The Potential Benefit of Monitoring Oxidative Stress and Inflammation in the Prevention of Non-Communicable Diseases (NCDs)

Abstract
:1. Introduction
2. Lifestyle Links to NCDs
3. Risk Scores and Lifestyle Behaviours
4. Oxidative Stress Is Upstream of Conventional NCD Biomarkers
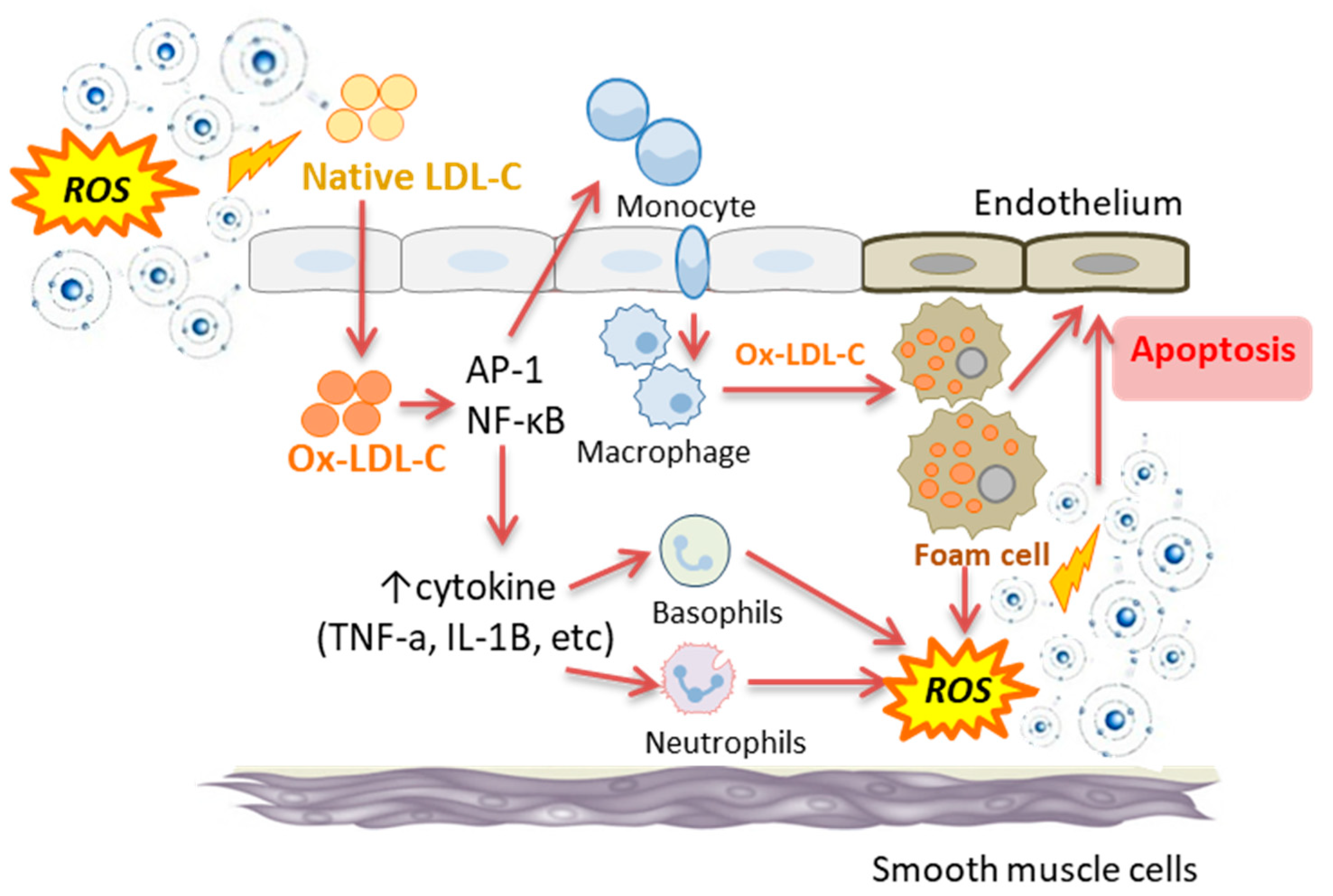
5. Association between Oxidative Stress and Inflammation
6. What Is Redox Imbalance and How Does It Relate to Oxidative Stress?
7. The Link between Lifestyle Behaviours, Oxidative Stress and Inflammation
7.1. Adiposity and Total and Regional Body Fat
7.2. Nutritional Behaviours
7.2.1. Energy Intake
7.2.2. Protein Intake
7.2.3. Processed Meat
7.2.4. Polyunsaturated Fatty Acids
7.2.5. Dietary Glycaemic Index
7.2.6. Fruits and Vegetables
7.2.7. Nutrient-Poor Foods
7.3. Common Social Drug Use
7.3.1. Alcohol Intake
7.3.2. Caffeine Intake
7.3.3. Cigarette Smoking
7.4. Physical Activity
7.5. Sleep and Tissue Oxygenation
7.6. Psychological Factors (Stress, Anxiety and Depression)
8. Evidence that Changes in Lifestyle-Linked Oxidative Stress Is Detectable in Disease-Free Humans
9. Clinical Application of Monitoring Oxidative Stress and Inflammation
10. Monitoring Oxidative Stress and Inflammation
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kvaavik, E.; Batty, G.D.; Ursin, G.; Huxley, R.; Gale, C.R. Influence of individual and combined health behaviors on total and cause-specific mortality in men and women: The United Kingdom Health and Lifestyle Survey. Arch. Intern. Med. 2010, 170, 711–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Non Communicable Diseases Progress Monitor 2017; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; Amann, M.; Anderson, H.R.; Andrews, K.G.; Aryee, M.; et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef] [Green Version]
- Naghavi, M.; Wang, H.; Lozano, R.; Davis, A.; Liang, X.; Zhou, M.; Vollset, S.E.; Abbasoglu Ozgoren, A.; Abdalla, S.; Abd-Allah, F.; et al. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 385, 117–171. [Google Scholar]
- Palotás, A.; Reis, H.J.; Bogáts, G.; Babik, B.; Racsmány, M.; Engvau, L.; Kecskeméti, E.; Juhász, A.; Vieira, L.B.; Teixeira, A.L.; et al. Coronary artery bypass surgery provokes alzheimer’s disease-like changes in the cerebrospinal fluid. J. Alzheimer’s Dis. 2010, 21, 1153–1164. [Google Scholar] [CrossRef]
- Ott, A.; Stolk, R.P.; Van Harskamp, F.; Pols, H.A.P.; Hofman, A. Breteler MMB. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology. 1999, 53, 1937–1942. [Google Scholar] [CrossRef]
- WHO. NCD Mortality and Morbidity; World Health Organization: Geneva, Switzerland, 2018; Available online: http://www.who.int/gho/ncd/mortality_morbidity/en/ (accessed on 8 October 2018).
- WHO. Global Action Plan for the Prevention and Control of Noncommunicable Diseases 2013–2020; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- Bloom, D.E.C.E.; Jane-Llopis, E.; Abrahams-Gessel, S.; Bloom, L.R.; Fathima, S.; Feigl, A.B.; Gaziano, T.; Mowafi, M.; Pandya, A.; Prettner, K.; et al. The Global Economic Burden of Non-Communicable Diseases; World Economic Forum: Geneva, Switzerland, 2011. [Google Scholar]
- Beard, J.R.; Officer, A.; De Carvalho, I.A.; Sadana, R.; Pot, A.M.; Michel, J.P.; Lloyd-Sherlock, P.; Epping-Jordan, J.E.; Peeters, G.M.E.E.; Mahanani, W.R.; et al. The World report on ageing and health: A policy framework for healthy ageing. Lancet 2016, 387, 2145–2154. [Google Scholar] [CrossRef] [Green Version]
- Ford, E.S.; Bergmann, M.M.; Kröger, J.; Schienkiewitz, A.; Weikert, C.; Boeing, H. Healthy living is the best revenge: Findings from the European prospective investigation into cancer and nutrition-potsdam study. Arch. Intern. Med. 2009, 169, 1355–1362. [Google Scholar]
- Siti, H.N.; Kamisah, Y.; Kamsiah, J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vasc. Pharmacol. 2015, 71, 40–56. [Google Scholar] [CrossRef]
- Devasagayam, T.P.A.; Tilak, J.C.; Boloor, K.K.; Sane, K.S.; Ghaskadbi, S.S.; Lele, R.D. Free radicals and antioxidants in human health: Current status and future prospects. J. Assoc. Physicians India 2004, 52, 794–804. [Google Scholar]
- Khansari, N.; Shakiba, Y.; Mahmoudi, M. Chronic inflammation and oxidative stress as a major cause of age-related diseases and cancer. Recent Pat. Inflamm. Allergy Drug Discov. 2009, 3, 73–80. [Google Scholar] [CrossRef]
- Camps, J.; García-Heredia, A. Introduction: Oxidation and inflammation, a molecular link between non-communicable diseases. Adv. Exp. Med. Biol. 2014, 824, 1–4. [Google Scholar] [PubMed]
- Peña-Oyarzun, D.; Bravo-Sagua, R.; Diaz-Vega, A.; Aleman, L.; Chiong, M.; Garcia, L.; Bambs, C.; Troncoso, R.; Cifuentes, M.; Morselli, E.; et al. Autophagy and oxidative stress in non-communicable diseases: A matter of the inflammatory state? Free Radic. Biol. Med. 2018, 124, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.G.J. Free Radicals in Biology and Disease; Oxford Science Pub.: Oxford, UK, 2007; Volume 219, pp. 21–23, 187–267. [Google Scholar]
- Yoon, J.H.; Kim, J.Y.; Park, J.K.; Ko, S.B. Oxidative damage markers are significantly associated with the carotid artery intima-media thickness after controlling for conventional risk factors of atherosclerosis in men. PLoS ONE 2015, 10, e0119731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Xia, S.; Kalionis, B.; Wan, W.; Sun, T. The Role of Oxidative Stress and Inflammation in Cardiovascular Aging. BioMed Res. Int. 2014, 2014, 615312. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, R.B., Sr.; Vasan, R.S.; Pencina, M.J.; Wolf, P.A.; Cobain, M.; Massaro, J.M.; Kannel, W.B. General cardiovascular risk profile for use in primary care: The Framingham heart study. Circulation 2008, 117, 743–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, P.W.F.; D’Agostino, R.B.; Levy, D.; Belanger, A.M.; Silbershatz, H.; Kannel, W.B. Prediction of coronary heart disease using risk factor categories. Circulation 1998, 97, 1837–1847. [Google Scholar] [CrossRef] [Green Version]
- Roberts, C.K.; Barnard, R.J. Effects of exercise and diet on chronic disease. J. Appl. Physiol. 2005, 98, 3–30. [Google Scholar] [CrossRef] [Green Version]
- Lichtenstein, P.; Holm, N.V.; Verkasalo, P.K.; Iliadou, A.; Kaprio, J.; Koskenvuo, M.; Pukkala, E.; Skytthe, A.; Hemminki, K. Environmental and heritable factors in the causation of cancer: Analyses of cohorts of twins from Sweden, Denmark, and Finland. New Engl. J. Med. 2000, 343, 78–85. [Google Scholar] [CrossRef]
- Katz, D.L.; Frates, E.P.; Bonnet, J.P.; Gupta, S.K.; Vartiainen, E.; Carmona, R.H. Lifestyle as Medicine: The Case for a True Health Initiative. Am. J. Health Promot. 2018, 32, 1452–1458. [Google Scholar] [CrossRef]
- Sagner, M.; Katz, D.; Egger, G.; Lianov, L.; Schulz, K.H.; Braman, M.; Behbod, B.; Phillips, E.; Dysinger, W.; Ornish, D. Lifestyle medicine potential for reversing a world of chronic disease epidemics: From cell to community. Int. J. Clin. Pract. 2014, 68, 1289–1292. [Google Scholar] [CrossRef]
- King, D.E.; Mainous, A.G., III; Geesey, M.E. Turning Back the Clock: Adopting a Healthy Lifestyle in Middle Age. Am. J. Med. 2007, 120, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, H.; Johnsen, N.F.; Christensen, J.; Frederiksen, K.; Overvad, K.; Tjønneland, A. Association of adherence to lifestyle recommendations and risk of colorectal cancer: A prospective Danish cohort study. BMJ (Online) 2010, 341, 978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsoukalas, D.; Sarandi, E. Micronutrient deficiencies in patients with COVID-19: How metabolomics can contribute to their prevention and replenishment. BMJ Nutr. Prev. Health 2020, 1–2. [Google Scholar]
- Lamichhane, S.; Kemppainen, E.; Trošt, K.; Siljander, H.; Hyöty, H.; Ilonen, J.; Toppari, J.; Veijola, R.; Hyötyläinen, T.; Knip, M.; et al. Circulating metabolites in progression to islet autoimmunity and type 1 diabetes. Diabetologia 2019, 62, 2287–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margină, D.; Ungurianu, A.; Purdel, C.; Tsoukalas, D.; Sarandi, E.; Thanasoula, M.; Tekos, F.; Mesnage, R.; Kouretas, D.; Tsatsakis, A. Chronic inflammation in the context of everyday life: Dietary changes as mitigating factors. Int. J. Environ. Res. Public Health 2020, 17, 4135. [Google Scholar] [CrossRef] [PubMed]
- Lakkur, S.; Bostick, R.M.; Roblin, D.; Ndirangu, M.; Okosun, I.; Annor, F.; Judd, S.; Dana Flanders, W.; Stevens, V.L.; Goodman, M. Oxidative balance score and oxidative stress biomarkers in a study of Whites, African Americans, and African immigrants. Biomarkers 2014, 19, 471–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotos-Prieto, M.; Bhupathiraju, S.N.; Falcon, L.M.; Gao, X.; Tucker, K.L.; Mattei, J. Association between a Healthy Lifestyle Score and inflammatory markers among Puerto Rican adults. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Devlin, H.M.; Smith, B.; Imperatore, G.; Thomas, W.; Lobelo, F.; Ali, M.K.; Norris, K.; Gruss, S.; Bardenheier, B.; et al. Effect of lifestyle interventions on cardiovascular risk factors among adults without impaired glucose tolerance or diabetes: A systematic review and metaanalysis. PLoS ONE 2017, 12, e0176436. [Google Scholar] [CrossRef]
- Ramachandran, A.; Snehalatha, C.; Mary, S.; Mukesh, B.; Bhaskar, A.D.; Vijay, V.; Programme, I.D.P. The Indian Diabetes Prevention Programme shows that lifestyle modification and metformin prevent type 2 diabetes in Asian Indian subjects with impaired glucose tolerance (IDPP-1). Diabetologia 2006, 49, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.M.; Odell, P.M.; Wilson, P.W.F.; Kannel, W.B. Cardiovascular disease risk profiles. Am. Heart J. 1991, 121, 293–298. [Google Scholar] [CrossRef]
