

Immunological Imbalances Associated with Epileptic Seizures in Type 2 Diabetes Mellitus

{kind=link}
{kind=link}
Abstract
:1. Introduction
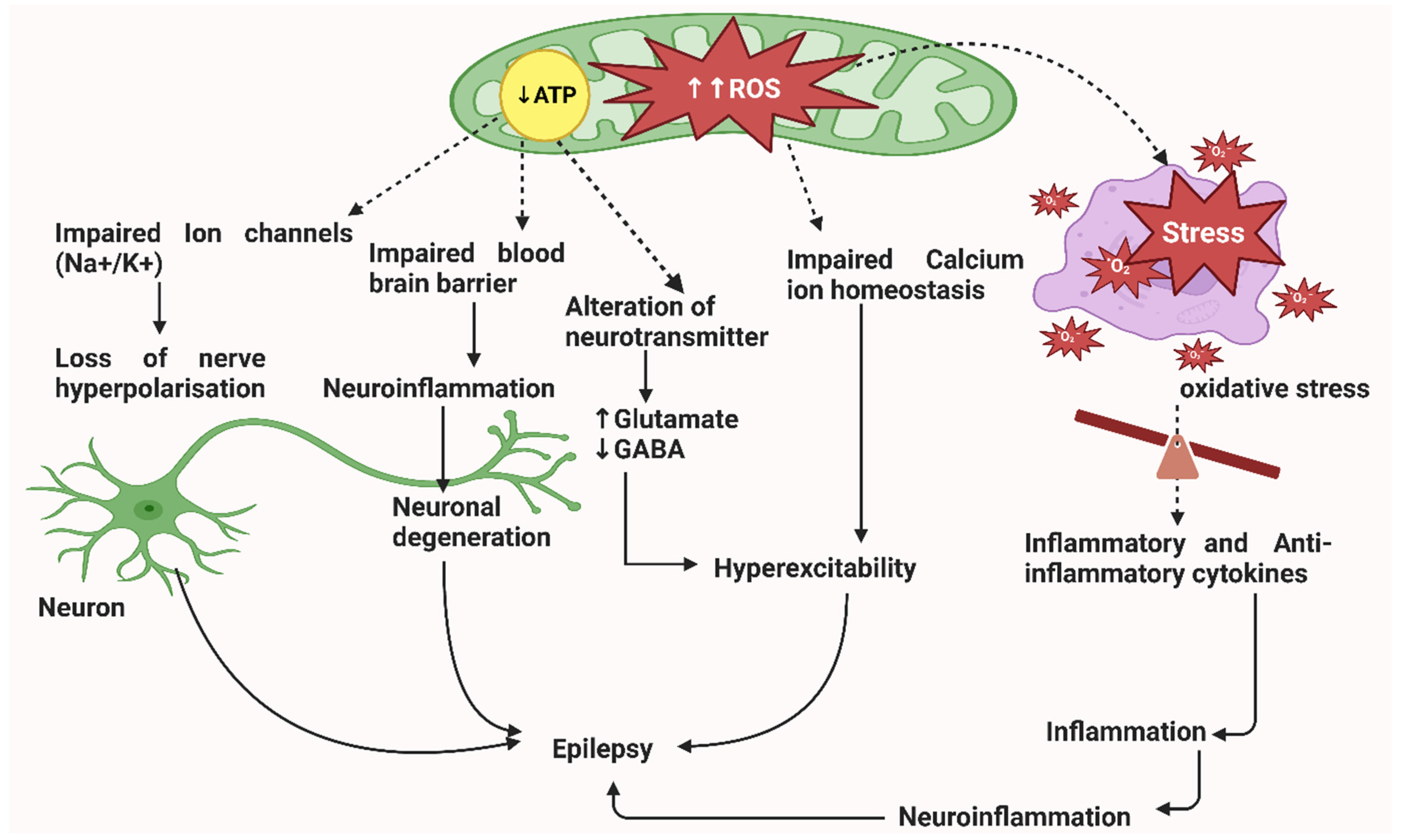
2. Pathophysiology of Epileptic Seizures
3. Immunological Expression in Epilepsy
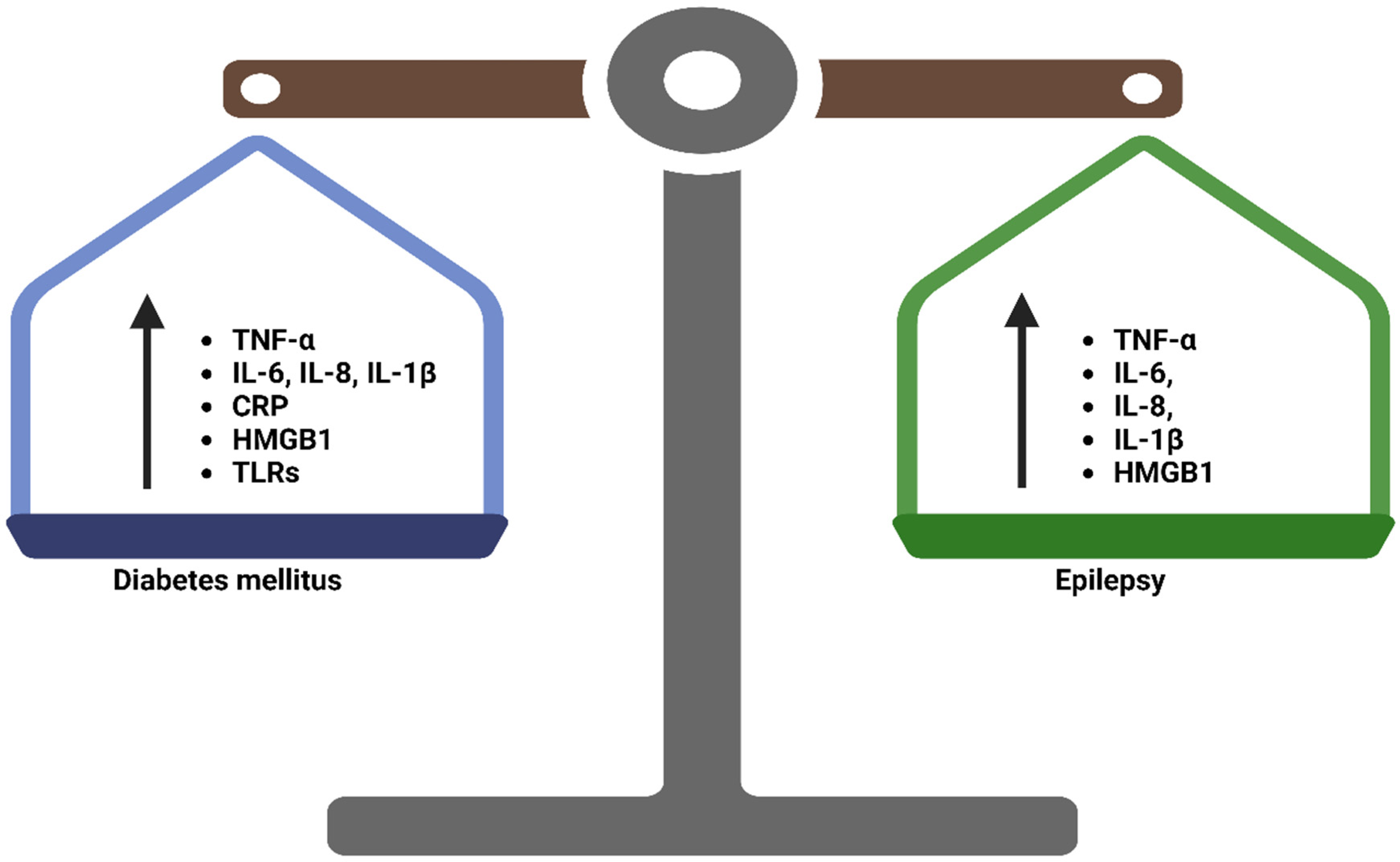
4. Inflammation in Epilepsy and Diabetes Mellitus
4.1. Central Inflammation in Epilepsy
4.2. Inflammation in T2DM
5. Brain Cells as a Primary Site for the Activation of Inflammatory Responses in Epileptic Seizures
5.1. Glial Cells
5.2. Microglia and Astrocytes
6. Peripheral Inflammatory Cytokines in Epilepsy
7. Immunological Factors Commonly Contribute to the Pathogenesis of Diabetes
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vinay, K.; Abbas, A.K.; Fauston, N. Robbins and Cotran Pathologic Basis of Disease: Saunders; Elsevier: Beijing, China, 2005; Volume 8, pp. 208–221. [Google Scholar]
- Association, A.D. 2. Classification and diagnosis of diabetes: Standards of medical care in diabetes—2019. Diabetes Care 2019, 42 (Suppl. S1), S13–S28. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, Y.; Galalain, A.; Yunusa, U. A modern overview on diabetes mellitus: A chronic endocrine disorder. Eur. J. Biol. 2020, 5, 1–14. [Google Scholar] [CrossRef]
- Wu, Y.; Ding, Y.; Tanaka, Y.; Zhang, W. Risk factors contributing to type 2 diabetes and recent advances in the treatment and prevention. Int. J. Med. Sci. 2014, 11, 1185. [Google Scholar] [CrossRef] [PubMed]
- Olaniyi, K.S.; Amusa, O.A.; Ajadi, I.O.; Alabi, B.Y.; Agunbiade, T.B.; Ajadi, M.B. Repression of HDAC5 by acetate restores hypothalamic-pituitary-ovarian function in type 2 diabetes mellitus. Reprod. Toxicol. 2021, 106, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Wild, S.H.; Roglic, G.; Green, A.; Sicree, R.; King, H. Global Prevalence of Diabetes: Estimates for the Year 2000 and Projections for 2030. Diabetes Care 2004, 27, 2569. [Google Scholar] [CrossRef]
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Magliano, D.J. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Magliano, D.; Zimmet, P. The worldwide epidemiology of type 2 diabetes mellitus—Present and future perspectives. Nat. Rev. Endocrinol. 2012, 8, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, T.I.; Metz, S.; Kilpeläinen, T.O. Do gene–environment interactions have implications for the precision prevention of type 2 diabetes? Diabetologia 2022, 65, 1804–1813. [Google Scholar] [CrossRef]
- He, Y.; Lakhani, C.M.; Rasooly, D.; Manrai, A.K.; Tzoulaki, I.; Patel, C.J. Comparisons of polyexposure, polygenic, and clinical risk scores in risk prediction of type 2 diabetes. Diabetes Care 2021, 44, 935–943. [Google Scholar] [CrossRef]
- Yun, C.; Xuefeng, W. Association between seizures and diabetes mellitus: A comprehensive review of literature. Curr. Diabetes Rev. 2013, 9, 350–354. [Google Scholar] [CrossRef]
- Garrahy, A.; Thompson, C.J. Management of central diabetes insipidus. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101385. [Google Scholar] [CrossRef]
