
Histochrome Attenuates Myocardial Ischemia-Reperfusion Injury by Inhibiting Ferroptosis-Induced Cardiomyocyte Death

 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Histochrome
2.2. Experimental Animals
2.3. Ischemia/Reperfusion Injury Model and In Vivo Drug Administration
2.4. Measurement of Myocardial Infarct Size
2.5. Evaluation of Heart Function by Echocardiography
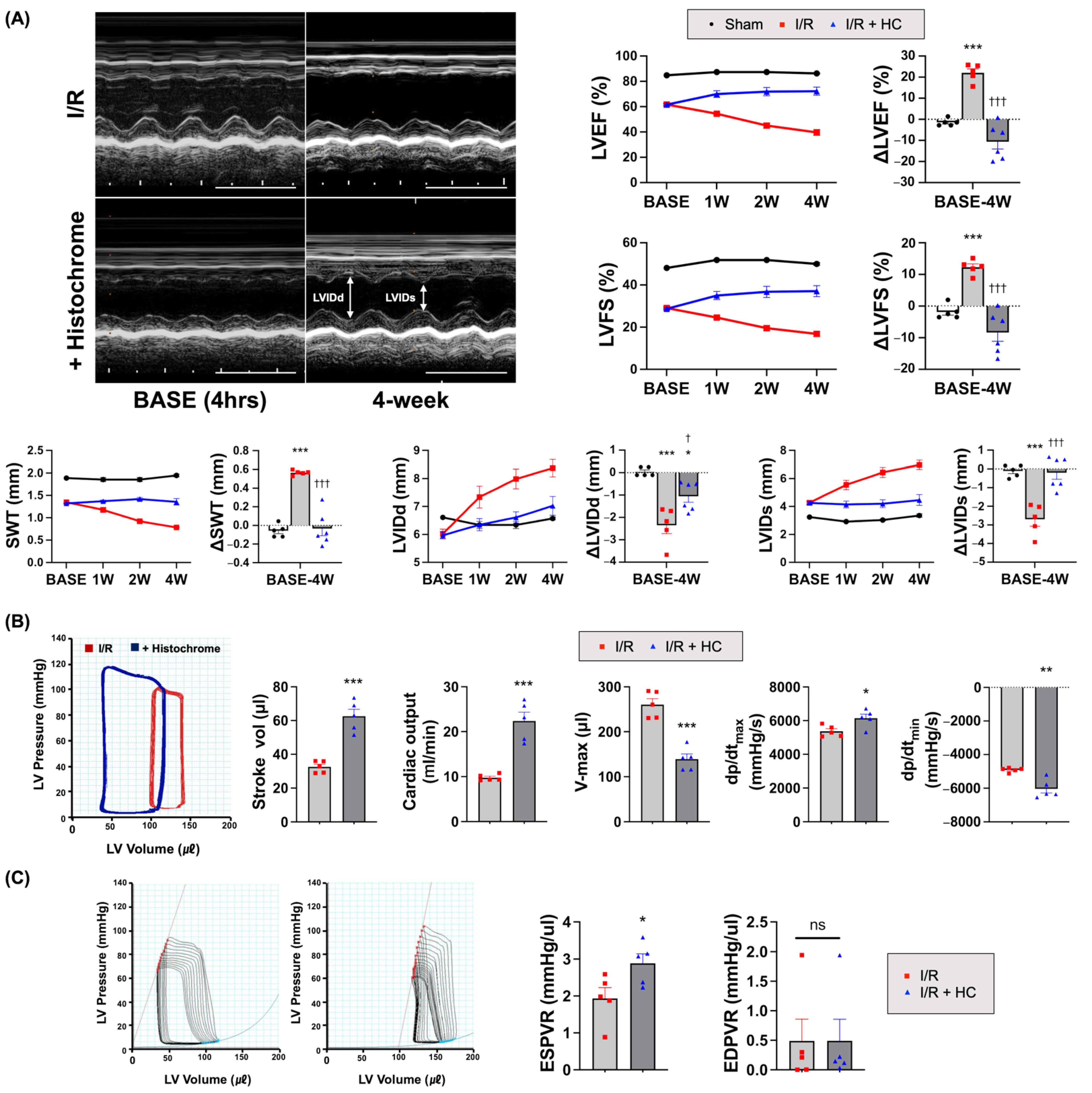
2.6. Hemodynamic Measurements
2.7. Measurement of Capillary Density by Immunohistochemical Staining
2.8. Measurement of Myocardial Infarct Size by Masson’s Trichrome Staining
2.9. Western Blotting
2.10. Evaluation of Mitochondrial Damage Scores by Transmission Electron Microscopy
2.11. Neonatal Rat Cardiomyocyte Isolation
2.12. Chemicals and Reagents
2.13. Cell Viability Assays
2.14. Quantitative Real-Time PCR with Reverse Transcription
2.15. Measurement of Cellular ROS and Mitochondrial Superoxide
2.16. Measurement of Lipid Peroxidation
2.17. Measurement of Glutathione Peroxidase Activity
2.18. Measurement of Glutathione (GSH) and Oxidized Glutathione (GSSG)
2.19. Detection of Intracellular Ferrous Ions (Fe2+)
2.20. Statistical Analysis
3. Results
3.1. Cardioprotective Effects of Histochrome on Myocardial I/R Injury
3.2. Prolonged Cardioprotective Effects of Histochrome on Myocardial I/R Injury
3.3. Histochrome Ameliorates Effect in Infarcted Hearts by Reducing Adverse Cardiac Remodeling after I/R
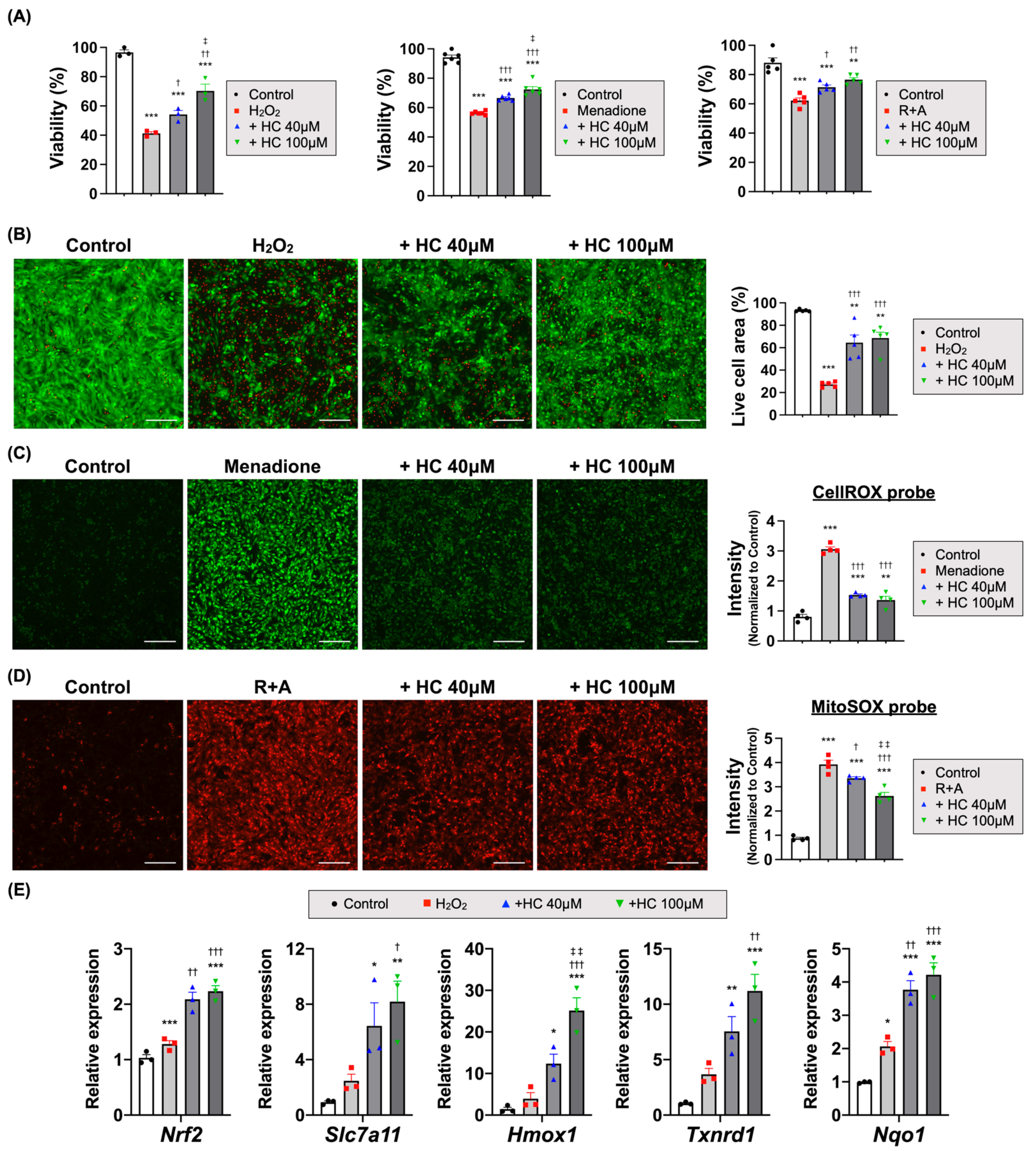
3.4. Histochrome Protects the Cardiomyocytes from I/R Injury via an Antioxidant Effect
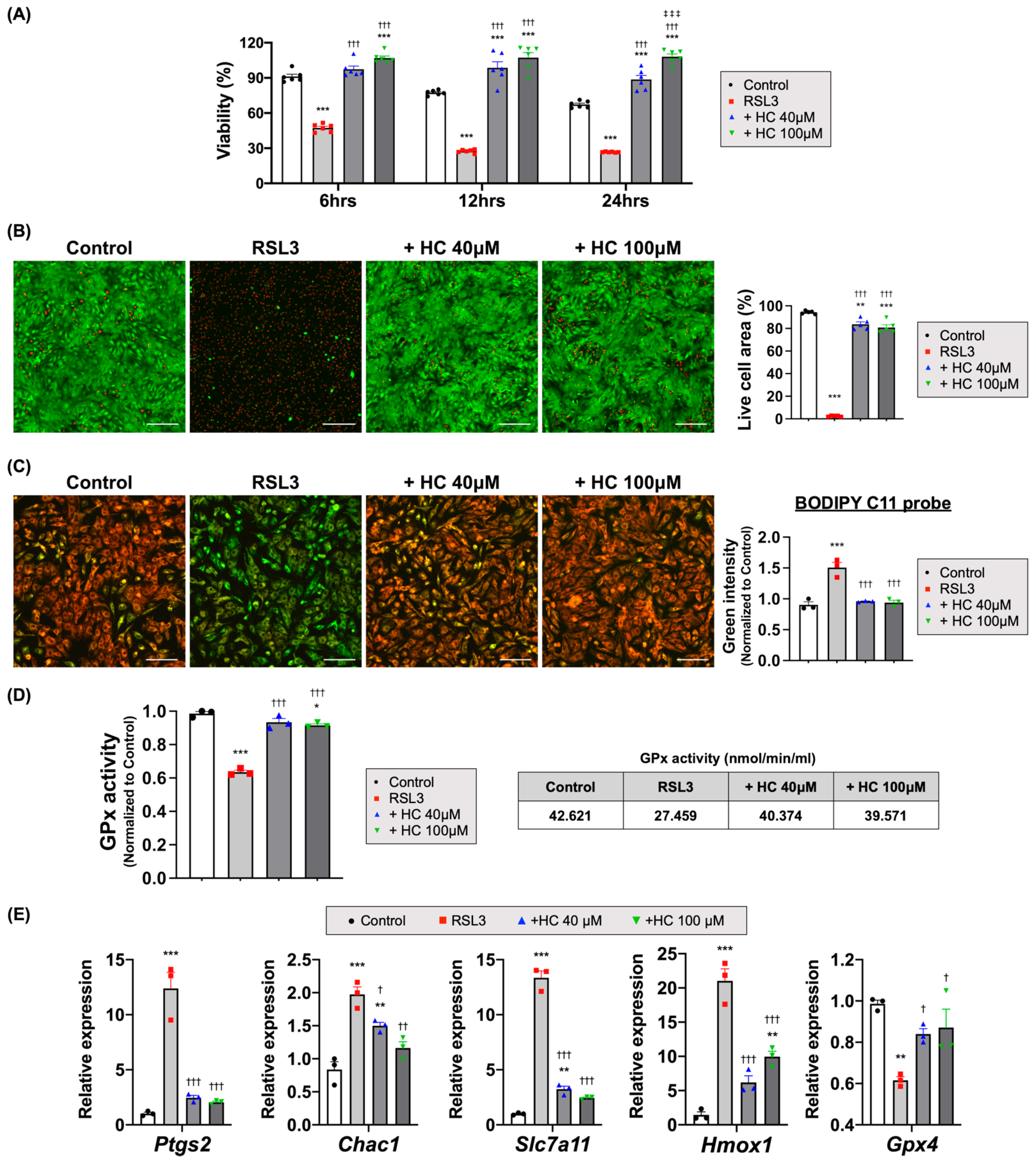
3.5. Histochrome Protected NRCMs from RSL3 and Erastin-Induced Ferroptotic Cell Death
3.6. Histochrome Attenuates Lipid Peroxidation by Upregulating Glutathione Peroxidase Activity and Iron-Chelating Capacity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Metra, M.; Teerlink, J.R. Heart failure. Lancet 2017, 390, 1981–1995. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of Ischemia/Reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [CrossRef] [Green Version]
- Heusch, G.; Libby, P.; Gersh, B.; Yellon, D.; Böhm, M.; Lopaschuk, G.; Opie, L. Cardiovascular remodelling in coronary artery disease and heart failure. Lancet 2014, 383, 1933–1943. [Google Scholar] [CrossRef] [Green Version]
