1 pathway to improve Alzheimer’s disease neuropathological deficits. Mol. Psychiatry 2017, 22, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Rehman, S.U.; Khan, A.; Badshah, H.; Abid, N.B.; Kim, M.W.; Jo, M.H.; Chung, S.S.; Lee, H.G.; Rutten, B.P.F.; et al. Adiponectin-mimetic novel nonapeptide rescues aberrant neuronal metabolic-associated memory deficits in Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 23. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Rehman, S.U.; Shah, F.A.; Kim, M.O. Acute dose of melatonin via Nrf2 dependently prevents acute ethanol-induced neurotoxicity in the developing rodent brain. J. Neuroinflamm. 2018, 15, 119. [Google Scholar] [CrossRef]
- Rehman, S.U.; Ahmad, A.; Yoon, G.H.; Khan, M.; Abid, M.N.; Kim, M.O. Inhibition of c-Jun N-terminal kinase protects against brain damage and improves learning and memory after traumatic brain injury in adult mice. Cereb. Cortex 2018, 28, 2854–2872. [Google Scholar] [CrossRef]
- Khan, M.; Ullah, R.; Rehman, S.U.; Shah, S.A.; Saeed, K.; Muhammad, T.; Park, H.Y.; Jo, M.H.; Choe, K.; Rutten, B.P.F.; et al. 17beta-estradiol modulates SIRT1 and halts oxidative stress-mediated cognitive impairment in a male aging mouse model. Cells 2019, 8, 928. [Google Scholar] [CrossRef] [Green Version]
- Stelmasiak, Z.; Dudkowska-Konopa, A.; Rejdak, K. Head trauma and neuroprotection. Med. Sci. Monit. 2000, 6, 426–432. [Google Scholar]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharm. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Ziebell, J.M.; Morganti-Kossmann, M.C. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 2010, 7, 22–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burda, J.E.; Bernstein, A.M.; Sofroniew, M.V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 2016, 275 Pt 3, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Houlton, J.; Abumaria, N.; Hinkley, S.F.R.; Clarkson, A.N. Therapeutic potential of neurotrophins for repair after brain injury: A helping hand from biomaterials. Front. Neurosci. 2019, 13, 790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skaper, S.D. The neurotrophin family of neurotrophic factors: An overview. Methods Mol. Biol. 2012, 846, 1–12. [Google Scholar] [CrossRef]
- Kaplan, G.B.; Vasterling, J.J.; Vedak, P.C. Brain-derived neurotrophic factor in traumatic brain injury, post-traumatic stress disorder, and their comorbid conditions: Role in pathogenesis and treatment. Behav. Pharm. 2010, 21, 427–437. [Google Scholar] [CrossRef]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Carling, D. The AMP-activated protein kinase cascade—A unifying system for energy control. Trends Biochem. Sci. 2004, 29, 18–24. [Google Scholar] [CrossRef]
- Manwani, B.; McCullough, L.D. Function of the master energy regulator adenosine monophosphate-activated protein kinase in stroke. J. Neurosci. Res. 2013, 91, 1018–1029. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.M.; Shu, H.; Wang, L.; Xu, J.J.; Niu, X.C.; Zhang, L. SIRT1-dependent AMPK pathway in the protection of estrogen against ischemic brain injury. CNS Neurosci. Ther. 2017, 23, 360–369. [Google Scholar] [CrossRef] [Green Version]
- Razmara, A.; Duckles, S.P.; Krause, D.N.; Procaccio, V. Estrogen suppresses brain mitochondrial oxidative stress in female and male rats. Brain Res. 2007, 1176, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Brandes, M.S.; Gray, N.E. NRF2 as a therapeutic target in neurodegenerative diseases. ASN Neuro 2020, 12, 1759091419899782. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 2013, 100, 30–47. [Google Scholar] [CrossRef] [Green Version]