- Yamada, Y.; Kitayama, K.; Oyachi, M.; Higuchi, S.; Kawakita, R.; Kanamori, Y.; Yorifuji, T. Nationwide survey of endogenous hyperinsulinemic hypoglycemia in Japan (2017–2018): Congenital hyperinsulinism, insulinoma, non-insulinoma pancreatogenous hypoglycemia syndrome and insulin autoimmune syndrome (Hirata’s disease). J. Diabetes Investig. 2020, 11, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.; Holmes, S. Diabetes in Pediatric ED Patients. Pediatr. Emerg. Med. Rep. 2020, 25. Available online: https://www.reliasmedia.com/articles/145939-diabetes-in-pediatric-ed-patients (accessed on 26 March 2023).
- Youn, Y.; Sung, I.K.; Lee, I.G. The role of cytokines in seizures: Interleukin (IL)-1β, IL-1Ra, IL-8, and IL-10. Korean J. Pediatr. 2013, 56, 271. [Google Scholar] [CrossRef]
- Patel, M. Mitochondrial dysfunction and oxidative stress: Cause and consequence of epileptic seizures. Free Radic. Biol. Med. 2004, 37, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Olowe, R.; Sandouka, S.; Saadi, A.; Shekh-Ahmad, T. Approaches for Reactive Oxygen Species and Oxidative Stress Quantification in Epilepsy. Antioxidants 2020, 9, 990. [Google Scholar] [CrossRef] [PubMed]
- Patel, M. Targeting Oxidative Stress in Central Nervous System Disorders. Trends Pharmacol. Sci. 2016, 37, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Asadi-Pooya, A.A.; Sperling, M.R. Antiepileptic Drugs: A Clinician’s Manual; Oxford University Press: Oxford, UK, 2016. [Google Scholar]
- Wesół-Kucharska, D.; Rokicki, D.; Jezela-Stanek, A. Epilepsy in Mitochondrial Diseases—Current State of Knowledge on Aetiology and Treatment. Children 2021, 8, 532. [Google Scholar] [CrossRef]
- Löscher, W.; Friedman, A. Structural, Molecular, and Functional Alterations of the Blood-Brain Barrier during Epileptogenesis and Epilepsy: A Cause, Consequence, or Both? Int. J. Mol. Sci. 2020, 21, 591. [Google Scholar] [CrossRef]
- Zamay, T.N.; Zamay, G.S.; Shnayder, N.A.; Dmitrenko, D.V.; Zamay, S.S.; Yushchenko, V.; Kolovskaya, O.S.; Susevski, V.; Berezovski, M.V.; Kichkailo, A.S. Nucleic Acid Aptamers for Molecular Therapy of Epilepsy and Blood-Brain Barrier Damages. Mol. Ther. Nucleic Acids 2020, 19, 157–167. [Google Scholar] [CrossRef]
- Nikolic, L.; Nobili, P.; Shen, W.; Audinat, E. Role of astrocyte purinergic signaling in epilepsy. Glia 2020, 68, 1677–1691. [Google Scholar] [CrossRef] [PubMed]
- Mula, M. Emerging drugs for focal epilepsy. Expert Opin. Emerg. Drugs 2013, 18, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Zhang, S.; Ren, L.; Zhang, J.; Zhao, Y.; Mao, X.; Gan, L.; Wang, H.; Ma, C.; Lin, Y.; et al. Electroacupuncture of the trigeminal nerve causes N-methyl-D-aspartate receptors to mediate blood-brain barrier opening and induces neuronal excitatory changes. Front. Cell. Neurosci. 2022, 16, 1020644. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.D.; YW Chang, A.; Chuang, Y.C. The potential role of mitochondrial dysfunction in seizure-associated cell death in the hippocampus and epileptogenesis. J. Bioenerg. Biomembr. 2010, 42, 461–465. [Google Scholar] [CrossRef]
- Vezzani, A.; Fujinami, R.S.; White, H.S.; Preux, P.-M.; Blümcke, I.; Sander, J.W.; Löscher, W. Infections, inflammation and epilepsy. Acta Neuropathol. 2016, 131, 211–234. [Google Scholar] [CrossRef]
- Fabene, P.F.; Mora, G.N.; Martinello, M.; Rossi, B.; Merigo, F.; Ottoboni, L.; Bach, S.; Angiari, S.; Benati, D.; Chakir, A.; et al. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat. Med. 2008, 14, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Zattoni, M.; Mura, M.L.; Deprez, F.; Schwendener, R.A.; Engelhardt, B.; Frei, K.; Fritschy, J.M. Brain infiltration of leukocytes contributes to the pathophysiology of temporal lobe epilepsy. J. Neurosci. 2011, 31, 4037–4050. [Google Scholar] [CrossRef] [PubMed]
