
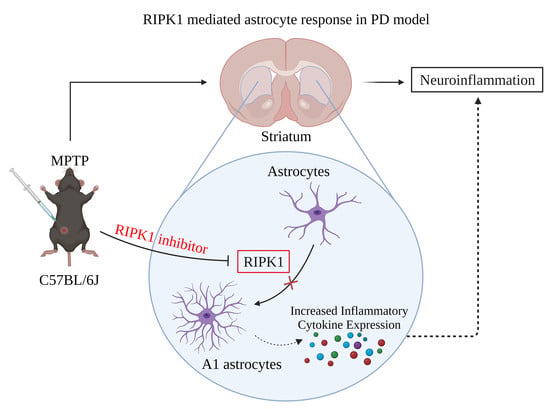
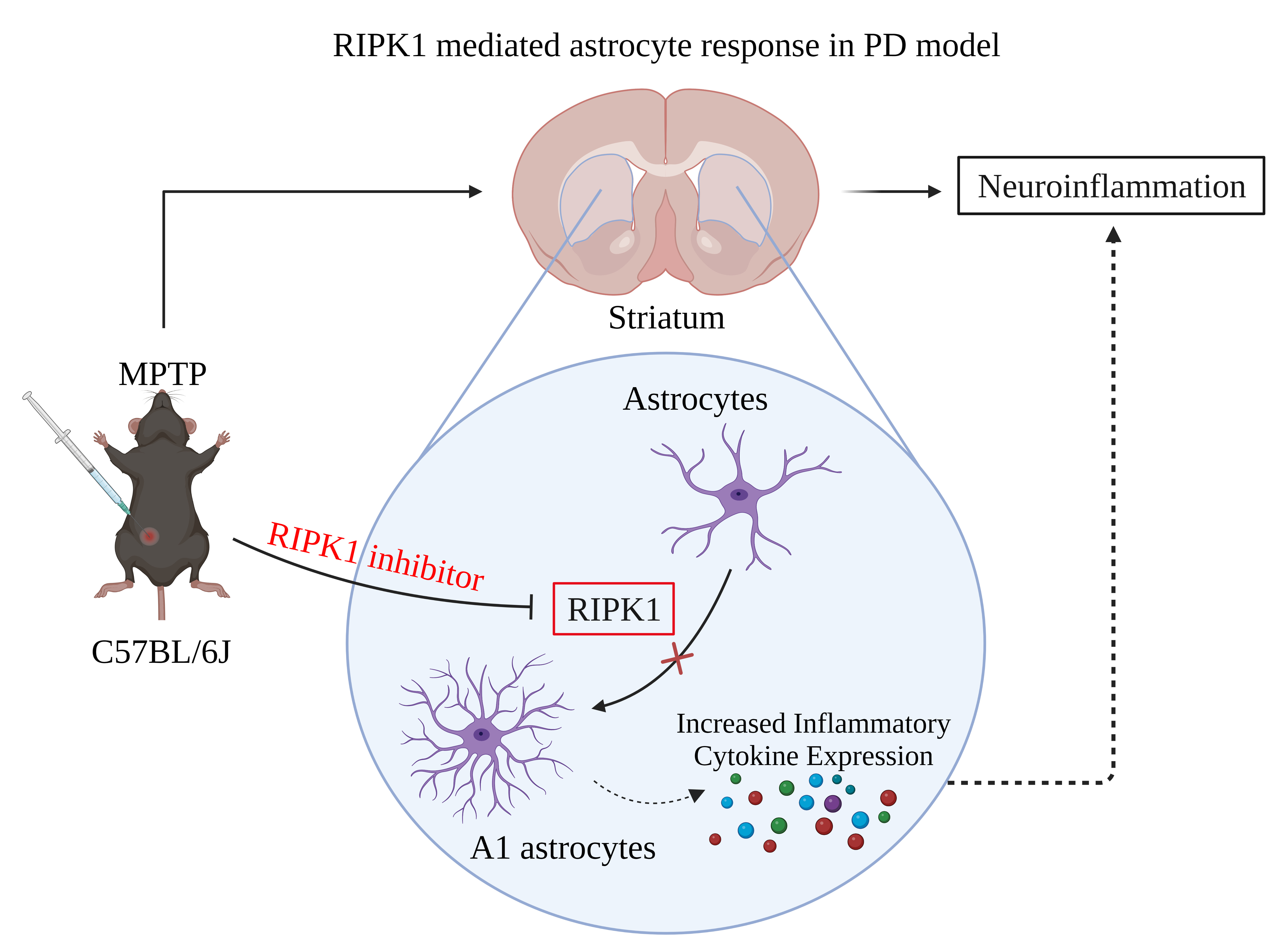
RIPK1-Induced A1 Reactive Astrocytes in Brain in MPTP-Treated Murine Model of Parkinson’s Disease
 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
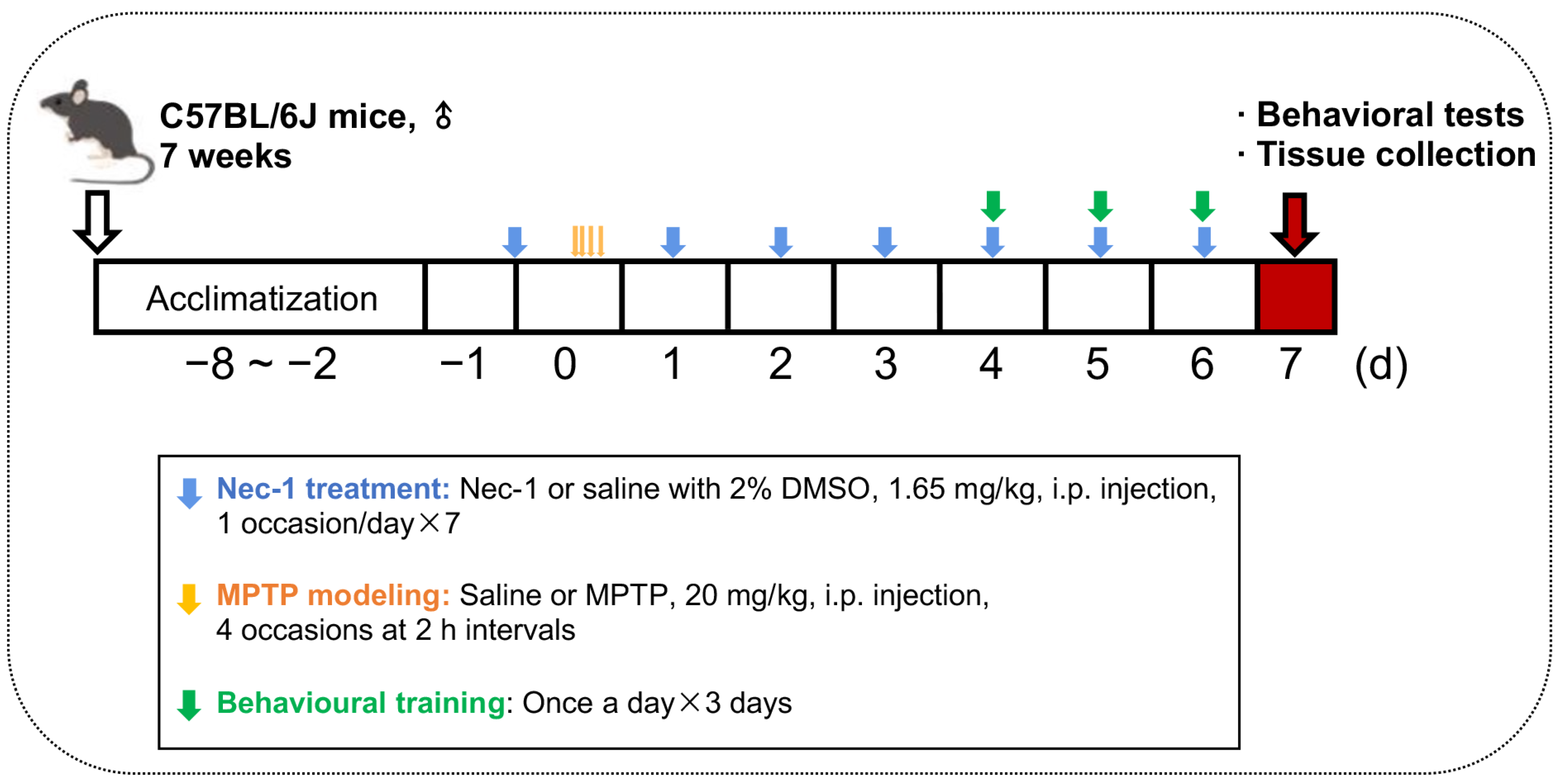
2.1. Animals and Treatments
2.2. Behavioral Tests

2.3. Tissue Sample Collection and Preparation
2.4. Quantitative Real-Time PCR Analysis
2.5. Western Blot Analysis
2.6. Immunofluorescence Staining
2.7. Statistical Analysis
3. Results
3.1. Nec-1 Inhibits RIPK1 Overexpression in MPTP-Treated Murine Model of PD
3.2. Inhibition of RIPK1 Improves MPTP Induced Motor and Non-Motor Dysfunctions in Mice
3.3. Inhibition of RIPK1 Rescues Dopaminergic Neurons in MPTP-Treated Murine Model of PD
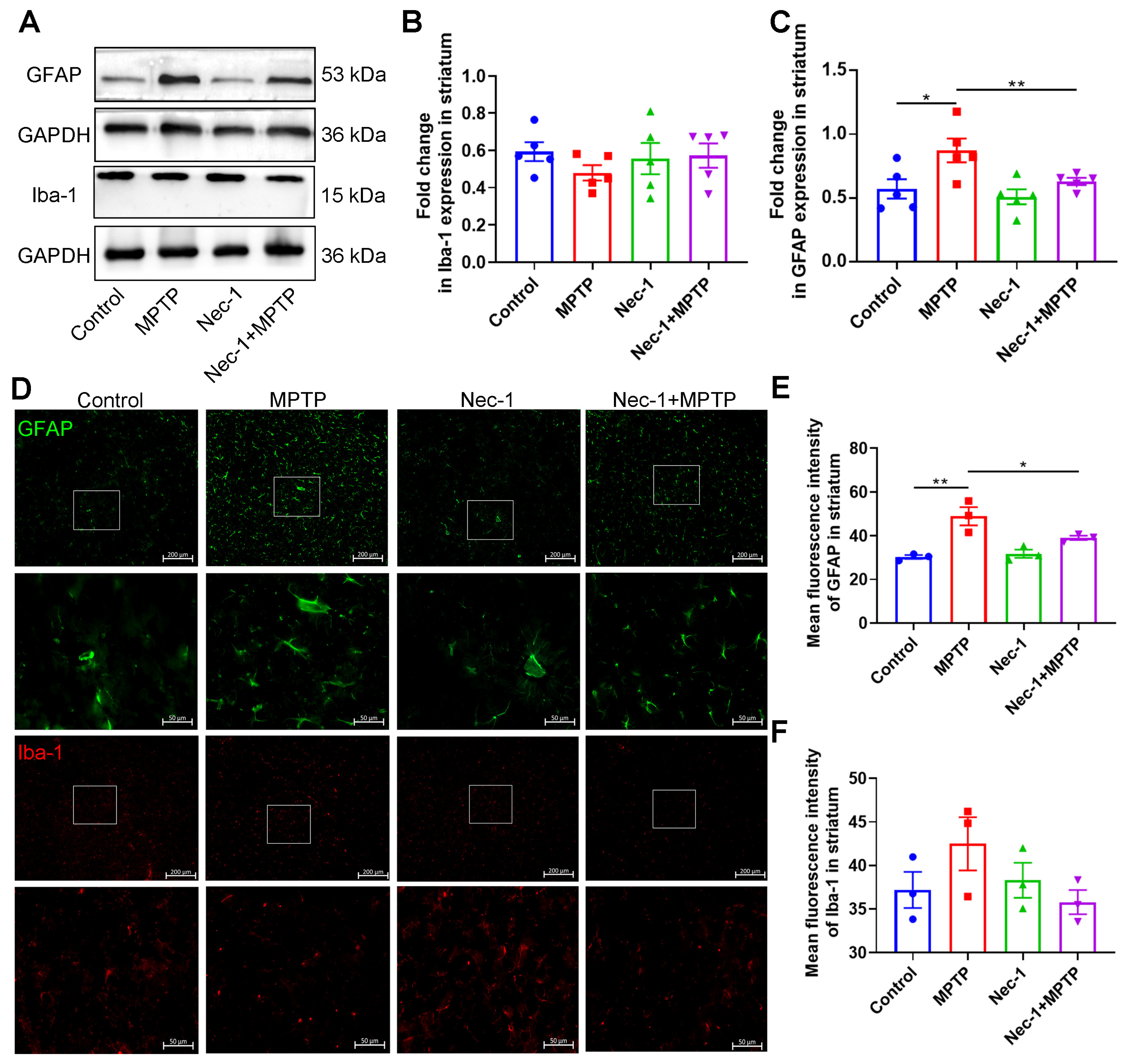
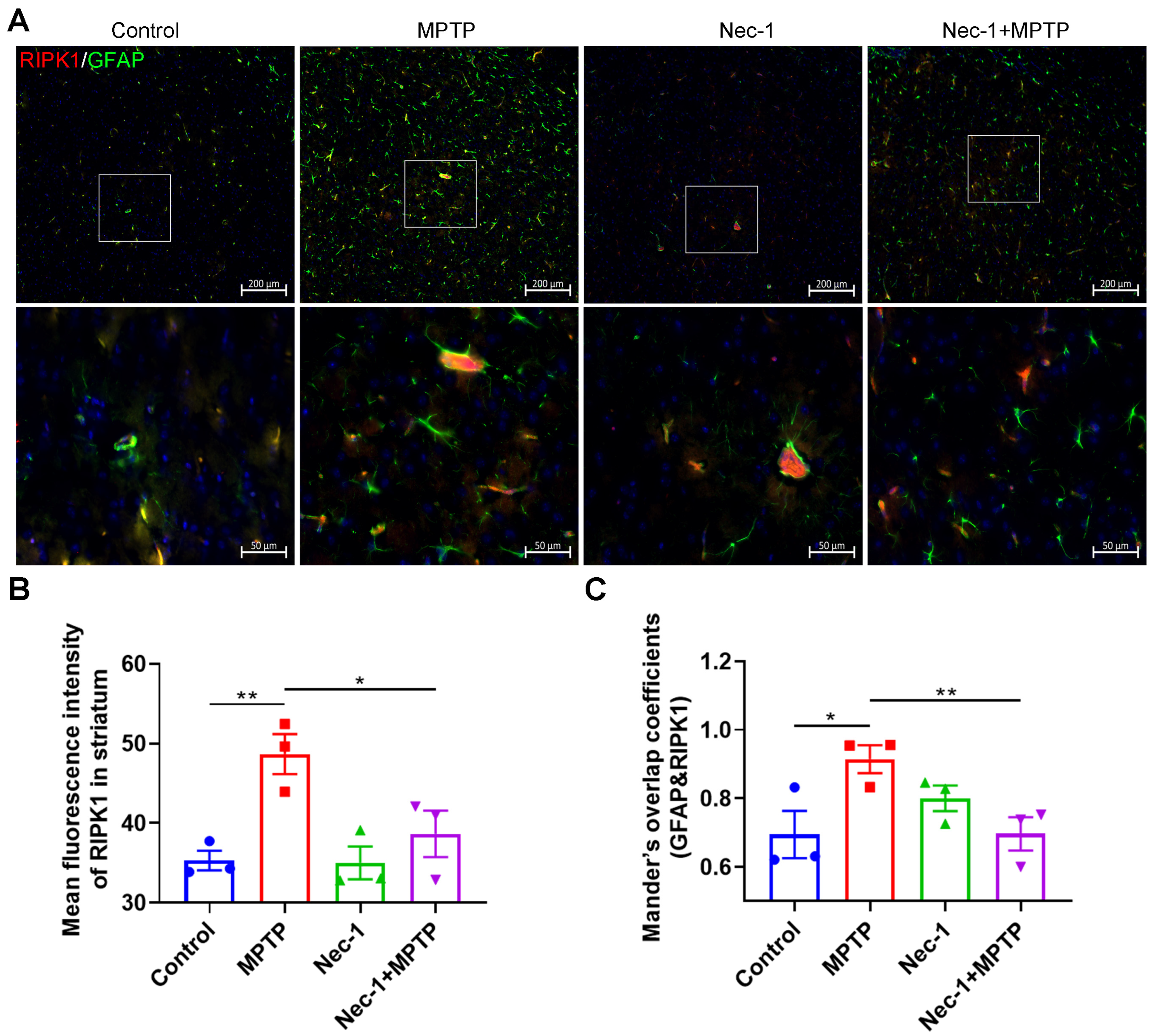
3.4. Inhibition of RIPK1 Reduces Astrocyte Activation in MPTP-Treated Murine Model of PD
3.5. RIPK1 Is a Key Mediator of Disease-Associated Astrocyte Response
3.6. Inhibition of RIPK1 Prevents the Polarization of Astrocytes into Neurotoxic A1 Type
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, A.; Gitler, A.D. Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 2016, 539, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Samii, A.; Nutt, J.G.; Ransom, B.R. Parkinson’s disease. Lancet 2004, 363, 1783–1793. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef]