- Goff, D.C., Jr.; Lloyd-Jones, D.M.; Bennett, G.; Coady, S.; D’Agostino, R.B., Sr.; Gibbons, R.; Greenland, P.; Lackland, D.T.; Levy, D.; O’Donnell, C.J.; et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: A report of the American college of cardiology/American heart association task force on practice guidelines. J. Am. Coll. Cardiol. 2014, 63, 2935–2959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deanfield, J.; Sattar, N.; Simpson, I.; Wood, D.; Bradbury, K.; Fox, K.; Boon, N.; Winocour, P.; Feher, M.; Doherty, P.; et al. Joint British Societies’ consensus recommendations for the prevention of cardiovascular disease (JBS3). Heart 2014, 100 (Suppl. 2), ii1–ii67. [Google Scholar]
- WHO. Prevention of Cardiovascular Disease: Guidelines for Assessment and Management of Total Cardiovascular Risk; World Health Organization: Geneva, Switzerland, 2007. [Google Scholar]
- Conroy, R.M.; Pyörälä, K.; Fitzgerald, A.P.; Sans, S.; Menotti, A.; De Backer, G.; De Bacquer, D.; Ducimetière, P.; Jousilahti, P.; Keil, U.; et al. Estimation of ten-year risk of fatal cardiovascular disease in Europe: The SCORE project. Eur. Heart J. 2003, 24, 987–1003. [Google Scholar] [CrossRef]
- Bansal, M.; Kasliwal, R.R.; Trehan, N. Comparative accuracy of different risk scores in assessing cardiovascular risk in Indians: A study in patients with first myocardial infarction. Indian Heart J. 2014, 66, 580–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, N.; Muduli, S.K.; Kapoor, A.; Tewari, S.; Kumar, S.; Khanna, R.; Goel, P.K. Comparison of different cardiovascular risk score calculators for cardiovascular risk prediction and guideline recommended statin uses. Indian Heart J. 2017, 69, 458–463. [Google Scholar] [CrossRef]
- Bhopal, R.; Fischbacher, C.; Vartiainen, E.; Unwin, N.; White, M.; Alberti, G. Predicted and observed cardiovascular disease in South Asians: Application of FINRISK, Framingham and SCORE models to Newcastle Heart Project data. J. Public Health 2005, 27, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Reissigová, J.; Zvárová, J. The Framingham risk function underestimated absolute coronary heart disease risk in Czech men. Methods Inf. Med. 2007, 46, 43–49. [Google Scholar]
- Otgontuya, D.; Oum, S.; Buckley, B.S.; Bonita, R. Assessment of total cardiovascular risk using WHO/ISH risk prediction charts in three low and middle income countries in Asia. BMC Public Health 2013, 13, 539. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.P.; Oeser, A.; Avalos, I.; Raggi, P.; Stein, C.M. Cardiovascular risk scores and the presence of subclinical coronary artery atherosclerosis in women with systemic lupus erythematosus. Lupus 2006, 15, 562–569. [Google Scholar] [CrossRef]
- Ernste, F.C.; Sánchez-Menéndez, M.; Wilton, K.M.; Crowson, C.S.; Matteson, E.L.; Maradit Kremers, H. Cardiovascular risk profile at the onset of psoriatic arthritis: A population-based cohort study. Arthritis Care Res. 2015, 67, 1015–1021. [Google Scholar] [CrossRef]
- Law, W.; Johnson, C.; Rushton, M.; Dent, S. The framingham risk score underestimates the risk of cardiovascular events in the HER2-positive breast cancer population. Curr. Oncol. 2017, 24, e348–e353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, R.; Kapoor, A.; Kumar, S.; Tewari, S.; Garg, N.; Goel, P.K. Metabolic syndrome & Framingham Risk Score: Observations from a coronary angiographic study in Indian patients. Indian J. Med Res. 2013, 137, 295–301. [Google Scholar] [PubMed]
- Huang, H.; Mai, W.; Liu, D.; Hao, Y.; Tao, J.; Dong, Y. The oxidation ratio of LDL: A predictor for coronary artery disease. Dis. Markers 2008, 24, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Chisolm, G.M.; Steinberg, D. The oxidative modification hypothesis of atherogenesis: An overview. Free Radic. Biol. Med. 2000, 28, 1815–1826. [Google Scholar] [CrossRef]
- Steinberg, D.; Witztum, J.L. Is the oxidative modification hypothesis relevant to human atherosclerosis? Do the antioxidant trials conducted to date refute the hypothesis? Circulation 2002, 105, 2107–2111. [Google Scholar] [CrossRef] [Green Version]
- Navab, M.; Berliner, J.A.; Watson, A.D.; Hama, S.Y.; Territo, M.C.; Lusis, A.J.; Shih, D.M.; Van Lenten, B.J.; Frank, J.S.; Demer, L.L.; et al. The Yin and Yang of oxidation in the development of the fatty streak: A review based on the 1994 George Lyman Duff memorial lecture. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 831–842. [Google Scholar] [CrossRef]
- Hulsmans, M.; Holvoet, P. The vicious circle between oxidative stress and inflammation in atherosclerosis. J. Cell. Mol. Med. 2010, 14, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Quinn, M.T.; Parthasarathy, S.; Fong, L.G.; Steinberg, D. Oxidatively modified low density lipoproteins: A potential role in recruitment and retention of monocyte/macrophages during atherogenesis. Proc. Natl. Acad. Sci. USA 1987, 84, 2995–2998. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, A.; Bonaterra, G.A.; Schwarzbach, H.; Kinscherf, R. Oxidized LDL-induced JAB1 influences NF-κB independent inflammatory signaling in human macrophages during foam cell formation. J. Biomed. Sci. 2017, 24, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ashfaq, S.; Abramson, J.L.; Jones, D.P.; Rhodes, S.D.; Weintraub, W.S.; Hooper, W.C.; Vaccarino, V.; Harrison, D.G.; Quyyumi, A.A. The relationship between plasma levels of oxidized and reduced thiols and early atherosclerosis in healthy adults. J. Am. Coll. Cardiol. 2006, 47, 1005–1011. [Google Scholar] [CrossRef] [Green Version]
- Shishehbor, M.H.; Zhang, R.; Medina, H.; Brennan, M.L.; Brennan, D.M.; Ellis, S.G.; Topol, E.J.; Hazen, S.L. Systemic elevations of free radical oxidation products of arachidonic acid are associated with angiographic evidence of coronary artery disease. Free Radic. Biol. Med. 2006, 41, 1678–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakai, N.A.; Katz, R.; Jenny, N.S.; Psaty, B.M.; Reiner, A.P.; Schwartz, S.M.; Cushman, M. Inflammation and hemostasis biomarkers and cardiovascular risk in the elderly: The Cardiovascular Health Study. J. Thromb. Haemost. 2007, 5, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Yeboah, J.; McClelland, R.L.; Polonsky, T.S.; Burke, G.L.; Sibley, C.T.; O’Leary, D.; Carr, J.J.; Goff, D.C., Jr. Greenland P, Herrington DM. Comparison of novel risk markers for improvement in cardiovascular risk assessment in intermediate-risk individuals. JAMA J. Am. Med Assoc. 2012, 308, 788–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, P.W.F.; Pencina, M.; Jacques, P.; Selhub, J.; D’Agostino, R.; O’Donnell, C.J. C-reactive protein and reclassification of cardiovascular risk in the Framingham Heart Study. Circ. Cardiovasc. Qual. Outcomes 2008, 1, 92–97. [Google Scholar] [CrossRef] [Green Version]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Massudi, H.; Grant, R.; Braidy, N.; Guest, J.; Farnsworth, B.; Guillemin, G.J. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS ONE 2012, 7, e42357. [Google Scholar] [CrossRef]
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age related changes in NAD+ metabolism oxidative stress and sirt1 activity in wistar rats. PLoS ONE. 2011, 6, e19194. [Google Scholar] [CrossRef]
- Head, E.; Liu, J.; Hagen, T.M.; Muggenburg, B.A.; Milgram, N.W.; Ames, B.N.; Cotman, C.W. Oxidative damage increases with age in a canine model of human brain aging. J. Neurochem. 2002, 82, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [Green Version]
- Closa, D.; Folch-Puy, E. Oxygen free radicals and the systemic inflammatory response. IUBMB Life 2004, 56, 185–191. [Google Scholar] [CrossRef]
- Janssen-Heininger, Y.M.W.; Mossman, B.T.; Heintz, N.H.; Forman, H.J.; Kalyanaraman, B.; Finkel, T.; Stamler, J.S.; Rhee, S.G.; van der Vliet, A. Redox-based regulation of signal transduction: Principles, pitfalls, and promises. Free Radic. Biol. Med. 2008, 45, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xanthoulea, S.; Curfs, D.M.J.; Hofker, M.H.; De Winther, M.P.J. Nuclear factor kappaB signaling in macrophage function and atherogenesis. Curr. Opin. Lipidol. 2005, 16, 536–542. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.P.; Bald, A.; Cassatella, M.A. Activation of the NF-≃B pathway by inflammatory stimuli in human neutrophils. Blood 1997, 89, 3421–3433. [Google Scholar] [CrossRef] [PubMed]
- Duque, G.A.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar]
- Davies, P.; Bonney, R.J.; Humes, J.L.; Kuehl, F.A. The role of macrophage secretory products in chronic inflammatory processes. J. Investig. Dermatol. 1980, 74, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Eklund, C.M. Chapter 5 Proinflammatory cytokines in CRP baseline regulation. Adv. Clin. Chem. 2009, 48, 111–136. [Google Scholar]
- Helming, L. Inflammation: Cell Recruitment versus local proliferation. Curr. Biol. 2011, 21, R548–R550. [Google Scholar] [CrossRef] [Green Version]
- Hold, G.L.; El-Omar, E.M. Genetic aspects of inflammation and cancer. Biochem. J. 2008, 410, 225–235. [Google Scholar] [CrossRef]
- Fenton, H. Oxidation of tartaric acid in presence of iron. J. Chem. Sot. 1984, 65, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Cadenas, E.; Davies, K.J.A. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007, 12, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Dansen, T.B.; Wirtz, K.W.A. The peroxisome in oxidative stress. IUBMB Life 2001, 51, 223–230. [Google Scholar] [PubMed]