- Librizzi, L.; Verde, D.V.; Colciaghi, F.; Deleo, F.; Regondi, M.C.; Costanza, M.; Cipelletti, B.; De Curtis, M. Peripheral blood mononuclear cell activation sustains seizure activity. Epilepsia 2021, 62, 1715–1728. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Adeloye, D.; George-Carey, R.; Kolčić, I.; Grant, L.; Chan, K.Y. An estimate of the prevalence of epilepsy in Sub–Saharan Africa: A systematic analysis. J. Glob. Health 2012, 2, 020405. [Google Scholar] [CrossRef]
- Costa, C.; Vecchio, F.; Romoli, M.; Miraglia, F.; Cesarini, E.N.; Alù, F.; Calabresi, P.; Rossini, P.M. Cognitive Decline Risk Stratification in People with Late-Onset Epilepsy of Unknown Etiology: An Electroencephalographic Connectivity and Graph Theory Pilot Study. J. Alzheimers Dis. 2022, 88, 893–901. [Google Scholar] [CrossRef]
- Aronica, E.; Crino, P.B. Inflammation in epilepsy: Clinical observations. Epilepsia 2011, 52, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef]
- Vezzani, A.; Lang, B.; Aronica, E. Immunity and inflammation in epilepsy. Cold Spring Harb. Perspect. Med. 2015, 6, a022699. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Ravizza, T.; Zurolo, E.; Vezzani, A. Astrocyte immune responses in epilepsy. Glia 2012, 60, 1258–1268. [Google Scholar] [CrossRef]
- Vezzani, A.; Maroso, M.; Balosso, S.; Sanchez, M.-A.; Bartfai, T. IL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav. Immun. 2011, 25, 1281–1289. [Google Scholar] [CrossRef]
- Mukhtar, I. Inflammatory and immune mechanisms underlying epileptogenesis and epilepsy: From pathogenesis to treatment target. Seizure 2020, 82, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Semple, B.D.; Dill, L.K.; O’Brien, T.J. Immune challenges and seizures: How do early life insults influence epileptogenesis? Front. Pharmacol. 2020, 11, 2. [Google Scholar] [CrossRef]
- Vezzani, A.; Granata, T. Brain Inflammation in Epilepsy: Experimental and Clinical Evidence. Epilepsia 2005, 46, 1724–1743. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xie, Y.; Shao, Y.; Chen, Y. LncRNA Snhg5 Attenuates Status Epilepticus Induced Inflammation through Regulating NF-κΒ Signaling Pathway. Biol. Pharm. Bull. 2022, 45, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wang, M.; Deng, X.; Chen, Y. Long non-coding RNA H19 alleviates hippocampal damage in convulsive status epilepticus rats through the nuclear factor-kappaB signaling pathway. Bioengineered 2022, 13, 12783–12793. [Google Scholar] [CrossRef]
- Cai, M.; Lin, W. The Function of NF-Kappa B During Epilepsy, a Potential Therapeutic Target. Front. Neurosci. 2022, 16, 851394. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Chen, Y. Inflammation: A Network in the Pathogenesis of Status Epilepticus. Front. Mol. Neurosci. 2018, 11, 341. [Google Scholar] [CrossRef]
- Rojas, A.; Jiang, J.; Ganesh, T.; Yang, M.-S.; Lelutiu, N.; Gueorguieva, P.; Dingledine, R. Cyclooxygenase-2 in epilepsy. Epilepsia 2014, 55, 17–25. [Google Scholar] [CrossRef]