- Heusch, G.; Gersh, B.J. The pathophysiology of acute myocardial infarction and strategies of protection beyond reperfusion: A continual challenge. Eur. Heart J. 2017, 38, 774–784. [Google Scholar] [CrossRef]
- Gross, G.J.; Auchampach, J.A. Reperfusion injury: Does it exist? J. Mol. Cell. Cardiol. 2007, 42, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Yellon, D.; Hausenloy, D. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Heusch, G. Myocardial ischaemia—Reperfusion injury and cardioprotection in perspective. Nat. Rev. Cardiol. 2020, 17, 773–789. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Avery, S.V. Molecular targets of oxidative stress. Biochem. J. 2011, 434, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Del Valle, N.R.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.K.; Adameova, A.; Hill, J.A.; Baines, C.; Kang, P.M.; Downey, J.M.; Narula, J.; Takahashi, M.; Abbate, A.; Piristine, H.; et al. Guidelines for evaluating myocardial cell death. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H891–H922. [Google Scholar] [CrossRef]
- Caricati-Neto, A.; Errante, P.R.; Menezes-Rodrigues, F.S. Recent advances in pharmacological and non-pharmacological strategies of cardioprotection. Int. J. Mol. Sci. 2019, 20, 4002. [Google Scholar] [CrossRef] [Green Version]
- Jovanović, A. Cardioprotective signalling: Past, present and future. Eur. J. Pharmacol. 2018, 833, 314–319. [Google Scholar] [CrossRef]
- Heusch, G. Molecular basis of cardioprotection: Signal transduction in ischemic pre-, post-, and remote conditioning. Circ. Res. 2015, 116, 674–699. [Google Scholar] [CrossRef]
- Heusch, G. Critical issues for the translation of cardioprotection. Circ. Res. 2017, 120, 1477–1486. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Chen, X.B.; Ying, M.D.; He, Q.J.; Cao, J.; Yang, B. The role of ferroptosis in cancer development and treatment response. Front. Pharmacol. 2018, 8, 992. [Google Scholar] [CrossRef]
- Guiney, S.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem. Int. 2017, 104, 34–48. [Google Scholar] [CrossRef]
- Tuo, Q.-Z.; Lei, P.; A Jackman, K.; Li, X.-L.; Xiong, H.; Liuyang, Z.-Y.; Roisman, L.C.; Zhang, S.-T.; Ayton, S.; Wang, Q.; et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 2017, 22, 1520–1530. [Google Scholar] [CrossRef]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed cell-death by ferroptosis: Antioxidants as mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef] [Green Version]