- Johnson, M.H.; Nasresfahani, M.H. Radical solutions and cultural problems: Could free oxygen radicals be responsible for the impaired development of preimplantation mammalian embryos in vitro? BioEssays 1994, 16, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Cao, Z.; Xu, X.; Meir, E.G.V.; Lambeth, J.D. Homologs of gp91phox: Cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 2001, 269, 131–140. [Google Scholar] [CrossRef]
- Albertolle, M.E.; Peter Guengerich, F. The relationships between cytochromes P450 and H2O2: Production, reaction, and inhibition. J. Inorg. Biochem. 2018, 186, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Fialkow, L.; Wang, Y.; Downey, G.P. Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radic. Biol. Med. 2007, 42, 153–164. [Google Scholar] [CrossRef]
- Sheppard, F.R.; Kelher, M.R.; Moore, E.E.; McLaughlin, N.J.D.; Banerjee, A.; Silliman, C.C. Structural organization of the neutrophil NADPH oxidase: Phosphorylation and translocation during priming and activation. J. Leukoc. Biol. 2005, 78, 1025–1042. [Google Scholar] [CrossRef] [Green Version]
- Sarsour, E.H.; Kumar, M.G.; Chaudhuri, L.; Kalen, A.L.; Goswami, P.C. Redox control of the cell cycle in health and disease. Antioxid. Redox Signal. 2009, 11, 2985–3011. [Google Scholar] [CrossRef]
- Aseervatham, G.S.; Sivasudha, T.; Jeyadevi, R.; Arul Ananth, D. Environmental factors and unhealthy lifestyle influence oxidative stress in humans--an overview. Environ. Sci. Pollut. Res. Int. 2013, 20, 4356–4369. [Google Scholar] [CrossRef]
- Black, C.N.; Bot, M.; Scheffer, P.G.; Penninx, B.W.J.H. Sociodemographic and Lifestyle Determinants of Plasma Oxidative Stress Markers 8-OHdG and F2-Isoprostanes and Associations with Metabolic Syndrome. Oxidative Med. Cell. Longev. 2016, 2016, 7530820. [Google Scholar] [CrossRef] [Green Version]
- Bowen, L.; Taylor, A.E.; Sullivan, R.; Ebrahim, S.; Kinra, S.; Krishna, K.R.; Kulkarni, B.; Ben-Shlomo, Y.; Ekelund, U.; Wells, J.C.; et al. Associations between diet, physical activity and body fat distribution: A cross sectional study in an Indian population. BMC Public Health 2015, 15, 281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondoh, T.; Takase, H.; Yamaguchi, T.F.; Ochiai, R.; Katashima, M.; Katsuragi, Y.; Sakane, N. Association of dietary factors with abdominal subcutaneous and visceral adiposity in Japanese men. Obes. Res. Clin. Pract. 2014, 8, e16–e25. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ebenezer, P.J.; Dasuri, K.; Fernandez-Kim, S.O.; Francis, J.; Mariappan, N.; Gao, Z.; Ye, J.; Bruce-Keller, A.J.; Keller, J.N. Aging is associated with hypoxia and oxidative stress in adipose tissue: Implications for adipose function. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E599–E607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef]
- Pasarica, M.; Sereda, O.R.; Redman, L.M.; Albarado, D.C.; Hymel, D.T.; Roan, L.E.; Rood, J.C.; Burk, D.H.; Smith, S.R. Reduced adipose tissue oxygenation in human obesity evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes 2009, 58, 718–725. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.O.Y.; Soro-Arnaiz, I.; Aragonés, J. Age-dependent obesity and mitochondrial dysfunction. Adipocyte. 2017, 6, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P. The role of hypoxia-induced factors in tumor progression. Oncologist 2004, 9 (Suppl. 5), 10–17. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- Payne, C.M.; Weber, C.; Crowley-Skillicorn, C.; Dvorak, K.; Bernstein, H.; Bernstein, C.; Holubec, H.; Dvorakova, B.; Garewal, H. Deoxycholate induces mitochondrial oxidative stress and activates NF-κB through multiple mechanisms in HCT-116 colon epithelial cells. Carcinogenesis 2007, 28, 215–222. [Google Scholar] [CrossRef]
- Visser, M.; Bouter, L.M.; McQuillan, G.M.; Wener, M.H.; Harris, T.B. Elevated C-reactive protein levels in overweight and obese adults. J. Am. Med Assoc. 1999, 282, 2131–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-α: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Fried, S.K.; Bunkin, D.A.; Greenberg, A.S. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: Depot difference and regulation by glucocorticoid. J. Clin. Endocrinol. Metab. 1998, 83, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Tangvarasittichai, S.; Pongthaisong, S.; Tangvarasittichai, O. Tumor Necrosis Factor-Α, Interleukin-6, C-Reactive Protein Levels and Insulin Resistance Associated with Type 2 Diabetes in Abdominal Obesity Women. Indian J. Clin. Biochem. 2016, 31, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, G.L.; Loeken, M.R. Hyperglycemia-induced oxidative stress in diabetic complications. Histochem. Cell Biol. 2004, 122, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Homko, C.; Barrero, C.A.; Stein, T.P.; Chen, X.; Cheung, P.; Fecchio, C.; Koller, S.; Merali, S. Excessive caloric intake acutely causes oxidative stress, GLUT4 carbonylation, and insulin resistance in healthy men. Sci. Transl. Med. 2015, 7, 304re7. [Google Scholar] [CrossRef] [Green Version]
- Dandona, P.; Mohanty, P.; Hamouda, W.; Ghanim, H.; Aljada, A.; Garg, R.; Kumar, V. Inhibitory effect of a two day fast on reactive oxygen species (ROS) generation by leucocytes and plasma ortho-tyrosine and meta-tyrosine concentrations. J. Clin. Endocrinol. Metab. 2001, 86, 2899–2902. [Google Scholar] [CrossRef]
- Dandona, P.; Mohanty, P.; Ghanim, H.; Aljada, A.; Browne, R.; Hamouda, W.; Prabhala, A.; Afzal, A.; Garg, R. The suppressive effect of dietary restriction and weight loss in the obese on the generation of reactive oxygen species by leukocytes, lipid peroxidation, and protein carbonylation. J. Clin. Endocrinol. Metab. 2001, 86, 355–362. [Google Scholar]
- Heilbronn, L.K.; De Jonge, L.; Frisard, M.I.; DeLany, J.P.; Larson-Meyer, D.E.; Rood, J.; Nguyen, T.; Martin, C.K.; Volaufova, J.; Most, M.M.; et al. Effect of 6-month calorie restriction on biomarkers of longevity, metabolic adaptation, and oxidative stress in overweight individuals: A randomized controlled trial. J. Am. Med. Assoc. 2006, 295, 1539–1548. [Google Scholar] [CrossRef]
- Buchowski, M.S.; Hongu, N.; Acra, S.; Wang, L.; Warolin, J.; Roberts, L.J., II. Effect of Modest Caloric Restriction on Oxidative Stress in Women, a Randomized Trial. PLoS ONE 2012, 7, e47079. [Google Scholar] [CrossRef] [Green Version]
- Khor, A.; Grant, R.; Tung, C.; Guest, J.; Pope, B.; Morris, M.; Bilgin, A. Postprandial oxidative stress is increased after a phytonutrient-poor food but not after a kilojoule-matched phytonutrient-rich food. Nutr. Res. 2014, 34, 391–400. [Google Scholar] [CrossRef]
- Kim, M.J.; Aiken, J.M.; Havighurst, T.; Hollander, J.; Ripple, M.O.; Weindruch, R. Adult-onset energy restriction of rhesus monkeys attenuates oxidative stress-induced cytokine expression by peripheral blood mononuclear cells. J. Nutr. 1997, 127, 2293–2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, L.; Villareal, D.T.; Weiss, E.P.; Racette, S.B.; Steger-May, K.; Klein, S.; Holloszy, J.O. Calorie restriction or exercise: Effects on coronary heart disease risk factors. A randomized, controlled trial. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E197–E202. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span-from yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercken, E.M.; Crosby, S.D.; Lamming, D.W.; Jebailey, L.; Krzysik-Walker, S.; Villareal, D.T.; Capri, M.; Franceschi, C.; Zhang, Y.; Becker, K.; et al. Calorie restriction in humans inhibits the PI3K/AKT pathway and induces a younger transcription profile. Aging Cell 2013, 12, 645–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speakman, J.R.; Mitchell, S.E.; Mazidi, M. Calories or protein? The effect of dietary restriction on lifespan in rodents is explained by calories alone. Exp. Gerontol. 2016, 86, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Fanson, B.G.; Weldon, C.W.; Pérez-Staples, D.; Simpson, S.J.; Taylor, P.W. Nutrients, not caloric restriction, extend lifespan in Queensland fruit flies (Bactrocera tryoni). Aging Cell 2009, 8, 514–523. [Google Scholar] [CrossRef]
- Baum, J.I.; Kim, I.Y.; Wolfe, R.R. Protein consumption and the elderly: What is the optimal level of intake? Nutrients 2016, 8, 359. [Google Scholar] [CrossRef] [Green Version]
- Pamplona, R.; Barja, G. Mitochondrial oxidative stress, aging and caloric restriction: The protein and methionine connection. Biochim. et Biophys. Acta Bioenerg. 2006, 1757, 496–508. [Google Scholar] [CrossRef] [Green Version]
- Mair, W.; Piper, M.D.W.; Partridge, L. Calories do not explain extension of life span by dietary restriction in Drosophila. PLoS Biol. 2005, 3, 1305–1311. [Google Scholar] [CrossRef] [Green Version]
- Levine, M.E.; Suarez, J.A.; Brandhorst, S.; Balasubramanian, P.; Cheng, C.W.; Madia, F.; Fontana, L.; Mirisola, M.G.; Guevara-Aguirre, J.; Wan, J.; et al. Low protein intake is associated with a major reduction in IGF-1, cancer, and overall mortality in the 65 and younger but not older population. Cell Metab. 2014, 19, 407–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solon-Biet, S.M.; McMahon, A.C.; Ballard, J.W.O.; Ruohonen, K.; Wu, L.E.; Cogger, V.C.; Warren, A.; Huang, X.; Pichaud, N.; Melvin, R.G.; et al. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum-fed mice. Cell Metab. 2014, 19, 418–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, L.; Cummings, N.E.; Arriola Apelo, S.I.; Neuman, J.C.; Kasza, I.; Schmidt, B.A.; Cava, E.; Spelta, F.; Tosti, V.; Syed, F.A.; et al. Decreased Consumption of Branched-Chain Amino Acids Improves Metabolic Health. Cell Rep. 2016, 16, 520–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, A.; Caro, P.; Barja, G. Protein restriction without strong caloric restriction decreases mitochondrial oxygen radical production and oxidative DNA damage in rat liver. J. Bioenerg. Biomembr. 2004, 36, 545–552. [Google Scholar] [CrossRef]