- Dhir, A. An update of cyclooxygenase (COX)-inhibitors in epilepsy disorders. Expert Opin. Investig. Drugs 2019, 28, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Yuhi, T.; Tsuji, S.; Yamashita, U. Cyclooxygenase-2 expression in the hippocampus of genetically epilepsy susceptible El mice was increased after seizure. Brain Res. 2001, 894, 332–335. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo-Pereira, M.E.; Rockwell, P.; Schmidt-Glenewinkel, T.; Serrano, P. Neuroinflammation and J2 prostaglandins: Linking impairment of the ubiquitin-proteasome pathway and mitochondria to neurodegeneration. Front. Mol. Neurosci. 2015, 7, 104. [Google Scholar] [CrossRef] [PubMed]
- Rawat, C.; Kukal, S.; Dahiya, U.R.; Kukreti, R. Cyclooxygenase-2 (COX-2) inhibitors: Future therapeutic strategies for epilepsy management. J. Neuroinflammation 2019, 16, 197. [Google Scholar] [CrossRef] [PubMed]
- Mesdaghinia, A.; Khazaee, P.; Heydari, A. The effect of cyclooxygenase-2 inhibition on pentylenetetrazole-induced seizure threshold in mice. KAUMS J. (FEYZ) 2018, 22, 258–266. [Google Scholar]
- Mokgalaboni, K.; Dludla, P.V.; Nyambuya, T.M.; Yakobi, S.H.; Mxinwa, V.; Nkambule, B.B. Monocyte-mediated inflammation and cardiovascular risk factors in type 2 diabetes mellitus: A systematic review and meta-analysis of pre-clinical and clinical studies. JRSM Cardiovasc. Dis. 2020, 9, 2048004019900748. [Google Scholar] [CrossRef]
- Araújo, L.S.; da Silva, M.V.; da Silva, C.A.; Borges, M.D.F.; Palhares, H.M.D.C.; Rocha, L.P.; Machado, J.R. Analysis of serum inflammatory mediators in type 2 diabetic patients and their influence on renal function. PLoS ONE 2020, 15, e0229765. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; Najjar, S.; De Lanerolle, N.C.; Rogawski, M.A. Glia and epilepsy: Excitability and inflammation. Trends Neurosci. 2013, 36, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, J.; Xin, C.; Xiao, J.; Liang, J.; Wu, X. Inflammatory response in epilepsy is mediated by glial cell gap junction pathway (Review). Mol. Med. Rep. 2021, 24, 493. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Liu, Y.; Guo, C.; Du, T.; Jiang, Y.; Wang, K.; Shao, X.; Meng, F.; Zhang, J. The lncRNA H19 binding to let-7b promotes hippocampal glial cell activation and epileptic seizures by targeting Stat3 in a rat model of temporal lobe epilepsy. Cell Prolif. 2020, 53, e12856. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shi, D.; Wang, L.; Wu, G. Chronic neuroinflammation regulates cAMP response element-binding protein in the formation of drug-resistant epilepsy by activating glial cells. J. Neurorestoratology 2022, 10, 100006. [Google Scholar] [CrossRef]
- Vezzani, A.; Aronica, E.; Mazarati, A.; Pittman, Q. Epilepsy and brain inflammation. Exp. Neurol. 2011, 244, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Raoof, R.M.; Al-Hamdany, M.; Noel, K.I. Role of High Mobility Group Box-1 in Status Epilepticus, From Pathophysiology to Biomarker and Therapeutic Potential. Ann. Coll. Med. Mosul 2022, 44, 37–41. [Google Scholar] [CrossRef]
- Gao, H.M.; Zhou, H.; Zhang, F.; Wilson, B.C.; Kam, W.; Hong, J.S. HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J. Neurosci. 2011, 31, 1081–1092. [Google Scholar] [CrossRef]
- Galic, M.A.; Riazi, K.; Pittman, Q.J. Cytokines and brain excitability. Front. Neuroendocr. 2012, 33, 116–125. [Google Scholar] [CrossRef]