- Yuan, J.; Amin, P.; Ofengeim, D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 2019, 20, 19–33. [Google Scholar] [CrossRef]
- Ofengeim, D.; Yuan, J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat. Rev. Mol. Cell Biol. 2013, 14, 727–736. [Google Scholar] [CrossRef]
- Rubinsztein, D.C. RIPK1 promotes inflammation and beta-amyloid accumulation in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 10813–10814. [Google Scholar] [CrossRef]
- Ofengeim, D.; Mazzitelli, S.; Ito, Y.; DeWitt, J.P.; Mifflin, L.; Zou, C.; Das, S.; Adiconis, X.; Chen, H.; Zhu, H.; et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E8788–E8797. [Google Scholar] [CrossRef]
- Najjar, M.; Saleh, D.; Zelic, M.; Nogusa, S.; Shah, S.; Tai, A.; Finger, J.N.; Polykratis, A.; Gough, P.J.; Bertin, J.; et al. RIPK1 and RIPK3 Kinases Promote Cell-Death-Independent Inflammation by Toll-like Receptor 4. Immunity 2016, 45, 46–59. [Google Scholar] [CrossRef]
- Zelic, M.; Pontarelli, F.; Woodworth, L.; Zhu, C.; Mahan, A.; Ren, Y.; LaMorte, M.; Gruber, R.; Keane, A.; Loring, P.; et al. RIPK1 activation mediates neuroinflammation and disease progression in multiple sclerosis. Cell Rep. 2021, 35, 109112. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Wang, J.; Zhang, H.; Cao, Y.; Qu, Y.; Huang, S.; Kong, X.; Song, C.; Li, J.; Li, Q.; et al. Inhibition of microglial receptor—Interacting protein kinase 1 ameliorates neuroinflammation following cerebral ischaemic stroke. J. Cell. Mol. Med. 2020, 24, 12585–12598. [Google Scholar] [CrossRef] [PubMed]