- Stamenkovic, A.; Pierce, G.N.; Ravandi, A. Phospholipid oxidation products in ferroptotic myocardial cell death. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H156–H163. [Google Scholar] [CrossRef]
- Sunitha, D. A review on antioxidant methods. Asian J. Pharm. Clin. Res. 2016, 9, 14–32. [Google Scholar] [CrossRef]
- Manal, A.A.; Kareem, S.D.; Mohammed, A.A. Antioxidant categories and mode of action. In Antioxidants; IntechOpen: London, UK, 2019. [Google Scholar]
- Anderson, H.A.; Mathieson, J.W.; Thomson, R.H. Distribution of spinochrome pigments in echinoids. Comp. Biochem. Physiol. 1969, 28, 333–345. [Google Scholar] [CrossRef]
- Mishchenko, N. Histochrome: A new original domestic drug. Pharm. Chem. J. 2003, 37, 48–52. [Google Scholar] [CrossRef]
- Utkina, N.K.; Pokhilo, N.D. Free radical scavenging activities of naturally occurring and synthetic analogues of sea urchin naphthazarin pigments. Nat. Prod. Commun. 2012, 7, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Lee, N.-K.; Lim, H.J.; Mazumder, S.; Rethineswaran, V.K.; Kim, Y.-J.; Jang, W.B.; Ji, S.T.; Kang, S.; Kim, D.Y.; et al. Therapeutic cell protective role of histochrome under oxidative stress in human cardiac progenitor cells. Mar. Drugs 2019, 17, 368. [Google Scholar] [CrossRef] [Green Version]
- Lebedev, A.V.; Ivanova, M.V.; Levitsky, D.O. Echinochrome, a naturally occurring iron chelator and free radical scavenger in artificial and natural membrane systems. Life Sci. 2005, 76, 863–875. [Google Scholar] [CrossRef]
- Mischenko, N.P.; Fedoreev, S.A.; Zapara, T.A.; Ratushnyak, A.S. Effects of histochrom and emoxypin on biophysical properties of electroexitable cells. Bull. Exp. Biol. Med. 2009, 147, 196–200. [Google Scholar] [CrossRef]
- Coates, C.J.; McCulloch, C.; Betts, J.; Whalley, T. Echinochrome a release by red spherule cells is an iron-withholding strategy of sea urchin innate immunity. J. Innate Immun. 2018, 10, 119–130. [Google Scholar] [CrossRef]