- Chanas, S.A.; Jiang, Q.; McMahon, M.; McWalter, G.K.; McLellan, L.I.; Elcombe, C.R.; Henderson, C.J.; Wolf, C.R.; Moffat, G.J.; Itoh, K.; et al. Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem. J. 2002, 365, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.J.; Thimmulappa, R.K.; Singh, A.; Blake, D.J.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic. Biol. Med. 2009, 46, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, H. Targeting the NF-E2-related factor 2 pathway: A novel strategy for traumatic brain injury. Mol. Neurobiol. 2018, 55, 1773–1785. [Google Scholar] [CrossRef]
- Dai, W.; Wang, H.; Fang, J.; Zhu, Y.; Zhou, J.; Wang, X.; Zhou, Y.; Zhou, M. Curcumin provides neuroprotection in model of traumatic brain injury via the Nrf2-ARE signaling pathway. Brain Res. Bull. 2018, 140, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, H.; Gao, Y.; Li, L.; Tang, C.; Wen, G.; Zhou, Y.; Zhou, M.; Mao, L.; Fan, Y. Protective effects of quercetin on mitochondrial biogenesis in experimental traumatic brain injury via the Nrf2 signaling pathway. PLoS ONE 2016, 11, e0164237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Wang, H.; Fan, Y.; Gao, Y.; Li, X.; Hu, Z.; Ding, K.; Wang, Y.; Wang, X. Fucoxanthin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE and Nrf2-autophagy pathways. Sci. Rep. 2017, 7, 46763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vegeto, E.; Benedusi, V.; Maggi, A. Estrogen anti-inflammatory activity in brain: A therapeutic opportunity for menopause and neurodegenerative diseases. Front. Neuroendocr. 2008, 29, 507–519. [Google Scholar] [CrossRef] [Green Version]
- Torihata, Y.; Asanuma, K.; Iijima, K.; Mikami, T.; Hamada, S.; Asano, N.; Koike, T.; Imatani, A.; Masamune, A.; Shimosegawa, T. Estrogen-dependent Nrf2 expression protects against reflux-induced esophagitis. Dig. Dis. Sci. 2018, 63, 345–355. [Google Scholar] [CrossRef]
- Wu, J.; Williams, D.; Walter, G.A.; Thompson, W.E.; Sidell, N. Estrogen increases Nrf2 activity through activation of the PI3K pathway in MCF-7 breast cancer cells. Exp. Cell Res. 2014, 328, 351–360. [Google Scholar] [CrossRef]
- Song, C.H.; Kim, N.; Kim, D.H.; Lee, H.N.; Surh, Y.J. 17-beta estradiol exerts anti-inflammatory effects through activation of Nrf2 in mouse embryonic fibroblasts. PLoS ONE 2019, 14, e0221650. [Google Scholar] [CrossRef]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system: A mixed blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef] [Green Version]
- Saito, M.; Saito, M.; Das, B.C. Involvement of AMP-activated protein kinase in neuroinflammation and neurodegeneration in the adult and developing brain. Int. J. Dev. Neurosci. 2019, 77, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, F.; Mander, K.A.; Leonard, A.V.; Vink, R. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J. Neuroinflamm. 2016, 13, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, L.; You, W.; Wang, H.; Meng, Y.; Feng, J.; Yang, X. Polarization of microglia to the M2 phenotype in a peroxisome proliferator-activated receptor gamma-dependent manner attenuates axonal injury induced by traumatic brain injury in mice. J. Neurotrauma 2018, 35, 2330–2340. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, J.; Hu, X.; Zhang, L.; Mao, L.; Jiang, X.; Liou, A.K.; Leak, R.K.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J. Cereb. Blood Flow Metab. 2013, 33, 1864–1874. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, Y.; Chiba, K. Diversity and plasticity of microglial cells in psychiatric and neurological disorders. Pharm. Ther. 2015, 154, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.Y.; Liu, L.; Yang, Q.W. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog. Neurobiol. 2016, 142, 23–44. [Google Scholar] [CrossRef]