- Pineda, E.; Shin, D.; You, S.J.; Auvin, S.; Sankar, R.; Mazarati, A. Maternal immune activation promotes hippocampal kindling epileptogenesis in mice. Ann. Neurol. 2013, 74, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Hiragi, T.; Ikegaya, Y.; Koyama, R. Microglia after Seizures and in Epilepsy. Cells 2018, 7, 26. [Google Scholar] [CrossRef]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.-T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [PubMed]
- Andoh, M.; Ikegaya, Y.; Koyama, R. Synaptic Pruning by Microglia in Epilepsy. J. Clin. Med. 2019, 8, 2170. [Google Scholar] [CrossRef]
- Benson, M.J.; Manzanero, S.; Borges, K. Complex alterations in microglial M1/M2 markers during the development of epilepsy in two mouse models. Epilepsia 2015, 56, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.K.; Miller, S.D. Microglia Initiate Central Nervous System Innate and Adaptive Immune Responses through Multiple TLRs. J. Immunol. 2004, 173, 3916–3924. [Google Scholar] [CrossRef]
- Kolek, M.J.; Carlquist, J.F.; Muhlestein, J.B.; Whiting, B.M.; Horne, B.D.; Bair, T.L.; Anderson, J.L. Toll–like receptor 4 gene Asp299Gly polymorphism is associated with reductions in vascular inflammation, angiographic coronary artery disease, and clinical diabetes. Am. Heart J. 2004, 148, 1034–1040. [Google Scholar] [CrossRef]
- Kim, J.J.; Sears, D.D. TLR4 and insulin resistance. Gastroenterol. Res. Pract. 2010, 2010, 212563. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Taha, I.M.; Allah, A.M.A.; El Gayed, E.M.A. Expression of toll-like receptor 4 and its connection with type 2 diabetes mellitus. Cell. Mol. Biol. 2018, 64, 15–20. [Google Scholar] [CrossRef]
- Fiebich, B.L.; Batista, C.R.A.; Saliba, S.W.; Yousif, N.M.; De Oliveira, A.C.P. Role of Microglia TLRs in Neurodegeneration. Front. Cell. Neurosci. 2018, 12, 329. [Google Scholar] [CrossRef]
- Chen, Y.G. Research progress in the pathogenesis of Alzheimer’s disease. Chin. Med. J. 2018, 131, 1618–1624. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Seo, H.I.; Cha, H.Y.; Yang, Y.J.; Kwon, S.H.; Yang, S.J. Diabetes and Alzheimer’s disease: Mechanisms and nutritional aspects. Clin. Nutr. Res. 2018, 7, 229–240. [Google Scholar] [CrossRef]
- Huang, N.-Q.; Jin, H.; Zhou, S.-Y.; Shi, J.-S.; Jin, F. TLR4 is a link between diabetes and Alzheimer’s disease. Behav. Brain Res. 2017, 316, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Xue, G.; Hölscher, C. The role of the TNFα-mediated astrocyte signaling pathway in epilepsy. Acta Epileptol. 2021, 3, 24. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, L.; Teng, J.; Miao, W. HMGB1 mediates microglia activation via the TLR4/NF-κB pathway in coriaria lactone induced epilepsy. Mol. Med. Rep. 2018, 17, 5125–5131. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Chen, J.; Guo, H.; Ding, L.; Zhang, Y.; Xu, Y. High Mobility Group Protein B1 (HMGB1) and Interleukin-1β as Prognostic Biomarkers of Epilepsy in Children. J. Child Neurol. 2018, 33, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Min, H.J.; Shin, J.-S. Increased levels of HMGB1 and pro-inflammatory cytokines in children with febrile seizures. J. Neuroinflammation 2011, 8, 135. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB1. Front. Immunol. 2020, 11, 484. [Google Scholar] [CrossRef] [PubMed]
- Zabielski, P.; Chacinska, M.; Charkiewicz, K.; Baranowski, M.; Gorski, J.; Blachnio-Zabielska, A.U. Effect of metformin on bioactive lipid metabolism in insulin-resistant muscle. J. Endocrinol. 2017, 233, 329–340. [Google Scholar] [CrossRef]