- Shvilkin, A.V.; Serebriakov, I.L.; Tskitishvili, O.V.; Sadretdinov, S.M.; Kol’Tsova, A.E.; Maksimov, O.B.; Mishchenko, N.P.; Novikov, V.L.; O Levitskiĭ, D.; Ruda, M.I. Effect of echinochrom on experimental myocardial reperfusion injury. Kardiologiia 1991, 31, 79–81. [Google Scholar]
- Anufriey, V.P.; Novikov, V.L.; Maximov, O.B.; Elyakov, G.B.; Levitsky, D.O.; Lebedev, A.V.; Sadretdinov, S.M.; Shvilkin, A.V.; Afonskaya, N.I.; Ruda, M.Y.; et al. Synthesis of some hydroxynaphthazarins and their cardioprotective effects under ischemia-reperfusion in vivo. Bioorg. Med. Chem. Lett. 1998, 8, 587–592. [Google Scholar] [CrossRef]
- Buĭmov, A.G.; Maksimov, I.V.; Perchatkin, A.V.; Repin, A.N.; Afanas’Ev, A.S.; Markov, A.V.; Karpov, R.S. Effect of the bioantioxidant histochrome on myocardial injury in reperfusion therapy on patients with myocardial infarction. Ter. Arkhiv 2002, 74, 12–16. [Google Scholar]
- Mischenko, N.P.; Fedoreyev, S.A.; Pokhilo, N.D.; Anufriev, V.P.; Denisenko, A.V.A.; Glazunov, V.P. Echinamines A and B, first aminated hydroxynaphthazarins from the sea urchin scaphechinus mirabilis. J. Nat. Prod. 2005, 68, 1390–1393. [Google Scholar] [CrossRef]
- Bøtker, H.E.; Hausenloy, D.; Andreadou, I.; Antonucci, S.; Boengler, K.; Davidson, S.; Deshwal, S.; Devaux, Y.; Di Lisa, F.; Di Sante, M.; et al. Practical guidelines for rigor and reproducibility in preclinical and clinical studies on cardioprotection. Basic Res. Cardiol. 2018, 113, 39. [Google Scholar] [CrossRef] [Green Version]
- Flameng, W.; Borgers, M.; Daenen, W.; Stalpaert, G. Ultrastructural and cytochemical correlates of myocardial protection by cardiac hypothermia in man. J. Thorac. Cardiovasc. Surg. 1980, 79, 413–424. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Andia, A.A.; Liu, H.; Csuka, J.M.; Hurlocker, B.; Vaiana, C.A.; Heindel, D.W.; Zuckerman, D.S.; Bos, P.H.; Reznik, E.; et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 2018, 14, 507–515. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.; Kagan, V.E.; et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Park, T.-J.; Park, J.H.; Lee, G.S.; Lee, J.-Y.; Shin, J.H.; Kim, M.W.; Kim, Y.S.; Kim, J.-Y.; Oh, K.-J.; Han, B.-S.; et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 2019, 10, 835. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Chen, P.; Zhai, B.; Zhang, M.; Xiang, Y.; Fang, J.; Xu, S.; Gao, Y.; Chen, X.; Sui, X.; et al. The emerging role of ferroptosis in inflammation. Biomed. Pharmacother. 