- Collmann, F.M.; Pijnenburg, R.; Hamzei-Taj, S.; Minassian, A.; Folz-Donahue, K.; Kukat, C.; Aswendt, M.; Hoehn, M. Individual in vivo profiles of microglia polarization after stroke, represented by the genes iNOS and Ym1. Front. Immunol. 2019, 10, 1236. [Google Scholar] [CrossRef]
- Akinrinmade, O.A.; Chetty, S.; Daramola, A.K.; Islam, M.U.; Thepen, T.; Barth, S. CD64: An attractive immunotherapeutic target for M1-type macrophage mediated chronic inflammatory diseases. Biomedicines 2017, 5, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Ore, M.V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Savman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef]
- Kanazawa, M.; Ninomiya, I.; Hatakeyama, M.; Takahashi, T.; Shimohata, T. Microglia and monocytes/macrophages polarization reveal novel therapeutic mechanism against stroke. Int. J. Mol. Sci. 2017, 18, 2135. [Google Scholar] [CrossRef]
- Villa, A.; Vegeto, E.; Poletti, A.; Maggi, A. Estrogens, neuroinflammation, and neurodegeneration. Endocr. Rev. 2016, 37, 372–402. [Google Scholar] [CrossRef] [Green Version]
- Aryanpour, R.; Pasbakhsh, P.; Zibara, K.; Namjoo, Z.; Beigi Boroujeni, F.; Shahbeigi, S.; Kashani, I.R.; Beyer, C.; Zendehdel, A. Progesterone therapy induces an M1 to M2 switch in microglia phenotype and suppresses NLRP3 inflammasome in a cuprizone-induced demyelination mouse model. Int. Immunopharmacol. 2017, 51, 131–139. [Google Scholar] [CrossRef]
- Thakkar, R.; Wang, R.; Wang, J.; Vadlamudi, R.K.; Brann, D.W. 17beta-estradiol regulates microglia activation and polarization in the hippocampus following global cerebral ischemia. Oxidative Med. Cell. Longev. 2018, 2018, 4248526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hou, Y.; Zhang, L.; Liu, M.; Zhao, J.; Zhang, Z.; Ma, Y.; Hou, W. Estrogen attenuates traumatic brain injury by inhibiting the activation of microglia and astrocyte-mediated neuroinflammatory responses. Mol. Neurobiol. 2021, 58, 1052–1061. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflammation 2014, 11, 98. [Google Scholar] [CrossRef] [Green Version]
- Gensel, J.C.; Zhang, B. Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res. 2015, 1619, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikram, M.; Jo, M.G.; Park, T.J.; Kim, M.W.; Khan, I.; Jo, M.H.; Kim, M.O. Oral administration of gintonin protects the brains of mice against aβ-induced Alzheimer disease pathology: Antioxidant and anti-inflammatory effects. Oxidative Med. Cell. Longev. 2021, 2021, 16. [Google Scholar] [CrossRef]
- Saijo, K.; Glass, C.K. Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Colton, C.A. Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroimmune Pharm. 2009, 4, 399–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Stoica, B.A.; Loane, D.J.; Yang, M.; Abulwerdi, G.; Khan, N.; Kumar, A.; Thom, S.R.; Faden, A.I. Microglial-derived microparticles mediate neuroinflammation after traumatic brain injury. J. Neuroinflamm. 2017, 14, 47. [Google Scholar] [CrossRef] [Green Version]
- Needham, E.J.; Helmy, A.; Zanier, E.R.; Jones, J.L.; Coles, A.J.; Menon, D.K. The immunological response to traumatic brain injury. J. Neuroimmunol. 2019, 332, 112–125. [Google Scholar] [CrossRef]
- Block, M.L.; Hong, J.S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef]
- Angeloni, C.; Prata, C.; Dalla Sega, F.V.; Piperno, R.; Hrelia, S. Traumatic brain injury and NADPH oxidase: A deep relationship. Oxidative Med. Cell. Longev. 2015, 2015, 370312. [Google Scholar] [CrossRef] [Green Version]