- De, A.K.; Chipalkatti, S.; Aiyar, A.S. Some biochemical parameters of ageing in relation to dietary protein. Mech. Ageing Dev. 1983, 21, 37–48. [Google Scholar] [CrossRef]
- Seyedsadjadi, N.; Berg, J.; Bilgin, A.A.; Braidy, N.; Salonikas, C.; Grant, R. High protein intake is associated with low plasma NAD+ levels in a healthy human cohort. PLoS ONE. 2018, 13, e0201968. [Google Scholar] [CrossRef]
- Hidiroglou, N.; Gilani, G.S.; Long, L.; Zhao, X.; Madere, R.; Cockell, K.; Belonge, B.; Ratnayake, W.M.N.; Peace, R. The influence of dietary vitamin E, fat, and methionine on blood cholesterol profile, homocysteine levels, and oxidizability of low density lipoprotein in the gerbil. J. Nutr. Biochem. 2004, 15, 730–740. [Google Scholar] [CrossRef]
- Mori, N.; Hirayama, K. Long-term consumption of a methionine-supplemented diet increases iron and lipid peroxide levels in rat liver. J. Nutr. 2000, 130, 2349–2355. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.S.M.; Lau, R.; Aune, D.; Vieira, R.; Greenwood, D.C.; Kampman, E.; Norat, T. Red and processed meat and colorectal cancer incidence: Meta-analysis of prospective studies. PLoS ONE 2011, 6, e20456. [Google Scholar] [CrossRef] [Green Version]
- Romeu, M.; Aranda, N.; Giralt, M.; Ribot, B.; Nogues, M.R.; Arija, V. Diet, iron biomarkers and oxidative stress in a representative sample of Mediterranean population. Nutr. J. 2013, 12, 102. [Google Scholar] [CrossRef] [Green Version]
- Mainous Iii, A.G.; Wells, B.; Carek, P.J.; Gill, J.M.; Geesey, M.E. The mortality risk of elevated serum transferrin saturation and consumption of dietary iron. Ann. Fam. Med. 2004, 2, 139–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, R.; Rothman, N.; Salmon, C.P.; Knize, M.G.; Brown, D.E.; Swanson, C.A.; Rhodes, D.; Rossi, S.; Felton, J.S.; Levander, O.A. Heterocyclic amine content in beef cooked by different methods to varying degrees of doneness and gravy made from meat drippings. Food Chem. Toxicol. 1998, 36, 279–287. [Google Scholar] [CrossRef]
- Chen, G.; Scott Smith, J. Determination of advanced glycation endproducts in cooked meat products. Food Chem. 2015, 168, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.M.; Miranda, A.M.; Santos, F.A.; Loureiro, A.P.M.; Fisberg, R.M.; Marchioni, D.M. High intake of heterocyclic amines from meat is associated with oxidative stress. Br. J. Nutr. 2015, 113, 1301–1307. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, S.I.; Maeda, S.; Matsui, T.; Ueda, S.; Fukami, K.; Okuda, S. Role of advanced glycation end products (AGEs) and oxidative stress in vascular complications in diabetes. Biochim. et Biophys. Acta Gen. Subj. 2012, 1820, 663–671. [Google Scholar] [CrossRef]
- Wilson, A.; McLean, C.; Kim, R.B. Trimethylamine-N-oxide: A link between the gut microbiome, bile acid metabolism, and atherosclerosis. Curr. Opin. Lipidol. 2016, 27, 148–154. [Google Scholar] [CrossRef]
- Sun, X.; Jiao, X.; Ma, Y.; Liu, Y.; Zhang, L.; He, Y.; Chen, Y. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 2016, 481, 63–70. [Google Scholar] [CrossRef]
- Wall, R.; Ross, R.P.; Fitzgerald, G.F.; Stanton, C. Fatty acids from fish: The anti-inflammatory potential of long-chain omega-3 fatty acids. Nutr. Rev. 2010, 68, 280–289. [Google Scholar] [CrossRef]
- Patterson, E.; Wall, R.; Fitzgerald, G.F.; Ross, R.P.; Stanton, C. Health implications of high dietary omega-6 polyunsaturated fatty acids. J. Nutr. Metab. 2012, 2012, 1–16. [Google Scholar] [CrossRef]
- Simopoulos, A.P. Omega-3 fatty acids in inflammation and autoimmune diseases. J. Am. Coll. Nutr. 2002, 21, 495–505. [Google Scholar] [CrossRef]
- Simopoulos, A.P. An increase in the Omega-6/Omega-3 fatty acid ratio increases the risk for obesity. Nutrients 2016, 8, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Micaelo, N.; González-Abuín, N.; Terra, X.; Richart, C.; Ardèvol, A.; Pinent, M.; Blay, M. Omega-3 docosahexaenoic acid and procyanidins inhibit cyclo-oxygenase activity and attenuate NF-κB activation through a p105/p50 regulatory mechanism in macrophage inflammation. Biochem. J. 2012, 441, 653–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Im, D.S. Omega-3 fatty acids in anti-inflammation (pro-resolution) and GPCRs. Prog. Lipid Res. 2012, 51, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Sakai, C.; Ishida, M.; Ohba, H.; Yamashita, H.; Uchida, H.; Yoshizumi, M.; Ishida, T. Fish oil omega-3 polyunsaturated fatty acids attenuate oxidative stress-induced DNA damage in vascular endothelial cells. PLoS ONE 2017, 12, e0187934. [Google Scholar] [CrossRef] [Green Version]
- Nalsen, C.; Vessby, B.; Berglund, L.; Uusitupa, M.; Hermansen, K.; Riccardi, G.; Rivellese, A.; Storlien, L.; Erkkila, A.; Yla-Herttuala, S.; et al. Dietary (n-3) fatty acids reduce plasma F2-isoprostanes but not prostaglandin F2α in healthy humans. J. Nutr. 2006, 136, 1222–1228. [Google Scholar]
- Shahidi, F.; Zhong, Y. Lipid oxidation and improving the oxidative stability. Chem. Soc. Rev. 2010, 39, 4067–4079. [Google Scholar] [CrossRef]
- Gueraud, F.; Atalay, M.; Bresgen, N.; Cipak, A.; Eckl, P.M.; Huc, L.; Jouanin, I.; Siems, W.; Uchida, K. Chemistry and biochemistry of lipid peroxidation products. Free Rad. Res. 2010, 44, 1098–1124. [Google Scholar] [CrossRef]
- Jira, W.; Spiteller, G.; Carson, W.; Schramm, A. Strong increase in hydroxy fatty acids derived from linoleic acid in human low density lipoproteins of atherosclerotic patients. Chem.Phys. Lipids 1997, 91, 1–11. [Google Scholar] [CrossRef]
- Jenkins, D.J.A.; Wolever, T.M.S.; Taylor, R.H. Glycemic index of foods: A physiological basis for carbohydrate exchange. Am. J. Clin. Nutr. 1981, 34, 362–366. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, S.; Hancock, D.P.; Petocz, P.; Ceriello, A.; Brand-Miller, J. High-glycemic index carbohydrate increases nuclear factor-κB activation in mononuclear cells of young, lean healthy subjects1-3. Am. J. Clin. Nutr. 2008, 87, 1188–1193. [Google Scholar]
- Hu, Y.; Block, G.; Norkus, E.P.; Morrow, J.D.; Dietrich, M.; Hudes, M. Relations of glycemic index and glycemic load with plasma oxidative stress markers. Am. J. Clin. Nut. 2006, 84, 70–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, C.; Milne, G.L.; Park, Y.M.M.; Sandler, D.P.; Nichols, H.B. Dietary glycemic index and glycemic load are positively associated with oxidative stress among premenopausal women. J. Nut. 2018, 148, 125–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.L.; Edelstein, D.; Rossetti, L.; Fantus, I.G.; Goldberg, H.; Ziyadeh, F.; Wu, J.; Brownlee, M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc. Natl. Acad. Sci. USA 2000, 97, 12222–12226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Jiao, X.; Lau, W.B.; Wang, Y.; Christopher, T.A.; Lopez, B.L.; RamachandraRao, S.P.; Tao, L.; Ma, X.L. Thioredoxin glycation: A novel posttranslational modification that inhibits its antioxidant and organ protective actions. Free Radic. Biol. Med. 2010, 49, 332–338. [Google Scholar] [CrossRef] [Green Version]
- Ceriello, A.; Bortolotti, N.; Motz, E.; Pieri, C.; Marra, M.; Tonutti, L.; Lizzio, S.; Feletto, F.; Catone, B.; Taboga, C. Meal-induced oxidative stress and low-density lipoprotein oxidation in diabetes: The possible role of hyperglycemia. Metab. Clin. Exp. 1999, 48, 1503–1508. [Google Scholar] [CrossRef]
- Pieper, G.M.; Riazul, H. Activation of nuclear factor-κb in cultured endothelial cells by increased glucose concentration: Prevention by calphostin C. J. Cardiovasc. Pharmacol. 1997, 30, 528–532. [Google Scholar] [CrossRef]
- Xia, P.; Kramer, R.M.; King, G.L. Identification of the mechanism for the inhibition of Na+,K+-adenosine triphosphatase by hyperglycemia involving activation of protein kinase C and cytosolic phospholipase A2. J. Clin. Investig. 1995, 96, 733–740. [Google Scholar] [CrossRef] [Green Version]
- Degenhardt, T.P.; Thorpe, S.R.; Baynes, J.W. Chemical modification of proteins by methylglyoxal. Cell. Mol. Biol. 1998, 44, 1139–1145. [Google Scholar]
- Qi, L.; Van Dam, R.M.; Liu, S.; Franz, M.; Mantzoros, C.; Hu, F.B. Whole-grain, bran, and cereal fiber intakes and markers of systemic inflammation in diabetic women. Diabetes Care 2006, 29, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Esfahani, A.; Wong, J.M.W.; Truan, J.; Villa, C.R.; Mirrahimi, A.; Srichaikul, K.; Kendall, C.W.C. Health Effects of Mixed Fruit and Vegetable Concentrates: A Systematic Review of the Clinical Interventions. J. Am. Coll. Nutr. 2011, 30, 285–294. [Google Scholar] [CrossRef]
- Fiedor, J.; Burda, K. Potential role of carotenoids as antioxidants in human health and disease. Nutrients 2014, 6, 466–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banjarnahor, S.D.S.; Artanti, N. Antioxidant properties of flavonoids. Med J. Indones. 2014, 23, 239–244. [Google Scholar] [CrossRef] [Green Version]