- Chmielewska, N.; Maciejak, P.; Osuch, B.; Kursa, M.B.; Szyndler, J. Pro-inflammatory cytokines, but not brain- and extracellular matrix-derived proteins, are increased in the plasma following electrically induced kindling of seizures. Pharmacol. Rep. 2021, 73, 506–515. [Google Scholar] [CrossRef]
- Huang, W.-S.; Zhu, L. MiR-134 expression and changes in inflammatory cytokines of rats with epileptic seizures. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3479–3484. [Google Scholar]
- Shin, H.-R.; Chu, K.; Lee, W.-J.; Lee, H.S.; Kim, E.Y.; Son, H.; Moon, J.; Kim, N.; Jung, K.-Y.; Jung, K.-H.; et al. Neuropsychiatric symptoms and seizure related with serum cytokine in epilepsy patients. Sci. Rep. 2022, 12, 7138. [Google Scholar] [CrossRef]
- Basnyat, P.; Peltola, M.; Raitanen, J.; Liimatainen, S.; Rainesalo, S.; Pesu, M.; Peltola, J. Elevated IL-6 plasma levels are associated with GAD antibodies-associated autoimmune epilepsy. Front. Cell. Neurosci. 2023, 17, 1129907. [Google Scholar] [CrossRef]
- Wang, N.; Liu, H.; Ma, B.; Zhao, T.; Chen, Y.; Yang, Y.; Zhao, P.; Han, X. CSF high-mobility group box 1 is associated with drug-resistance and symptomatic etiology in adult patients with epilepsy. Epilepsy Res. 2021, 177, 106767. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Sepehri, Z.; Kiani, Z.; Nasiri, A.A.; Kohan, F. Toll-like receptor 2 and type 2 diabetes. Cell. Mol. Biol. Lett. 2016, 21, 2. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Muñoz-Planillo, R.; Núñez, G. Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 2012, 13, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Menu, P.; Vince, J.E. The NLRP3 inflammasome in health and disease: The good, the bad and the ugly. Clin. Exp. Immunol. 2011, 166, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Boyden, E.D.; Dietrich, W.F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 2006, 38, 240–244. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, J.; Shi, J.; Gong, Y.-N.; Lu, Q.; Xu, H.; Liu, L.; Shao, F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 2011, 477, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Yu, J.W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Pressman, B.C. Biological Applications of Ionophores. Annu. Rev. Biochem. 1976, 45, 501–530. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Puerto, A.D.; Wandosell, F.; Garrido, J.J. Neuronal and glial purinergic receptors functions in neuron development and brain disease. Front. Cell. Neurosci. 2013, 7, 197. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Pankratov, Y.; Lalo, U.; Nedergaard, M. P2X receptors in neuroglia. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2012, 1, 151–161. [Google Scholar] [CrossRef]
- Avignone, E.; Ulmann, L.; Levavasseur, F.; Rassendren, F.; Audinat, E. Status Epilepticus Induces a Particular Microglial Activation State Characterized by Enhanced Purinergic Signaling. J. Neurosci. 2008, 28, 9133–9144. [Google Scholar] [CrossRef]