- Gunther, M.; Plantman, S.; Davidsson, J.; Angeria, M.; Mathiesen, T.; Risling, M. COX-2 regulation and TUNEL-positive cell death differ between genders in the secondary inflammatory response following experimental penetrating focal brain injury in rats. Acta Neurochir. 2015, 157, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Dehlaghi Jadid, K.; Davidsson, J.; Lidin, E.; Hanell, A.; Angeria, M.; Mathiesen, T.; Risling, M.; Gunther, M. COX-2 inhibition by diclofenac is associated with decreased apoptosis and lesion area after experimental focal penetrating traumatic brain injury in rats. Front. Neurol. 2019, 10, 811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Dai, X.; Zhou, L.; Li, X.; Pan, D. Edaravone plays protective effects on LPS-induced microglia by switching M1/M2 phenotypes and regulating NLRP3 inflammasome activation. Front. Pharm. 2021, 12, 691773. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Subramanian, S.; Dziennis, S.; Jia, J.; Uchida, M.; Akiyoshi, K.; Migliati, E.; Lewis, A.D.; Vandenbark, A.A.; Offner, H.; et al. Estradiol and G1 reduce infarct size and improve immunosuppression after experimental stroke. J. Immunol. 2010, 184, 4087–4094. [Google Scholar] [CrossRef] [Green Version]
- Gibson, C.L.; Constantin, D.; Prior, M.J.; Bath, P.M.; Murphy, S.P. Progesterone suppresses the inflammatory response and nitric oxide synthase-2 expression following cerebral ischemia. Exp. Neurol. 2005, 193, 522–530. [Google Scholar] [CrossRef]
- Kipp, M.; Berger, K.; Clarner, T.; Dang, J.; Beyer, C. Sex steroids control neuroinflammatory processes in the brain: Relevance for acute ischaemia and degenerative demyelination. J. Neuroendocr. 2012, 24, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Nadkarni, S.; McArthur, S. Oestrogen and immunomodulation: New mechanisms that impact on peripheral and central immunity. Curr. Opin. Pharm. 2013, 13, 576–581. [Google Scholar] [CrossRef]
- Plesnila, N.; von Baumgarten, L.; Retiounskaia, M.; Engel, D.; Ardeshiri, A.; Zimmermann, R.; Hoffmann, F.; Landshamer, S.; Wagner, E.; Culmsee, C. Delayed neuronal death after brain trauma involves p53-dependent inhibition of NF-kappaB transcriptional activity. Cell Death Differ. 2007, 14, 1529–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.Y.; Chu, Y.H.; Tweedie, D.; Yu, Q.S.; Pick, C.G.; Hoffer, B.J.; Greig, N.H.; Wang, J.Y. Post-trauma administration of the pifithrin-alpha oxygen analog improves histological and functional outcomes after experimental traumatic brain injury. Exp. Neurol. 2015, 269, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Filichia, E.; Shen, H.; Zhou, X.; Qi, X.; Jin, K.; Greig, N.; Hoffer, B.; Luo, Y. Forebrain neuronal specific ablation of p53 gene provides protection in a cortical ischemic stroke model. Neuroscience 2015, 295, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Haldar, S. The relationship between BcI2, Bax and p53: Consequences for cell cycle progression and cell death. Mol. Hum. Reprod. 1998, 4, 1099–1109. [Google Scholar] [CrossRef]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Gudkov, A.V.; Komarova, E.A. Pathologies associated with the p53 response. Cold Spring Harb. Perspect. Biol. 2010, 2, a001180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borras, C.; Gambini, J.; Lopez-Grueso, R.; Pallardo, F.V.; Vina, J. Direct antioxidant and protective effect of estradiol on isolated mitochondria. Biochim. Biophys. Acta 2010, 1802, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.C.; Lee, J.H.; Hong, K.W.; Lee, K.S. 17 Beta-estradiol prevents focal cerebral ischemic damages via activation of Akt and CREB in association with reduced PTEN phosphorylation in rats. Fundam. Clin. Pharm. 2004, 18, 547–557. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlo, L.; Cimino, F.; Angileri, F.F.; La Torre, D.; Conti, A.; Cardali, S.M.; Saija, A.; Germano, A. Alteration in synaptic junction proteins following traumatic brain injury. J. Neurotrauma 2014, 31, 1375–1385. [Google Scholar] [CrossRef]