- Leeds, A.R.; Ferris, E.A.E.; Staley, J.; Ayesh, R.; Ross, F. Availability of micronutrients from dried, encapsulated fruit and vegetable preparations: A study in healthy volunteers. J. Human Nutr. Diet. 2000, 13, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Thompson, H.J.; Heimendinger, J.; Sedlacek, S.; Haegele, A.; Diker, A.; O’Neill, C.; Meinecke, B.; Wolfe, P.; Zhu, Z.; Jiang, W. 8-Isoprostane F2α excretion is reduced in women by increased vegetable and fruit intake. Am. J. Clin. Nutr. 2005, 82, 768–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruel, G.; Pomerleau, S.; Couture, P.; Lamarche, B.; Couillard, C. Changes in plasma antioxidant capacity and oxidized low-density lipoprotein levels in men after short-term cranberry juice consumption. Metab. Clin. Exp. 2005, 54, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.J.; Heimendinger, J.; Gillette, C.; Sedlacek, S.M.; Haegele, A.; O‘Neill, C.; Wolfe, P. In vivo investigation of changes in biomarkers of oxidative stress induced by plant food rich diets. J. Agric. Food Chem. 2005, 53, 6126–6132. [Google Scholar] [CrossRef] [PubMed]
- Hermsdorff, H.H.M.; Zulet, M.A.; Puchau, B.; Martínez, J.A. Fruit and vegetable consumption and proinflammatory gene expression from peripheral blood mononuclear cells in young adults: A translational study. Nutr. Metab. 2010, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Yeum, K.J.; Booth, S.L.; Sadowski, J.A.; Liu, C.; Tang, G.; Krinsky, N.I.; Russell, R.M. Human plasma carotenoid response to the ingestion of controlled diets high in fruits and vegetables. Am. J. Clin. Nutr. 1996, 64, 594–602. [Google Scholar] [CrossRef]
- Harasym, J.; Oledzki, R. Effect of fruit and vegetable antioxidants on total antioxidant capacity of blood plasma. Nutrition 2014, 30, 511–517. [Google Scholar] [CrossRef]
- Gruszecki, W.I.; Strzałka, K. Carotenoids as modulators of lipid membrane physical properties. Biochim. et Biophys. Acta Mol. Basis Dis. 2005, 1740, 108–115. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Rao, A.V. Tomato lycopene and low density lipoprotein oxidation: A human dietary intervention study. Lipids 1998, 33, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Visioli, F.; Riso, P.; Grande, S.; Galli, C.; Porrini, M. Protective activity of tomato products on in vivo markers of lipid oxidation. Eur. J. Nutr. 2003, 42, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Asgard, R.; Rytter, E.; Basu, S.; Abramsson-Zetterberg, L.; Moller, L.; Vessby, B. High intake of fruit and vegetables is related to low oxidative stress and inflammation in a group of patients with type 2 diabetes. Scand. J. Food Nutr. 2007, 51, 149–158. [Google Scholar] [CrossRef]
- Myles, I.A. Fast food fever: Reviewing the impacts of the Western diet on immunity. Nutr. J. 2014, 13, 61. [Google Scholar] [CrossRef] [Green Version]
- McAuliffe, S.; Ray, S.; Fallon, E.; Bradfield, J.; Eden, T.; Kohlmeier, M. Dietary micronutrients in the wake of COVID-19: An appraisal of evidence with a focus on high-risk groups and preventative healthcare. BMJ Nutr. Prev. Health 2020, 3, 93. [Google Scholar] [CrossRef]
- Crabb, D.W.; Matsumoto, M.; Chang, D.; You, M. Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc. Nutr. Soc. 2004, 63, 49–63. [Google Scholar] [CrossRef] [Green Version]
- Montoliu, C.; Sancho-Tello, M.; Azorin, I.; Burgal, M.; Vallés, S.; Renau-Piqueras, J.; Guerri, C. Ethanol Increases Cytochrome P4502E1 and Induces Oxidative Stress in Astrocytes. J. Neurochem. 1995, 65, 2561–2570. [Google Scholar] [CrossRef]
- Lu, Y.; Cederbaum, A.I. CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008, 44, 723–738. [Google Scholar] [CrossRef] [Green Version]
- Roig, R.; Cascón, E.; Arola, L.; Bladé, C.; Salvadó, M.J. Effects of chronic wine and alcohol intake on glutathione and malondialdehyde levels in rats. Nutr. Res. 2000, 20, 1547–1555. [Google Scholar] [CrossRef]
- Hirano, T. Alcohol consumption and oxidative DNA damage. Int. J. Environ. Res. Public Health 2011, 8, 2895–2906. [Google Scholar] [CrossRef] [Green Version]
- Schrieks, I.C.; van den Berg, R.; Sierksma, A.; Beulens, J.W.J.; Vaes, W.H.J.; Hendriks, N.F.J. Effect of red wine consumption on biomarkers of oxidative stress. Alcohol Alcohol. 2013, 48, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zima, T.; Fialová, L.; Mestek, O.; Janebová, M.; Crkovská, J.; Malbohan, I.; Štípek, S.; Mikulíková, L.; Popov, P. Oxidative stress, metabolism of ethanol and alcohol-related diseases. J. Biomed. Sci. 2001, 8, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Marta, K.; Tomáš, Z.; Petr, P.; Pavel, Š.; Martin, B.; Jiřina, S.; Květa, P.; Rosemarie, K.E. Advanced glycation end-products in patients with chronic alcohol misuse. Alcohol Alcohol. 2004, 39, 316–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemela, O. Aldehyde-protein adducts in the liver as a result of ethanol-induced oxidative stress. Front. Biosci. J. Virtual Libr. 1999, 4, D506–D513. [Google Scholar] [CrossRef] [Green Version]
- Guest, J.; Guillemin, G.J.; Heng, B.; Grant, R. Lycopene pretreatment ameliorates acute ethanol induced NAD+depletion in human astroglial cells. Oxidative Med. Cell. Longev. 2015, 2015, 741612. [Google Scholar] [CrossRef]
- Frary, C.D.; Johnson, R.K.; Wang, M.Q. Food sources and intakes of caffeine in the diets of persons in the United States. J. Am. Diet. Assoc. 2005, 105, 110–113. [Google Scholar] [CrossRef]
- Smit, H.J.; Gaffan, E.A.; Rogers, P.J. Methylxanthines are the psycho-pharmacologically active constituents of chocolate. Psychopharmacology 2004, 176, 412–419. [Google Scholar] [CrossRef]
- Chu, Y.F.; Chen, Y.; Brown, P.H.; Lyle, B.J.; Black, R.M.; Cheng, I.H.; Ou, B.; Prior, R.L. Bioactivities of crude caffeine: Antioxidant activity, cyclooxygenase-2 inhibition, and enhanced glucose uptake. Food Chem. 2012, 131, 564–568. [Google Scholar] [CrossRef]
- Acheson, K.J.; Gremaud, G.; Meirim, I.; Montigon, F.; Krebs, Y.; Fay, L.B.; Gay, L.-J.; Schneiter, P.; Schindler, C.; Tappy, L. Metabolic effects of caffeine in humans: Lipid oxidation or futile cycling? Am. J. Clin. Nutr. 2004, 79, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Zeraatpishe, A.; Malekirad, A.A.; Nik-Kherad, J.; Jafari, A.; Babadi, S.Y.; Tanwir, F.; Espanani, H.R. The effects of caffeine supplements on exercise-induced oxidative damages. Asian J. Sports Med. 2015, 6, e23020. [Google Scholar] [CrossRef] [Green Version]
- Al-Basher, G.I.; Aljabal, H.; Almeer, R.S.; Allam, A.A.; Mahmoud, A.M. Perinatal exposure to energy drink induces oxidative damage in the liver, kidney and brain, and behavioral alterations in mice offspring. Biomed. Pharmacother. 2018, 102, 798–811. [Google Scholar] [CrossRef] [PubMed]
- Zampelas, A.; Panagiotakos, D.B.; Pitsavos, C.; Chrysohoou, C.; Stefanadis, C. Associations between coffee consumption and inflammatory markers in healthy persons: The ATTICA study. Am. J. Clin. Nutr. 2004, 80, 862–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez Valdez, R.; Ahlawat, R.; Wills-Karp, M.; Nathan, A.; Ezell, T.; Gauda, E.B. Correlation between serum caffeine levels and changes in cytokine profile in a cohort of preterm infants. J. Pediatrics 2011, 158, 57–64.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correa, T.A.F.; Rogero, M.M.; Mioto, B.M.; Tarasoutchi, D.; Tuda, V.L.; Cesar, L.A.M.; Torres EAFS. Paper-filtered coffee increases cholesterol and inflammation biomarkers independent of roasting degree: A clinical trial. Nutrition 2013, 29, 977–981. [Google Scholar] [CrossRef] [Green Version]
- Chavez-Valdez, R.; Ahlawat, R.; Wills-Karp, M.; Gauda, E.B. Mechanisms of modulation of cytokine release by human cord blood monocytes exposed to high concentrations of caffeine. Pediatric Res. 2016, 80, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Karki, R.; Igwe, O.J. Toll-Like Receptor 4–Mediated Nuclear Factor Kappa B Activation Is Essential for Sensing Exogenous Oxidants to Propagate and Maintain Oxidative/Nitrosative Cellular Stress. PLOS ONE 2013, 8, e73840. [Google Scholar] [CrossRef]
- Burns, D.M. Cigarettes and cigarette smoking. Clin. Chest Med. 1991, 12, 631–642. [Google Scholar]
- Rohrer, J.; Wuertz, B.R.K.; Ondrey, F. Cigarette smoke condensate induces nuclear factor kappa-B activity and proangiogenic growth factors in aerodigestive cells. Laryngoscope 2010, 120, 1609–1613. [Google Scholar] [CrossRef]
- Csordas, A.; Wick, G.; Laufer, G.; Bernhard, D. An evaluation of the clinical evidence on the role of inflammation and oxidative stress in smoking-mediated cardiovascular disease. Biomark. Insights 2008, 2008, 127–139. [Google Scholar] [CrossRef]
- Teixeira De Lemos, E.; Oliveira, J.; Páscoa Pinheiro, J.; Reis, F. Regular physical exercise as a strategy to improve antioxidant and anti-inflammatory status: Benefits in type 2 diabetes mellitus. Oxidative Med. Cell. Longev. 2012, 2012, 741545. [Google Scholar] [CrossRef]
- Gomez-Cabrera, M.C.; Domenech, E.; Viña, J. Moderate exercise is an antioxidant: Upregulation of antioxidant genes by training. Free Radic. Biol. Med. 2008, 44, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Finaud, J.; Lac, G.; Filaire, E. Oxidative stress: Relationship with exercise and training. Sports Med. 2006, 36, 327–358. [Google Scholar] [CrossRef] [PubMed]
- Caimi, G.; Canino, B.; Amodeo, G.; Montana, M.; Presti, R.L. Lipid peroxidation and total antioxidant status in unprofessional athletes before and after a cardiopulmonary test. Clin. Hemorheol. Microcirc. 2009, 43, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.D.; Sureda, A.; Batle, J.M.; Tauler, P.; Tur, J.A.; Pons, A. Scuba diving enhances endogenous antioxidant defenses in lymphocytes and neutrophils. Free Radic. Res. 2007, 41, 274–281. [Google Scholar] [CrossRef]