- Rappold, P.; Lynd-Balta, E.; Joseph, S. P2X7 receptor immunoreactive profile confined to resting and activated microglia in the epileptic brain. Brain Res. 2006, 1089, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Pacheco, A.; Diaz-Hernandez, M.; Arribas-Blázquez, M.; Sanz-Rodriguez, A.; Olivos-Oré, L.A.; Artalejo, A.R.; Henshall, D.C. Transient P2X7 receptor antagonism produces lasting reductions in spontaneous seizures and gliosis in experimental temporal lobe epilepsy. J. Neurosci. 2016, 36, 5920–5932. [Google Scholar] [CrossRef]
- Kim, J.-E.; Kang, T.-C. The P2X7 receptor–pannexin-1 complex decreases muscarinic acetylcholine receptor–mediated seizure susceptibility in mice. J. Clin. Investig. 2011, 121, 2037–2047. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhang, Z.; Li, Y. Relevance of the Pyroptosis-Related Inflammasome Pathway in the Pathogenesis of Diabetic Kidney Disease. Front. Immunol. 2021, 12, 603416. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Gou, X.; Zhang, L.; Gao, H.; Wei, Y.; Yu, X.; Pang, B.; Tian, J.; Tong, X.; Li, M. Interactions Between Gut Microbiota, Host, and Herbal Medicines: A Review of New Insights Into the Pathogenesis and Treatment of Type 2 Diabetes. Front. Cell. Infect. Microbiol. 2020, 10, 360. [Google Scholar] [CrossRef]
- Koushki, K.; Shahbaz, S.K.; Mashayekhi, K.; Sadeghi, M.; Zayeri, Z.D.; Taba, M.Y.; Sahebkar, A. Anti-inflammatory action of statins in cardiovascular disease: The role of inflammasome and toll-like receptor pathways. Clin. Rev. Allergy Immunol. 2021, 60, 175–199. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, H.; Su, Y.; Zhang, B. Application and prospect of targeting innate immune sensors in the treatment of autoimmune diseases. Cell Biosci. 2022, 12, 68. [Google Scholar] [CrossRef]
- Dasu, M.R.; Ramirez, S.; Isseroff, R.R. Toll-like receptors and diabetes: A therapeutic perspective. Clin. Sci. 2012, 122, 203–214. [Google Scholar] [CrossRef]
- Liu, T.; Zheng, W.; Wang, L.; Wang, L.; Zhang, Y. TLR4/NF-κB signaling pathway participates in the protective effects of apocynin on g0estational diabetes mellitus induced placental oxidative stress and inflammation. Reprod. Sci. 2020, 27, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Qu, H.; Deng, H. Plasma HMGB-1 Levels in Subjects with Obesity and Type 2 Diabetes: A Cross-Sectional Study in China. PLoS ONE 2015, 10, e0136564. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Lee, D.H.; Song, J. HMGB1 signaling pathway in diabetes-related dementia: Blood-brain barrier breakdown, brain insulin resistance, and Aβ accumulation. Biomed. Pharmacother. 2022, 150, 112933. [Google Scholar] [CrossRef] [PubMed]
- Dokken, B.B. The Pathophysiology of Cardiovascular Disease and Diabetes: Beyond Blood Pressure and Lipids. Diabetes Spectr. 2008, 21, 160–165. [Google Scholar] [CrossRef]
- Banks, W.A.; Rhea, E.M. The Blood–Brain Barrier, Oxidative Stress, and Insulin Resistance. Antioxidants 2021, 10, 1695. [Google Scholar] [CrossRef]
- Choi, S.E.; Roy, B.; Freeby, M.; Mullur, R.; Woo, M.A.; Kumar, R. Prefrontal cortex brain damage and glycemic control in patients with type 2 diabetes. J. Diabetes 2020, 12, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Mendes, N.F.; Pansani, A.P.; Carmanhães, E.R.F.; Tange, P.; Meireles, J.V.; Ochikubo, M.; Le Sueur-Maluf, L. The blood-brain barrier breakdown during acute phase of the pilocarpine model of epilepsy is dynamic and time-dependent. Front. Neurol. 2019, 10, 382. [Google Scholar] [CrossRef]