- Jackson-Lewis, V.; Przedborski, S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat. Protoc. 2007, 2, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.S.; Chen, P.; Wang, W.X.; Lin, C.C.; Zhou, Y.; Yu, L.H.; Lin, Y.X.; Xu, Y.F.; Kang, D.Z. RIP1/RIP3/MLKL mediates dopaminergic neuron necroptosis in a mouse model of Parkinson disease. Lab. Investig. 2020, 100, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Qiao, C.M.; Sun, M.F.; Jia, X.B.; Li, Y.; Zhang, B.P.; Zhao, L.P.; Shi, Y.; Zhou, Z.L.; Zhu, Y.L.; Cui, C.; et al. Sodium Butyrate Exacerbates Parkinson’s Disease by Aggravating Neuroinflammation and Colonic Inflammation in MPTP-Induced Mice Model. Neurochem. Res. 2020, 45, 2128–2142. [Google Scholar] [CrossRef]
- Cao, L.; Mu, W. Necrostatin-1 and necroptosis inhibition: Pathophysiology and therapeutic implications. Pharmacol. Res. 2021, 163, 105297. [Google Scholar] [CrossRef]
- Shi, Y.; Qiao, C.M.; Zhou, Y.; Wu, J.; Cui, C.; Hong, H.; Jia, X.B.; Huang, S.B.; Yao, L.; Zhao, W.J.; et al. Protective effects of prucalopride in MPTP-induced Parkinson’s disease mice: Neurochemistry, motor function and gut barrier. Biochem. Biophys. Res. Commun. 2021, 556, 16–22. [Google Scholar] [CrossRef]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Saijo, K.; Winner, B.; Carson, C.T.; Collier, J.G.; Boyer, L.; Rosenfeld, M.G.; Gage, F.H.; Glass, C.K. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 2009, 137, 47–59. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef]