- Niess, A.M.; Passek, F.; Lorenz, I.; Schneider, E.M.; Dickhuth, H.H.; Northoff, H.; Fehrenbach, E. Expression of the antioxidant stress protein heme oxygenase-1 (HO-1) in human leukocytes: Acute and adaptational responses to endurance exercise. Free Radic. Biol. Med. 1999, 26, 184–192. [Google Scholar] [CrossRef]
- Galassetti, P.R.; Nemet, D.; Pescatello, A.; Rose-Gottron, C.; Larson, J.; Cooper, D.M. Exercise, caloric restriction, and systemic oxidative stress. J. Investig. Med. 2006, 54, 67–75. [Google Scholar] [CrossRef]
- Martin-Cordero, L.; Garcia, J.J.; Giraldo, E.; De la Fuente, M.; Manso, R.; Ortega, E. Influence of exercise on the circulating levels and macrophage production of IL-1β and IFNγ affected by metabolic syndrome: An obese Zucker rat experimental animal model. Eur. J. Appl.Physiol. 2009, 107, 535–543. [Google Scholar] [CrossRef]
- Mattusch, F.; Dufaux, B.; Heine, O.; Mertens, I.; Rost, R. Reduction of the plasma concentration of C-reactive protein following nine months of endurance training. Int. J. Sports Med. 2000, 21, 21–24. [Google Scholar] [CrossRef]
- Smith, J.K.; Dykes, R.; Douglas, J.E.; Krishnaswamy, G.; Berk, S. Long-term exercise and atherogenic activity of blood mononuclear cells in persons at risk of developing ischemic heart disease. J. Am. Med. Assoc. 1999, 281, 1722–1727. [Google Scholar] [CrossRef]
- Reimund, E. The free radical flux theory of sleep. Med Hypotheses 1994, 43, 231–233. [Google Scholar] [CrossRef]
- Ikeda, M.; Ikeda-Sagara, M.; Okada, T.; Clement, P.; Urade, Y.; Nagai, T.; Sugiyama, T.; Yoshioka, T.; Honda, K.; Inoué, S. Brain oxidation is an initial process in sleep induction. Neuroscience 2005, 130, 1029–1040. [Google Scholar] [CrossRef] [PubMed]
- Boudjeltia, K.; Faraut, B.; Esposito, M.J.; Stenuit, P.; Dyzma, M.; van Antwerpen, P.; Brohée, D.; Vanhamme, L.; Moguilevsky, N.; Vanhaeverbeek, M.; et al. Temporal dissociation between myeloperoxidase (MPO)-modified LDL and MPO elevations during chronic sleep restriction and recovery in healthy young men. PLoS ONE. 2011, 6, e28230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villafuerte, G.; Miguel-Puga, A.; Murillo Rodríguez, E.; Machado, S.; Manjarrez, E.; Arias-Carrión, O. Sleep deprivation and oxidative stress in animal models: A systematic review. Oxidative Med. Cell. Longev. 2015, 2015, 234952. [Google Scholar] [CrossRef] [PubMed]
- Alzoubi, K.H.; Khabour, O.F.; Salah, H.A.; Abu Rashid, B.E. The combined effect of sleep deprivation and western diet on spatial learning and memory: Role of BDNF and oxidative stress. J. Mol. Neurosci. 2013, 50, 124–133. [Google Scholar] [CrossRef]
- van Leeuwen, W.M.A.; Lehto, M.; Karisola, P.; Lindholm, H.; Luukkonen, R.; Sallinen, M.; Härmä, M.; Porkka-Heiskanen, T.; Alenius, H. Sleep restriction increases the risk of developing cardiovascular diseases by augmenting proinflammatory responses through IL-17 and CRP. PLoS ONE 2009, 4, e4589. [Google Scholar] [CrossRef] [Green Version]
- Meier-Ewert, H.K.; Ridker, P.M.; Rifai, N.; Regan, M.M.; Price, N.J.; Dinges, D.F.; Mullington, J.M. Effect of sleep loss on C-Reactive protein, an inflammatory marker of cardiovascular risk. J. Am. Coll. Cardiol. 2004, 43, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Eisele, H.J.; Markart, P.; Schulz, R. Obstructive sleep apnea, oxidative stress, and cardiovascular disease: Evidence from human studies. Oxidative Med. Cell. Longev. 2015, 2015, 608438. [Google Scholar] [CrossRef] [Green Version]
- Younes, M. Role of respiratory control mechanisms in the pathogenesis of obstructive sleep disorders. J. Appl. Physiol. 2008, 105, 1389–1405. [Google Scholar] [CrossRef] [Green Version]
- Schulz, R.; Mahmoudi, S.; Hattar, K.; Sibelius, U.L.F.; Olschewski, H.; Mayer, K.; Seeger, W.; Grimminger, F. Enhanced release of superoxide from polymorphonuclear neutrophils in obstructive sleep apnea: Impact of continuous positive airway pressure therapy. Am. J. Respir. Crit. Care Med. 2000, 162, 566–670. [Google Scholar] [CrossRef]
- Alonso-Fernández, A.; García-Río, F.; Arias, M.A.; Hernanz, Á.; De La Peña, M.; Piérola, J.; Barceló, A.; López-Collazo, E.; Agustí, A. Effects of CPAP on oxidative stress and nitrate efficiency in sleep apnoea: A randomised trial. Thorax 2009, 64, 581–586. [Google Scholar] [CrossRef] [Green Version]
- Lavie, L.; Vishnevsky, A.; Lavie, P. Evidence for lipid peroxidation in obstructive sleep apnea. Sleep 2004, 27, 123–128. [Google Scholar] [PubMed]
- Yamauchi, M.; Nakano, H.; Maekawa, J.; Okamoto, J.; Ohnishi, Y.; Suzuki, T.; Kimura, H. Oxidative stress in obstructive sleep apnea. Chest 2005, 127, 1674–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozben, S.; Huseyinoglu, N.; Hanikoglu, F.; Guvenc, T.S.; Yildirim, B.Z.; Cort, A.; Ozdem, S.; Ozben, T. Advanced oxidation protein products and ischaemia-modified albumin in obstructive sleep apnea. Eur. J. Clin. Investig. 2014, 44, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Barceló, A.; Barbé, F.; de la Peña, M.; Vila, M.; Pérez, G.; Piérola, J.; Durán, J.; Agustí, A.G.N. Antioxidant status in patients with sleep apnoea and impact of continuous positive airway pressure treatment. Eur. Respir. J. 2006, 27, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Wysocka, E.; Cofta, S.; Cymerys, M.; Gozdzik, J.; Torlinski, L.; Batura-Gabryel, H. The impact of the sleep apnea syndrome on oxidant-antioxidant balance in the blood of overweight and obese patients. J. Physiol. Pharmacol. 2008, 59 (Suppl. 6), 761–769. [Google Scholar]
- Unnikrishnan, D.; Jun, J.; Polotsky, V. Inflammation in sleep apnea: An update. Rev. Endocr. Metab. Disord. 2015, 16, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Ryan, S.; Taylor, C.T.; McNicholas, W.T. Predictors of elevated nuclear factor-κB-dependent genes in obstructive sleep apnea syndrome. Am. J. Respir. Crit. Care Med. 2006, 174, 824–830. [Google Scholar] [CrossRef]
- Gross, C.; Hen, R. The developmental origins of anxiety. Nat. Rev. Neurosci. 2004, 5, 545–552. [Google Scholar] [CrossRef]
- Weinberger, D.R. Anxiety at the frontier of molecular medicine. New Engl. J. Med. 2001, 344, 1247–1249. [Google Scholar] [CrossRef]
- Anderson, G.; Maes, M. Oxidative/nitrosative stress and immuno-inflammatory pathways in depression: Treatment implications. Curr. Pharm. Des. 2014, 20, 3812–3847. [Google Scholar] [CrossRef]
- Bouayed, J.; Rammal, H.; Soulimani, R. Oxidative stress and anxiety: Relationship and cellular pathways. Oxidative Med. Cell. Longev. 2009, 2, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Rammal, H.; Bouayed, J.; Younos, C.; Soulimani, R. The impact of high anxiety level on the oxidative status of mouse peripheral blood lymphocytes, granulocytes and monocytes. Eur. J. Pharmacol. 2008, 589, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Guney, E.; Fatih Ceylan, M.; Tektas, A.; Alisik, M.; Ergin, M.; Goker, Z.; Senses Dinc, G.; Ozturk, O.; Korkmaz, A.; Eker, S.; et al. Oxidative stress in children and adolescents with anxiety disorders. J. Affect. Disord. 2014, 156, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Gałecki, P.; Szemraj, J.; Bieńkiewicz, M.; Florkowski, A.; Gałecka, E. Lipid peroxidation and antioxidant protection in patients during acute depressive episodes and in remission after fluoxetine treatment. Pharmacol. Rep. 2009, 61, 436–447. [Google Scholar] [CrossRef]
- Dimopoulos, N.; Piperi, C.; Psarra, V.; Lea, R.W.; Kalofoutis, A. Increased plasma levels of 8-iso-PGF2α and IL-6 in an elderly population with depression. Psychiatry Res. 2008, 161, 59–66. [Google Scholar] [CrossRef]
- Maes, M.; Mihaylova, I.; Kubera, M.; Uytterhoeven, M.; Vrydags, N.; Bosmans, E. Increased 8-hydroxy-deoxyguanosine, a marker of oxidative damage to DNA, in major depression and myalgic encephalomyelitis / chronic fatigue syndrome. Neuroendocrinol. Lett. 2009, 30, 715–722. [Google Scholar]
- Arranz, L.; Guayerbas, N.; Siboni, L.; De la Fuente, M. Effect of acupuncture treatment on the immune function impairment found in anxious women. Am. J. Chin. Med. 2007, 35, 35–51. [Google Scholar] [CrossRef]
- Szego, É.M.; Janáky, T.; Szabó, Z.; Csorba, A.; Kompagne, H.; Müller, G.; Lévay, G.; Simor, A.; Juhász, G.; Kékesi, K.A. A mouse model of anxiety molecularly characterized by altered protein networks in the brain proteome. Eur. Neuropsychopharmacol. 2010, 20, 96–111. [Google Scholar] [CrossRef]
- Maes, M.; De Vos, N.; Pioli, R.; Demedts, P.; Wauters, A.; Neels, H.; Christophe, A. Lower serum vitamin E concentrations in major depression. Another marker of lowered antioxidant defenses in that illness. J. Affect. Disord. 2000, 58, 241–246. [Google Scholar] [CrossRef]
- Kodydková, J.; Vávrová, L.; Zeman, M.; Jirák, R.; Macášek, J.; Staňková, B.; Tvrzická, E.; Žák, A. Antioxidative enzymes and increased oxidative stress in depressive women. Clin. Biochem. 2009, 42, 1368–1374. [Google Scholar] [CrossRef]
- Maes, M.; Mihaylova, I.; Kubera, M.; Uytterhoeven, M.; Vrydags, N.; Bosmans, E. Lower plasma Coenzyme Q10 in depression: A marker for treatment resistance and chronic fatigue in depression and a risk factor to cardiovascular disorder in that illness. Neuroendocrinol. Lett. 2009, 30, 462–469. [Google Scholar] [PubMed]
- Vogelzangs, N.; Beekman, A.T.F.; De Jonge, P.; Penninx, B.W.J.H. Anxiety disorders and inflammation in a large adult cohort. Transl. Psychiatry 2013, 3, e249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.J.; Zhao, X.H.; Chen, W.; Bo, N.; Wang, X.J.; Chi, Z.F.; Wu, W. Sirtuin 1 activation enhances the PGC-1á/mitochondrial antioxidant system pathway in status epilepticus. Mol. Med. Rep. 2015, 11, 521–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christmas, D.M.; Potokar, J.P.; Davies, S.J.C. A biological pathway linking inflammation and depression: Activation of indoleamine 2,3-dioxygenase. Neuropsychiatr. Dis. Treat. 2011, 7, 431–439. [Google Scholar] [PubMed] [Green Version]