- Van Vliet, E.A.; Aronica, E.; Gorter, J.A. Blood–Brain Barrier Dysfunction, Seizures and Epilepsy. In Seminars in Cell Developmental Biology; Academic Press: Cambridge, MA, USA, 2015; Volume 38, pp. 26–34. [Google Scholar]
- Löscher, W. Epilepsy and Alterations of the Blood–Brain Barrier: Cause or Consequence of Epileptic Seizures or Both? In Physiology, Pharmacology and Pathology of the Blood-Brain Barrier; Cader, Z., Neuhaus, W., Eds.; Springer International Publishing: Berlin/Heidelberg, Germany; Cham, Switzerland, 2022; pp. 331–350. [Google Scholar]
- Chen, T.-S.; Huang, T.-H.; Lai, M.-C.; Huang, C.-W. The Role of Glutamate Receptors in Epilepsy. Biomedicines 2023, 11, 783. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.D.; Manson, J.E.; Rifai, N.; Buring, J.E.; Ridker, P.M. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 2001, 286, 327–334. [Google Scholar] [CrossRef]
- Laishram, V.; Debnath, S.; Hijam, D.; Das, T.; Saha, S. Evaluation of serum interleukin 6 in type II diabetes mellitus. Int. J. Med. Res. Prof. 2017, 3, 156–160. [Google Scholar]
- Thamer, M.M.; Wong, C.F.; Ghali, Z.H. Estimation of Interleukin-4 (IL-4) and Interleukin-6 (IL-6) Levels in Sera From Patients with Type 2 Diabetes Mellitus. Indian J. Forensic Med. Toxicol. 2021, 15, 1732–1739. [Google Scholar]
- Chanda, D.; Ray, S.; Chakraborti, D.; Sen, S.; Mitra, A. Interleukin-6 Levels in Patients With Diabetic Polyneuropathy. Cureus 2022, 14, 21952. [Google Scholar] [CrossRef] [PubMed]
- Phosat, C.; Panprathip, P.; Chumpathat, N.; Prangthip, P.; Chantratita, N.; Soonthornworasiri, N.; Puduang, S.; Kwanbunjan, K. Elevated C-reactive protein, interleukin 6, tumor necrosis factor alpha and glycemic load associated with type 2 diabetes mellitus in rural Thais: A cross-sectional study. BMC Endocr. Disord. 2017, 17, 44. [Google Scholar] [CrossRef] [PubMed]
- Bosek, I.; Kuczerowski, R.; Miłek, T.; Rabijewski, M.; Kaleta, B.; Kniotek, M.; Piątkiewicz, P. The levels of interleukin-2 and interleukin-10 in patients with type 2 diabetes and colon cancer. Clin. Diabetol. 2018, 7, 114–121. [Google Scholar] [CrossRef]
- Suri, S.; Mitra, P.; Abhilasha, A.; Saxena, I.; Garg, M.K.; Bohra, G.K.; Sharma, P. Role of interleukin-2 and interleukin-18 in newly diagnosed type 2 diabetes mellitus. J. Basic Clin. Physiol. Pharmacol. 2021, 33, 185–190. [Google Scholar] [CrossRef] [PubMed]
- AlZamil, H. Elevated Serum TNF-α Is Related to Obesity in Type 2 Diabetes Mellitus and Is Associated with Glycemic Control and Insulin Resistance. J. Obes. 2020, 2020, 5076858. [Google Scholar] [CrossRef] [PubMed]
- Swaroop, J.J.; Naidu, J.; Rajarajeswari, D. Association of TNF-α with insulin resistance in type 2 diabetes mellitus. Indian J. Med. Res. 2012, 135, 127–130. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phoswa, W.N.; Mokgalaboni, K. Immunological Imbalances Associated with Epileptic Seizures in Type 2 Diabetes Mellitus. Brain Sci. 2023, 13, 732. https://doi.org/10.3390/brainsci13050732
Phoswa WN, Mokgalaboni K. Immunological Imbalances Associated with Epileptic Seizures in Type 2 Diabetes Mellitus. Brain Sciences. 2023; 13(5):732. https://doi.org/10.3390/brainsci13050732
Chicago/Turabian StylePhoswa, Wendy N., and Kabelo Mokgalaboni. 2023. "Immunological Imbalances Associated with Epileptic Seizures in Type 2 Diabetes Mellitus" Brain Sciences 13, no. 5: 732. https://doi.org/10.3390/brainsci13050732