- Huang, D.; Xu, J.; Wang, J.; Tong, J.; Bai, X.; Li, H.; Wang, Z.; Huang, Y.; Wu, Y.; Yu, M.; et al. Dynamic Changes in the Nigrostriatal Pathway in the MPTP Mouse Model of Parkinson’s Disease. Parkinsons. Dis. 2017, 2017, 9349487. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, D.; Xu, J.; Tong, J.; Wang, Z.; Huang, L.; Yang, Y.; Bai, X.; Wang, P.; Suo, H.; et al. Tiagabine Protects Dopaminergic Neurons against Neurotoxins by Inhibiting Microglial Activation. Sci. Rep. 2015, 5, 15720. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Chou, T.W.; Chang, N.P.; Krishnagiri, M.; Patel, A.P.; Lindman, M.; Angel, J.P.; Kung, P.L.; Atkins, C.; Daniels, B.P. Fibrillar alpha-synuclein induces neurotoxic astrocyte activation via RIP kinase signaling and NF-kappaB. Cell Death Dis. 2021, 12, 756. [Google Scholar] [CrossRef] [PubMed]
- Murray, H.; Qiu, B.; Ho, S.Y.; Wang, X. Complement Factor B Mediates Ocular Angiogenesis through Regulating the VEGF Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 9580. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Wang, Y.; He, Y.; Wang, T.; Huang, X.H.; Zhao, C.M.; Zhang, L.; Li, S.W.; Wang, C.; Qu, Y.N.; et al. A1 astrocytes contribute to murine depression-like behavior and cognitive dysfunction, which can be alleviated by IL-10 or fluorocitrate treatment. J. Neuroinflam. 2020, 17, 200. [Google Scholar] [CrossRef]