- Maes, M.; Leonard, B.E.; Myint, A.M.; Kubera, M.; Verkerk, R. The new ‘5-HT’ hypothesis of depression: Cell-mediated immune activation induces indoleamine 2,3-dioxygenase, which leads to lower plasma tryptophan and an increased synthesis of detrimental tryptophan catabolites (TRYCATs), both of which contribute to the onset of depression. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2011, 35, 702–721. [Google Scholar]
- Kong, S.Y.; Goodman, M.; Judd, S.; Bostick, R.M.; Flanders, W.D.; McClellan, W. Oxidative balance score as predictor of all-cause, cancer, and noncancer mortality in a biracial US cohort. Ann. Epidemiol. 2015, 25, 256–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geybels, M.S.; Verhage, B.A.J.; van Schooten, F.J.; van den Brandt, P.A. Measures of combined antioxidant and pro-oxidant exposures and risk of overall and advanced stage prostate cancer. Ann. Epidemiol. 2012, 22, 814–820. [Google Scholar] [CrossRef]
- Ilori, T.O.; Wang, X.; Huang, M.; Gutierrez, O.M.; Narayan, K.M.V.; Goodman, M.; McClellan, W.; Plantinga, L.; Ojo, A.O. Oxidative Balance Score and the Risk of End-Stage Renal Disease and Cardiovascular Disease. Am. J. Nephrol. 2017, 45, 338–345. [Google Scholar] [CrossRef]
- Agalliu, I.; Kirsh, V.A.; Kreiger, N.; Soskolne, C.L.; Rohan, T.E. Oxidative balance score and risk of prostate cancer: Results from a case-cohort study. Cancer Epidemiol. 2011, 35, 353–361. [Google Scholar] [CrossRef]
- Colpani, V.; Baena, C.P.; Jaspers, L.; van Dijk, G.M.; Farajzadegan, Z.; Dhana, K.; Tielemans, M.J.; Voortman, T.; Freak-Poli, R.; Veloso, G.G.V.; et al. Lifestyle factors, cardiovascular disease and all-cause mortality in middle-aged and elderly women: A systematic review and meta-analysis. Eur. J. Epidemiol. 2018, 33, 831–845. [Google Scholar] [CrossRef]
- Lakkur, S.; Judd, S.; Bostick, R.M.; McClellan, W.; Flanders, W.D.; Stevens, V.L.; Goodman, M. Oxidative stress, inflammation, and markers of cardiovascular health. Atherosclerosis 2015, 243, 38–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odegaard, A.O.; Koh, W.P.; Gross, M.D.; Yuan, J.M.; Pereira, M.A. Combined lifestyle factors and cardiovascular disease mortality in chinese men and women: The singapore chinese health study. Circulation 2011, 124, 2847–2854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abuja, P.M.; Albertini, R. Methods for monitoring oxidative stress, lipid peroxidation and oxidation resistance of lipoproteins. Clin. Chim. Acta 2001, 306, 1–17. [Google Scholar] [CrossRef]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxidative Med. Cell. Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef] [PubMed]
- D’Arena, G.; Vitale, C.; Perbellini, O.; Coscia, M.; La Rocca, F.; Ruggieri, V.; Visco, C.; Di Minno, N.M.D.; Innocenti, I.; Pizza, V.; et al. Prognostic relevance of oxidative stress measurement in chronic lymphocytic leukaemia. Eur. J. Haematol. 2017, 99, 306–314. [Google Scholar] [CrossRef]
- Ho, E.; Karimi Galougahi, K.; Liu, C.C.; Bhindi, R.; Figtree, G.A. Biological markers of oxidative stress: Applications to cardiovascular research and practice. Redox Biol. 2013, 1, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Montuschi, P.; Barnes, P.J.; Roberts Ii, L.J. Isoprostanes: Markers and mediators of oxidative stress. FASEB J. 2004, 18, 1791–1800. [Google Scholar] [CrossRef]
- Stephens, J.W.; Khanolkar, M.P.; Bain, S.C. The biological relevance and measurement of plasma markers of oxidative stress in diabetes and cardiovascular disease. Atherosclerosis 2009, 202, 321–329. [Google Scholar] [CrossRef]
- Seyedsadjadi, N.; Berg, J.; Bilgin, A.A.; Tung, C.; Grant, R. Significant relationships between a simple marker of redox balance and lifestyle behaviours; Relevance to the Framingham risk score. PLoS ONE 2017, 12, e0187713. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B.; Chirico, S.; Crawford, M.A.; Bjerve, K.S.; Gey, K.F. Lipid peroxidation: Its mechanism, measurement, and significance. Am. J. Clin. Nutr. 1993, 57 (Suppl. 5), 715S–725S. [Google Scholar] [CrossRef] [Green Version]
- Repetto, M.; Semprine, J.; Boveris, A. Lipid Peroxidation: Chemical Mechanism, Biological Implications and Analytical Determination; IntechOpen: London, UK, 2012. [Google Scholar]
- Orhan, H.; Gurer-Orhan, H.; Vriese, E.; Vermeulen, N.P.E.; Meerman, J.H.N. Application of lipid peroxidation and protein oxidation biomarkers for oxidative damage in mammalian cells. A Comp. Two Fluoresc. Probes. Toxicol. Vitr. 2006, 20, 1005–1013. [Google Scholar]
- Ghiselli, A.; Serafini, M.; Natella, F.; Scaccini, C. Total antioxidant capacity as a tool to assess redox status: Critical view and experimental data. Free Radic. Biol. Med. 2000, 29, 1106–1114. [Google Scholar] [CrossRef]
- Brunelli, E.; La Russa, D.; Pellegrino, D. Impaired oxidative status is strongly associated with cardiovascular risk factors. Oxidative Med. Cell. Longev. 2017, 2017, 6480145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbasi, A.; Corpeleijn, E.; Postmus, D.; Gansevoort, R.T.; de Jong, P.E.; Gans, R.O.; Struck, J.; Schulte, J.; Hillege, H.L.; van der Harst, P.; et al. Peroxiredoxin 4, a novel circulating biomarker for oxidative stress and the risk of incident cardiovascular disease and all-cause mortality. J. Am. Heart Assoc. 2012, 1, e002956. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.N.; Kim, K.M.; Lee, D.J.; Joo, N.S. Serum gamma-glutamyltransferase concentration correlates with Framingham risk score in Koreans. J. Korean Med. Sci. 2011, 26, 1305–1309. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.M.; Kim, B.T.; Park, S.B.; Cho, D.Y.; Je, S.H.; Kim, K.N. Serum Total Bilirubin Concentration Is Inversely Correlated with Framingham Risk Score in Koreans. Arch. Med Res. 2012, 43, 288–293. [Google Scholar] [CrossRef]
- Ashfaq, S.; Abramson, J.L.; Jones, D.P.; Rhodes, S.D.; Weintraub, W.S.; Hooper, W.C.; Vaccarino, V.; Alexander, R.W.; Harrison, D.G.; Quyyumi, A.A. Endothelial function and aminothiol biomarkers of oxidative stress in healthy adults. Hypertension 2008, 52, 80–85. [Google Scholar] [CrossRef]
- Taleb, A.; Witztum, J.L.; Tsimikas, S. Oxidized phospholipids on apoB-100-containing lipoproteins: A biomarker predicting cardiovascular disease and cardiovascular events. Biomark. Med. 2011, 5, 673–694. [Google Scholar] [CrossRef] [Green Version]
- Rama Krishna Reddy, Y.V.; Mahendra, J.; Gurumurthy, P.; Jayamathi Babu, S. Identification of predictable biomarkers in conjunction to framingham risk score to predict the risk for cardiovascular disease (CVD) in non cardiac subjects. J. Clin. Diagn. Res. 2015, 9, BC23–BC27. [Google Scholar]
- Okada, K.; Maeda, N.; Tatsukawa, M.; Shimizu, C.; Sawayama, Y.; Hayashi, J. The influence of lifestyle modification on carotid artery intima-media thickness in a suburban Japanese population. Atherosclerosis 2004, 173, 329–337. [Google Scholar] [CrossRef]
- Giannini, C.; Diesse, L.; D’Adamo, E.; Chiavaroli, V.; de Giorgis, T.; Di Iorio, C.; Chiarelli, F.; Mohn, A. Influence of the Mediterranean diet on carotid intima-media thickness in hypercholesterolaemic children: A 12-month intervention study. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Marshall, D.; Walizer, E.; Vernalis, M. The effect of a one-year lifestyle intervention program on carotid intima media thickness. Mil. Med. 2011, 176, 798–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagi, H.; Sumino, H.; Yoshida, K.; Aoki, T.; Tsunekawa, K.; Araki, O.; Kimura, T.; Nara, M.; Nakajima, K.; Murakami, M. Biological antioxidant potential negatively correlates with carotid artery intima-media thickness. Int. Heart J. 2016, 57, 220–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyedsadjadi, N.; Berg, J.; Bilgin, A.A.; Grant, R. A pilot study providing evidence for a relationship between a composite lifestyle score and risk of higher carotid intima-media thickness: Is there a link to oxidative stress? Oxidative Med. Cell. Longev. 2018, 2018, 4504079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira, M.C.; Schoffen, J.P.F. Oxidative stress action in cellular aging. Braz. Arch. Biol. Technol. 2010, 53, 1333–1342. [Google Scholar] [CrossRef] [Green Version]
- Griendling, K.K.; Touyz, R.M.; Zweier, J.L.; Dikalov, S.; Chilian, W.; Chen, Y.R.; Harrison, D.G.; Bhatnagar, A. Measurement of Reactive Oxygen Species, Reactive Nitrogen Species, and Redox-Dependent Signaling in the Cardiovascular System: A Scientific Statement from the American Heart Association. Circ. Res. 2016, 119, e39–e75. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Rossi, R.; Colombo, R.; Giustarini, D.; Milzani, A. Biomarkers of oxidative damage in human disease. Clin. Chem. 2006, 52, 601–623. [Google Scholar] [CrossRef]
- Milne, G.L.; Dai, Q.; Roberts, L.J. The isoprostanes—25 years later. Biochim. et Biophys. Acta Mol. Cell Biol. Lipids 2015, 1851, 433–445. [Google Scholar] [CrossRef] [Green Version]
- Vassalle, C.; Landi, P.; Boni, C.; Zucchelli, G. Oxidative stress evaluated using an automated method for hydroperoxide estimation in patients with coronary artery disease. Clin. Chem. Lab. Med. 2007, 45, 367–371. [Google Scholar] [CrossRef]