2020, 127, 110108. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Dai, E.; Zhang, W.; Cong, D.; Kang, R.; Wang, J.; Tang, D. AIFM2 blocks ferroptosis independent of ubiquinol metabolism. Biochem. Biophys. Res. Commun. 2020, 523, 966–971. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; Da, S.M.; Ingold, I.; Goya, G.A.; Xavier, D.S.T.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, F.; Yin, H.-L.; Huang, Z.-J.; Lin, Z.-T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Xie, M.; Kang, R.; Fan, Y.; Niu, X.; Wang, H.; Cao, L.; Tang, D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015, 34, 5617–5625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, Z.; Zou, P.; Liu, F.; Xia, Z.; Li, J. Ferroptosis is a potential novel diagnostic and therapeutic target for patients with cardiomyopathy. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Li, S.; Rozanski, G.J.; Zheng, M.-Q. Glutathione homeostasis in ventricular myocytes from rat hearts with chronic myocardial infarction. Exp. Physiol. 2009, 94, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Humeres, C.; Frangogiannis, N.G. Fibroblasts in the infarcted, remodeling, and failing heart. JACC Basic Transl. Sci. 2019, 4, 449–467. [Google Scholar] [CrossRef]
- Fan, D.; Takawale, A.; Lee, J.; Kassiri, Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair 2012, 5, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction—From repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loor, G.; Kondapalli, J.; Schriewer, J.M.; Chandel, N.S.; Hoek, T.L.V.; Schumacker, P.T. Menadione triggers cell death through ROS-dependent mechanisms involving PARP activation without requiring apoptosis. Free Radic. Biol. Med. 2010, 49, 1925–1936. [Google Scholar] [CrossRef] [Green Version]
- Heinzel, F.R.; Luo, Y.; Li, X.; Boengler, K.; Buechert, A.; García-Dorado, D.; Di Lisa, F.; Schulz, R.; Heusch, G. Impairment of diazoxide-induced formation of reactive oxygen species and loss of cardioprotection in connexin 43 deficient mice. Circ. Res. 2005, 97, 583–586. [Google Scholar] [CrossRef] [Green Version]
- Turrens, J.F.; Boveris, A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem. J. 1980, 191, 421–427. [Google Scholar] [CrossRef]