- Veerhuis, R.; Janssen, I.; Hoozemans, J.J.; De Groot, C.J.; Hack, C.E.; Eikelenboom, P. Complement C1-inhibitor expression in Alzheimer’s disease. Acta Neuropathol. 1998, 96, 287–296. [Google Scholar] [CrossRef]
- Perriard, G.; Mathias, A.; Enz, L.; Canales, M.; Schluep, M.; Gentner, M.; Schaeren-Wiemers, N.; Du Pasquier, R.A. Interleukin-22 is increased in multiple sclerosis patients and targets astrocytes. J. Neuroinflamm. 2015, 12, 119. [Google Scholar] [CrossRef]
- Ito, Y.; Ofengeim, D.; Najafov, A.; Das, S.; Saberi, S.; Li, Y.; Hitomi, J.; Zhu, H.; Chen, H.; Mayo, L.; et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 2016, 353, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Re, D.B.; Le Verche, V.; Yu, C.H.; Amoroso, M.W.; Politi, K.A.; Phani, S.; Ikiz, B.; Hoffmann, L.; Koolen, M.; Nagata, T.; et al. Necroptosis Drives Motor Neuron Death in Models of Both Sporadic and Familial ALS. Neuron 2014, 81, 1001–1008. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Y.; Li, D.; Wu, J.; Si, W.; Wu, Y. Necrostatin-1 Attenuates Inflammatory Response and Improves Cognitive Function in Chronic Ischemic Stroke Mice. Medicines 2016, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Iannielli, A.; Bido, S.; Folladori, L.; Segnali, A.; Cancellieri, C.; Maresca, A.; Massimino, L.; Rubio, A.; Morabito, G.; Caporali, L.; et al. Pharmacological Inhibition of Necroptosis Protects from Dopaminergic Neuronal Cell Death in Parkinson’s Disease Models. Cell Rep. 2018, 22, 2066–2079. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Leem, Y.H.; Park, J.S.; Park, J.E.; Park, J.M.; Kang, J.L.; Kim, H.S. RIPK1 Regulates Microglial Activation in Lipopolysaccharide-Induced Neuroinflammation and MPTP-Induced Parkinson’s Disease Mouse Models. Cells 2023, 12, 417. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.B.; Zhang, Y.F.; Wang, H.; Ren, R.J.; Cui, H.L.; Huang, W.Y.; Cheng, Q.; Chen, H.Z.; Wang, G. miR-425 deficiency promotes necroptosis and dopaminergic neurodegeneration in Parkinson’s disease. Cell Death Dis. 2019, 10, 589. [Google Scholar] [CrossRef]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef]
- Brodacki, B.; Staszewski, J.; Toczylowska, B.; Kozlowska, E.; Drela, N.; Chalimoniuk, M.; Stepien, A. Serum interleukin (IL-2, IL-10, IL-6, IL-4), TNFalpha, and INFgamma concentrations are elevated in patients with atypical and idiopathic parkinsonism. Neurosci. Lett. 2008, 441, 158–162. [Google Scholar] [CrossRef]
- Reale, M.; Iarlori, C.; Thomas, A.; Gambi, D.; Perfetti, B.; Di Nicola, M.; Onofrj, M. Peripheral cytokines profile in Parkinson’s disease. Brain Behav. Immun. 2009, 23, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.L.; Jia, X.B.; Sun, M.F.; Zhu, Y.L.; Qiao, C.M.; Zhang, B.P.; Zhao, L.P.; Yang, Q.; Cui, C.; Chen, X.; et al. Neuroprotection of Fasting Mimicking Diet on MPTP-Induced Parkinson’s Disease Mice via Gut Microbiota and Metabolites. Neurotherapeutics 2019, 16, 741–760. [Google Scholar] [CrossRef]