- Teixeira, D.; Fernandes, R.; Prudêncio, C.; Vieira, M. 3-Nitrotyrosine quantification methods: Current concepts and future challenges. Biochimie 2016, 125, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ndrepepa, G. Myeloperoxidase—A bridge linking inflammation and oxidative stress with cardiovascular disease. Clin. Chim. Acta 2019, 493, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Roustazadeh, A.; Alipoor, B. Ox-LDL Particles: Modified Components, Cellular Uptake, Biological roles and clinical assessments. Cardiovasc. Hematol. Disord. Drug Targets 2011, 11, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, N.; Vitaglione, P.; Granato, D.; Fogliano, V. Twenty-five years of total antioxidant capacity measurement of foods and biological fluids: Merits and limitations. J. Sci. Food Agric. 2020, 100, 5064–5078. [Google Scholar] [CrossRef] [PubMed]
- Natoli, G.; Ghisletti, S.; Barozzi, I. The genomic landscapes of inflammation. Genes Dev. 2011, 25, 101–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monastero, R.N.; Pentyala, S. Cytokines as Biomarkers and Their Respective Clinical Cutoff Levels. Int. J. Inflamm. 2017, 2017, 4309485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davalos, D.; Akassoglou, K. Fibrinogen as a key regulator of inflammation in disease. Semin. Immunopathol. 2012, 34, 43–62. [Google Scholar] [CrossRef]
- Maeda, S.; Takeya, Y.; Oguro, R.; Akasaka, H.; Ryuno, H.; Kabayama, M.; Yokoyama, S.; Nagasawa, M.; Fujimoto, T.; Takeda, M.; et al. Serum albumin/globulin ratio is associated with cognitive function in community-dwelling older people: The Septuagenarians, Octogenarians, Nonagenarians Investigation with Centenarians study. Geriatr. Gerontol. Int. 2019, 19, 967–971. [Google Scholar] [CrossRef]
- Suh, B.; Park, S.; Shin, D.W.; Yun, J.M.; Keam, B.; Yang, H.K.; Ahn, E.; Lee, H.; Park, J.H.; Cho, B. Low albumin-to-globulin ratio associated with cancer incidence and mortality in generally healthy adults. Ann. Oncol. 2014, 25, 2260–2266. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, D.; Song, P.; Zou, M.H. Tryptophan-kynurenine pathway is dysregulated in inflammation, and immune activation. Front. Biosci. Landmark 2015, 20, 1116–1143. [Google Scholar]
- Gieseg, S.P.; Baxter-Parker, G.; Lindsay, A. Neopterin, inflammation, and oxidative stress: What could we be missing? Antioxidants 2018, 7, 80. [Google Scholar] [CrossRef] [Green Version]
- Del Giudice, M.; Gangestad, S.W. Rethinking IL-6 and CRP: Why they are more than inflammatory biomarkers, and why it matters. Brain Behav. Immun. 2018, 70, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Dinicolantonio, J.J.; O’Keefe, J.H. Importance of maintaining a low omega-6/omega-3 ratio for reducing inflammation. Open Heart 2018, 5, 1–4. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OS Marker | Advantages | Disadvantages | Reference |
|---|---|---|---|
| IsoPros | Can be detected in various samples (serum, plasma urine) and has been shown to be elevated in the presence of a range of cardiovascular risk factors and cancer | Evidence in clinical disease still developing. Test available in specialist labs only | [275] |
| MDA | Easy to quantify in serum, plasma or urine. Studies show MDA can predict progression of coronary artery disease and carotid atherosclerosis at 3 years. | TBARS assay is non-specific (can detect aldehydes other than MDA) therefore potential for interferences. Test available in specialist labs only. | [252] |
| Hydroperoxides (HPX) | Lipid hydroperoxides are non-radical intermediates of lipid peroxidation. If frozen on collection, relatively stable in serum/plasma and can be transported. Growing body of evidence for correlation with clinical disease. Commercial test kits available | Can undergo reductive degradation if not stored properly which will decrease levels. | [276] |
| Nitrotyrosine | Human studies have demonstrated association with coronary artery disease independent of traditional risk factors | Circulating levels not equivalent to tissue levels. Current detection methods are expensive and impractical for scaling up to the clinical setting. | [277] |
| MPO | Measured in serum or plasma. Commercial assays available. Strong evidence that MPO correlates with cardiovascular disease risk | Influenced by sample storage and time to analysis. Test available in specialist labs only. | [278] |
| oxLDL | Serum or plasma levels routinely available as a clinical marker in many larger routine pathology labs. Elevated in coronary artery disease, increasing oxLDL correlates with increasing clinical severity. Also is predictive of future coronary artery disease in healthy population. Good reproducibility from frozen samples | Ox LDL has been studied mostly in relation to CVD so results not generalisable to other NCDs at this stage | [279] |
| Total antioxidant capacity (TAC) | Plasma levels considered a good measure of the activity of the low molecular weight, antioxidants, including albumin, Vitamin C, SH group containing antioxidants (glutathione), polyphenolic and carotenoid compounds and bilirubin. Has been correlated to a variety of disease states. Commercial testing kits available | The specific contribution of antioxidant enzymes such as catalase and SOD are excluded | [280] |
| Gene expression | The change in expression of various genes may reflect altered redox balance and can be measured simultaneously using microarray technology | Expensive and It is unclear if expression profiles of cells in biological samples reflect that in clinical tissues. Test only available form specialist labs | [281] |
| Inflammation Linked Marker | Description/Advantage | Disadvantages | Reference |
|---|---|---|---|
| IL-6 | Early release pro-inflammatory cytokine produced in response to infections and tissue injuries | Half life of 10 min. Therefore, only elevated during active inflammatory activity. Can also have anti-inflammatory effects. Test only available in specialist labs | [282] |
| TNF-α | Pro-inflammatory cytokine produced by macrophages and lymphocytes | Half life of 18 min. Therefore, only elevated during active inflammatory activity. Test only available in specialist labs | [282] |
| IL-1β | Pro-inflammatory cytokine and lymphocyte/monocyte activating factor | Half life of <4 h. Therefore, only elevated during active inflammatory activity. Test only available in specialist labs | [282] |
| IL-8 | Pro-inflammatory cytokine produced by polymorphonuclear (PMN) cells | Half life of ~4 h. Therefore, only elevated during active inflammation due largely to bacterial infections. Test only available in specialist labs | [282] |
| IL-10, IL-4 | Anti-inflammatory cytokines. IL-4 produced by activated T cells, mast cells, basophils, eosinophils, and Natural killer T cells. IL-10 produced by macrophages, dendritic cells (DC),3 B cells, and various subsets of CD4+ and CD8+ T cells | Half life of <4.5 h. Therefore, only elevated during active inflammatory activity. Test only available in specialist labs | [282] |
| Fibrinogen | Pro-inflammatory regulation. High levels well documented risk for many inflammatory conditions. Test routinely available in pathology labs | The mechanistic links between coagulation and inflammation involving fibrinogen activation and signalling are not yet fully known | [283] |
| Alb/Glob ratio | Decreased in inflammation. Sensitive indicator of inflammatory disease, in particular kidney disease. Test available in routine pathology labs | Ratio may change for reasons other than inflammation, e.g., nutritional deficiencies such as low protein intakes | [284,285] |
| Tryp/Kyn ratio | Sensitive indicator of T-helper 1 (i.e., IFN-γ) immune responses. Reduced ratio reported in a range of inflammatory associated conditions including aging, cancer, rheumatoid arthritis and CVD. Altered Kyn:Tryp ratio remains longer and fluctuates less than individual cytokines. | Test not routinely available outside of research labs | [286] |
| Neopterin | Macrophage activation product dependent on IFN-γ activation. Levels remain elevated for longer after IFN-γ levels return to normal. | Produced secondary to IFN-γ activation of macrophages. Test only available in specialist labs. | [287] |
| hs-CRP | Acute phase protein produced by liver following IL-6 activation. Widely used as a clinical marker of inflammation. Levels remain elevated longer than IL-6 and other cytokines. Test widely available in routine pathology labs. | As an acute phase protein levels rise by 2 h peak by 48 h. Half life 18 h. However, one of the 2 isoforms of CRP may have anti-inflammatory effects | [288] |
| Omega-3 Index/ratio | Primary ant-inflammatory modulators. Large body of evidence linking reduced omega-3 levels to conditions associated with chronic inflammation including rheumatoid arthritis, CAD and depression. | Indirect marker of inflammation potential. Test only available in specialist labs | [289] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seyedsadjadi, N.; Grant, R. The Potential Benefit of Monitoring Oxidative Stress and Inflammation in the Prevention of Non-Communicable Diseases (NCDs). Antioxidants 2021, 10, 15. https://doi.org/10.3390/antiox10010015
Seyedsadjadi N, Grant R. The Potential Benefit of Monitoring Oxidative Stress and Inflammation in the Prevention of Non-Communicable Diseases (NCDs). Antioxidants. 2021; 10(1):15. https://doi.org/10.3390/antiox10010015
Chicago/Turabian StyleSeyedsadjadi, Neda, and Ross Grant. 2021. "The Potential Benefit of Monitoring Oxidative Stress and Inflammation in the Prevention of Non-Communicable Diseases (NCDs)" Antioxidants 10, no. 1: 15. https://doi.org/10.3390/antiox10010015