- Turrens, J.F. Superoxide production by the mitochondrial respiratory chain. Biosci. Rep. 1997, 17, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Cai, Z.; Wang, H.; Han, D.; Cheng, Q.; Zhang, P.; Gao, F.; Yu, Y.; Song, Z.; Wu, Q.; et al. Loss of cardiac ferritin H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ. Res. 2020, 127, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M.; Valente, M.; Ferri, L.; Gregolin, C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim. Biophys. Acta 1982, 710, 197–211. [Google Scholar] [CrossRef]
- Conrad, M.; Kagan, V.E.; Bayir, H.; Pagnussat, G.; Head, B.; Traber, M.; Stockwell, B.R. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018, 32, 602–619. [Google Scholar] [CrossRef] [Green Version]
- Forcina, G.C.; Dixon, S.J. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics 2019, 19, e1800311. [Google Scholar] [CrossRef]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. 2018, 38, 12–13. [Google Scholar] [CrossRef] [Green Version]
- Conrad, M.; Sato, H. The oxidative stress-inducible cystine/glutamate antiporter, system xc−: Cystine supplier and beyond. Amino Acids 2011, 42, 231–246. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system xc−in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [Green Version]
- Nishizawa, H.; Matsumoto, M.; Shindo, T.; Saigusa, D.; Kato, H.; Suzuki, K.; Sato, M.; Ishii, Y.; Shimokawa, H.; Igarashi, K. Ferroptosis is controlled by the coordinated transcriptional regulation of glutathione and labile iron metabolism by the transcription factor BACH. J. Biol. Chem. 2020, 295, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Sun, L.; Wu, W.; Wu, J.; Sun, Z.; Ren, J. USP22 Protects against myocardial ischemia—Reperfusion Injury via the SIRT1-p53/SLC7A11—Dependent inhibition of ferroptosis—Induced cardiomyocyte death. Front. Physiol. 2020, 11, 551318. [Google Scholar] [CrossRef]
- Soetan, K.; Olaiya, C.; Oyewole, O. The importance of mineral elements for humans, domestic animals and plants—A review. Afr. J. Food Sci. 2010, 4, 200–222. [Google Scholar] [CrossRef]
- Frank, A.; Bonney, M.; Bonney, S.; Weitzel, L.; Koeppen, M.; Eckle, T. Myocardial ischemia reperfusion injury: From basic science to clinical bedside. Semin. Cardiothorac. Vasc. Anesth. 2012, 16, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.-F. Clinical manifestations and basic mechanisms of myocardial ischemia/reperfusion injury. Tzu Chi Med. J. 2018, 30, 209–215. [Google Scholar] [CrossRef]