- Simola, N.; Morelli, M.; Carta, A.R. The 6-hydroxydopamine model of Parkinson’s disease. Neurotox Res. 2007, 11, 151–167. [Google Scholar] [CrossRef]
- Cui, C.; Hong, H.; Shi, Y.; Zhou, Y.; Qiao, C.M.; Zhao, W.J.; Zhao, L.P.; Wu, J.; Quan, W.; Niu, G.Y.; et al. Vancomycin Pretreatment on MPTP-Induced Parkinson’s Disease Mice Exerts Neuroprotection by Suppressing Inflammation Both in Brain and Gut. J. Neuroimmune Pharm. 2022. [Google Scholar] [CrossRef]
- Langston, J.W.; Forno, L.S.; Tetrud, J.; Reeves, A.G.; Kaplan, J.A.; Karluk, D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann. Neurol. 1999, 46, 598–605. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Forward and Reverse Sequences |
|---|---|
| GAPDH | Forward: 5′-AGGTCGGTGTGAACGGATTTG-3′ Reverse: 5′-TGTAGACCATGTAGTTGAGGTCA-3′ |
| CFB | Forward: 5′-TGCTATGATGGTTACGTTCTCCG-3′ Reverse: 5′-TCCCAATAGGAATACCGGGATT-3′ |
| H2-T23 | Forward: 5′-AGAGTAACGACGAATCTCACACG-3′ Reverse: 5′-CTTGCAGGTATGCCCTCTGTT-3′ |
| SERPING1 | Forward: 5′-ATCCAAAGGTGTCACTTCTGTG-3′ Reverse: 5′-GCGGATCTTATGGTTGGTGTTC-3′ |
| S100A10 | Forward: 5′-TGGAAACCATGATGCTTACGTT-3′ Reverse: 5′-GAAGCCCACTTTGCCATCTC-3′ |
| EMP1 | Forward: 5′-TGAAGATGCTATCAAGGCAGTG-3′ Reverse: 5′-CTGGAACACGAAGACCACAAG-3′ |