- Ansari, M.A.; Roberts, K.N.; Scheff, S.W. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic. Biol. Med. 2008, 45, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Meyer, E.M.; Simpkins, J.W. The effect of ovariectomy and estradiol replacement on brain-derived neurotrophic factor messenger ribonucleic acid expression in cortical and hippocampal brain regions of female Sprague-Dawley rats. Endocrinology 1995, 136, 2320–2324. [Google Scholar] [CrossRef]
- Sohrabji, F.; Miranda, R.C.; Toran-Allerand, C.D. Identification of a putative estrogen response element in the gene encoding brain-derived neurotrophic factor. Proc. Natl. Acad. Sci. USA 1995, 92, 11110–11114. [Google Scholar] [CrossRef] [Green Version]
- Jezierski, M.K.; Sohrabji, F. Region- and peptide-specific regulation of the neurotrophins by estrogen. Brain Res. Mol. Brain Res. 2000, 85, 77–84. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Host | Product Identifier | Application (Conc.) | Manufacturer |
|---|---|---|---|---|
| p-AMPK | Rabbit | #2535S | WB (1:1000), IF (1:100) | Cell Signaling |
| AMPK | = | #2603S | WB (1:1000) | = |
| Nrf2 | Mouse | SC-722 | WB (1:1000) | Santa Cruz |
| Histon H3 | Rabbit | ab1791 | WB (1:1000) | Abcam |
| HO-1 | Mouse | SC-136961 | WB (1:1000) | Santa Cruz |
| GFAP | = | SC-33673 | WB (1:1000) | = |
| Iba-1 | = | SC-32725 | WB (1:1000) | = |
| Iba-1 | Rabbit | PA5-27436 | IF (1:100), IHC (1:100) | Thermo Fisher |
| iNOS | Mouse | SC-7271 | WB (1:1000) | Santa Cruz |
| CD64 | = | SC-515431 | WB (1:10,000) | = |
| TGF-β | Rabbit | MA5-15065 | WB (1:1000) | Thermo Fisher |
| IGF-1 | Rabbit | ab133542 | WB (1:1000) | Abcam |
| IL1-β | Mouse | SC-32294 | WB (1:1000) | Santa Cruz |
| TNF-α | = | SC-52746 | WB (1:1000) | = |
| COX-2 | = | SC-376861 | WB (1:1000), IF (1:100) | = |
| p-TrkB | = | SC-8058 | WB (1:1000) | = |
| BDNF | = | SC-65514 | WB (1:1000) | = |
| SNAP-23 | = | SC-374215 | WB (1:1000) | = |
| SYP | = | SC 17750 | WB (1:1000) | = |
| p-53 | = | SC-126 | WB (1:1000) | = |
| Bax | Rabbit | 2772S | WB (1:1000) | Cell Signaling |
| Bcl-2 | Mouse | SC: 7382 | WB (1:1000) | Santa Cruz |
| Caspase-3 | = | SC-7272 | WB (1:1000), IF (1:100) | = |
| β-Actin | Mouse | SC-47778 | WB (1:1000) | = |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saeed, K.; Jo, M.H.; Park, J.S.; Alam, S.I.; Khan, I.; Ahmad, R.; Khan, A.; Ullah, R.; Kim, M.O. 17β-Estradiol Abrogates Oxidative Stress and Neuroinflammation after Cortical Stab Wound Injury. Antioxidants 2021, 10, 1682. https://doi.org/10.3390/antiox10111682
Saeed K, Jo MH, Park JS, Alam SI, Khan I, Ahmad R, Khan A, Ullah R, Kim MO. 17β-Estradiol Abrogates Oxidative Stress and Neuroinflammation after Cortical Stab Wound Injury. Antioxidants. 2021; 10(11):1682. https://doi.org/10.3390/antiox10111682
Chicago/Turabian StyleSaeed, Kamran, Myeung Hoon Jo, Jun Sung Park, Sayed Ibrar Alam, Ibrahim Khan, Riaz Ahmad, Amjad Khan, Rahat Ullah, and Myeong Ok Kim. 2021. "17β-Estradiol Abrogates Oxidative Stress and Neuroinflammation after Cortical Stab Wound Injury" Antioxidants 10, no. 11: 1682. https://doi.org/10.3390/antiox10111682