- Alim, I.; Caulfield, J.T.; Chen, Y.; Swarup, V.; Geschwind, D.H.; Ivanova, E.; Seravalli, J.; Ai, Y.; Sansing, L.H.; Ste.Marie, E.J.; et al. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell 2019, 177, 1262–1279.e25. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Li, Q.-Q.; Jia, J.-N.; Sun, Q.-Y.; Zhou, H.-H.; Jin, W.-L.; Mao, X.-Y. Baicalein Exerts neuroprotective effects in fecl3-induced posttraumatic epileptic seizures via suppressing ferroptosis. Front. Pharmacol. 2019, 10, 638. [Google Scholar] [CrossRef]
- Liu, B.; Zhao, C.; Li, H.; Chen, X.; Ding, Y.; Xu, S. Puerarin protects against heart failure induced by pressure overload through mitigation of ferroptosis. Biochem. Biophys. Res. Commun. 2018, 497, 233–240. [Google Scholar] [CrossRef]
- Vomund, S.; Schäfer, A.; Parnham, M.J.; Brüne, B.; Von Knethen, A. Nrf2, the master regulator of anti-oxidative responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [Green Version]
- Panieri, E.; Telkoparan-Akillilar, P.; Suzen, S.; Saso, L. The NRF2/KEAP1 axis in the regulation of tumor metabolism: Mechanisms and therapeutic perspectives. Biomolecules 2020, 10, 791. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine—Glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.-K.; Chen, S.-E.; Chang, L.-C. A dual role of heme oxygenase-1 in cancer cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [Green Version]
- Bulluck, H.; Rosmini, S.; Abdel-Gadir, A.; White, S.K.; Bhuva, A.N.; Treibel, T.A.; Fontana, M.; Ramlall, M.; Hamarneh, A.; Sirker, A.; et al. Residual Myocardial iron following intramyocardial hemorrhage during the convalescent phase of reperfused st-segment—Elevation myocardial infarction and adverse left ventricular remodeling. Circ. Cardiovasc. Imaging 2016, 9, e004940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roghi, A.; Poggiali, E.; Duca, L.; Mafrici, A.; Pedrotti, P.; Paccagnini, S.; Brenna, S.; Galli, A.; Consonni, D.; Cappellini, M.D. Role of non-transferrin-bound iron in the pathogenesis of cardiotoxicity in patients with ST-elevation myocardial infarction assessed by cardiac magnetic resonance imaging. Int. J. Cardiol. 