| PTX3 | Forward: 5′-CGCAGGTTGTGAAACAGCAAT-3′ Reverse: 5′-GGGTTCCACTTTGTGCCATAAG-3′ |
| IL-1β | Forward: 5′-GAAATGCCACCTTTTGACAGTG-3′ Reverse: 5′-TGGATGCTCTCATCAGGACAG-3′ |
| TNF-α | Forward: 5′-CCTGTAGCCCACGTCGTAG-3′ Reverse: 5′-GGGAGTAGACAAGGTACAACCC-3′ |
| CCL2 | Forward: 5′-TTAAAAACCTGGATCGGAACCAA-3′ Reverse: 5′-GCATTAGCTTCAGATTTACGGGT-3′ |
| IL-10 | Forward: 5′-GCTGGACAACATACTGCTAACC-3′ Reverse: 5′-ATTTCCGATAAGGCTTGGCAA-3′ |
| IL-22 | Forward: 5′-ATGAGTTTTTCCCTTATGGGGAC-3′ Reverse: 5′-GCTGGAAGTTGGACACCTCAA-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiao, C.; Niu, G.; Zhao, W.; Quan, W.; Zhou, Y.; Zhang, M.; Li, T.; Zhou, S.; Huang, W.; Zhao, L.; et al. RIPK1-Induced A1 Reactive Astrocytes in Brain in MPTP-Treated Murine Model of Parkinson’s Disease. Brain Sci. 2023, 13, 733. https://doi.org/10.3390/brainsci13050733
Qiao C, Niu G, Zhao W, Quan W, Zhou Y, Zhang M, Li T, Zhou S, Huang W, Zhao L, et al. RIPK1-Induced A1 Reactive Astrocytes in Brain in MPTP-Treated Murine Model of Parkinson’s Disease. Brain Sciences. 2023; 13(5):733. https://doi.org/10.3390/brainsci13050733
Chicago/Turabian StyleQiao, Chenmeng, Guyu Niu, Weijiang Zhao, Wei Quan, Yu Zhou, Meixuan Zhang, Ting Li, Shengyang Zhou, Wenyan Huang, Liping Zhao, and et al. 2023. "RIPK1-Induced A1 Reactive Astrocytes in Brain in MPTP-Treated Murine Model of Parkinson’s Disease" Brain Sciences 13, no. 5: 733. https://doi.org/10.3390/brainsci13050733