2015, 199, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.; Taylor, A.J.; Ellims, A.H.; Lefkovits, L.; Wong, C.; Kingwell, B.A.; Natoli, A.; Croft, K.; Mori, T.; Kaye, D.M.; et al. Effect of iron chelation on myocardial infarct size and oxidative stress in ST-elevation—Myocardial infarction. Circ. Cardiovasc. Interv. 2012, 5, 270–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitenberg, A.; Paycha, F.; Ledoux, S.; Sachs, R.; Attali, J.-R.; Valensi, P. Coronary artery responses to physiological stimuli are improved by deferoxamine but not by l -arginine in non-insulin-dependent diabetic patients with angiographically normal coronary arteries and no other risk factors. Circulation 1998, 97, 736–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egorov, A.E.; Alekhina, A.V.; Volobueva, T.M.; Fedoreev, A.S.; Mishchenko, N.P.; Kol’Tsova, A.E. Histochrome, a new antioxidant, in the treatment of ocular diseases. Vestn. Oftalmol. 1999, 115, 34–35. [Google Scholar] [PubMed]
- Martins-Marques, T.; Hausenloy, D.J.; Sluijter, J.P.G.; Leybaert, L.; Girao, H. Intercellular communication in the heart: Therapeutic opportunities for cardiac ischemia. Trends Mol. Med. 2020, 27, 248–262. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, J.-W.; Park, J.-H.; Park, B.-W.; Kim, H.; Kim, J.-J.; Sim, W.-S.; Mishchenko, N.P.; Fedoreyev, S.A.; Vasileva, E.A.; Ban, K.; et al. Histochrome Attenuates Myocardial Ischemia-Reperfusion Injury by Inhibiting Ferroptosis-Induced Cardiomyocyte Death. Antioxidants 2021, 10, 1624. https://doi.org/10.3390/antiox10101624
Hwang J-W, Park J-H, Park B-W, Kim H, Kim J-J, Sim W-S, Mishchenko NP, Fedoreyev SA, Vasileva EA, Ban K, et al. Histochrome Attenuates Myocardial Ischemia-Reperfusion Injury by Inhibiting Ferroptosis-Induced Cardiomyocyte Death. Antioxidants. 2021; 10(10):1624. https://doi.org/10.3390/antiox10101624
Chicago/Turabian StyleHwang, Ji-Won, Jae-Hyun Park, Bong-Woo Park, Hyeok Kim, Jin-Ju Kim, Woo-Sup Sim, Natalia P. Mishchenko, Sergey A. Fedoreyev, Elena A. Vasileva, Kiwon Ban, and et al. 2021. "Histochrome Attenuates Myocardial Ischemia-Reperfusion Injury by Inhibiting Ferroptosis-Induced Cardiomyocyte Death" Antioxidants 10, no. 10: 1624. https://doi.org/10.3390/antiox10101624