1. Introduction
At least 40 known
SPTAN1 mutations with confirmed Mendelian inheritance patterns are described to date [
1]. These mutations are linked to a spectrum of mild to severe phenotypes involving either central or peripheral neuropathology or both [
2]. In their most severe form, epileptic encephalopathies (West syndrome) with developmental delay, accompanied by cerebellar atrophy, hypomyelination, and microcephaly, often lead to early infantile death [
2]. Other phenotypes related to
SPTAN1 mutations include cerebellar ataxias, hereditary-motor and sensory-motor neuropathies, and spastic paraplegia [
3,
4]. Several molecular mechanisms were proposed to account for the large phenotypic spectrum of
SPTAN1 mutations. The nonsense-mediated decay of mRNA encoding mutated
SPTAN1 allele causes a decrease in the total expression level of α-II, believed to contribute to disease etiology [
3]. Another mechanism, recognized in numerous dominant-negative
SPTAN1 mutations prevents the proper heterodimerization of α-II with beta spectrins and promotes the accumulation of cytotoxic α-II intracellular aggregates [
5]. Another group of recently identified de novo missense
SPTAN1 mutations involve positively charged amino acids located at positions, interconnecting the three-helix bundle of spectrin repeats [
1]. This set of mutations shares an otherwise unrecognized feature of affecting spectrins’ scaffold bending and flexibility.
Strong evolutionary conservation of spectrins’ primary amino-acid sequences makes mice relevant animal models of inherited human spectrinopathies [
6,
7]. Indeed, almost all studied loss-of-function variants of murine homologs of
SPTBN1,
SPTBN2,
SPTBN3, and
SPTAN1 present phenotypes similar to the clinical manifestations in affected patients [
8]. For instance, although homozygous
SPTBN1 mutant mice die in the midterm of gestation, the heterozygous neuron-restricted
SPTBN1 mutation in mice presents a number of characteristics observed in patients carrying heterozygous, dominant-negative
SPTBN1 variants, including impaired motor abilities, social and learning deficits (i.e., hyperactivity attention-deficit/hyperactivity disorder and autism spectrum disorder), facial dysmorphisms, arrested growth, and smaller weight [
9]. The homozygous
SPTBN2 deficiency in mice does not cause premature embryonic death of animals, but it appears in a form of progressive gait abnormalities, tremors, Purkinje cell loss, and cerebellar atrophy (molecular layer thinning), recapitulating features of spinocerebellar ataxia type 5 in humans [
10]. No ataxic phenotype, however, is observed in heterozygous mutant mice, arguing against haploinsufficiency as a disease mechanism. The “quivering” mice carrying recessively inherited loss-of-function mutations of murine
SPTBN3 homolog present auditory and motor neuropathies, similar to symptoms observed in
SPTBN3 homozygous or compound heterozygous patients suffering from congenital muscular hypotonia, neuropathy, and deafness [
11,
12]. General or organ-restricted gene ablation of
SPTAN1 ortholog in rodents uncovered its essential roles in the development and function of neural and cardiac systems and cochlear hair cell function [
6,
7,
8,
13].
The homozygous loss-of-function mutation of murine
Spna2 (
SPTAN1) is lethal during embryonic development due to retarded intrauterine growth and craniofacial, neural tube, and cardiac anomalies [
14]. In contrast to heterozygous
SPTBN1 mutant mice, the
SPTAN1 heterozygotes present no obvious phenotype, precluding their use as models for human pathogenic
SPTAN1 variants, whose phenotypic effects appear after birth. In this context, the identification of a spontaneous dominant-negative missense
SPTAN1 R1098Q variant in C57Bl/6J mice offers an experimental model of
SPTAN1 spectrinopathy unfolding during postnatal life [
15].
Here, we aimed at further extending our initial observations on motor incoordination of R1098Q mice, and additionally assess their muscle strength, memory performance, and seizure episodes at different ages. Our analyses uncovered a number of phenotypic features of R1098Q mice that can be directly compared to clinical presentations in patients carrying pathogenic SPTAN1 variants. Thus, R1098Q mice constitute a unique pre-clinical model for studying phenotypic and molecular aspects of dominant-types missense α-II mutations affecting spectrin’s repeat stability.
2. Materials and Methods
2.1. Mice
Animal experimentation whose severity qualified it as a procedure according to European Union directive on the protection of animals used for scientific purposes (2010/63/EU) was approved by the Local Ethical Committee in Wroclaw (Poland) under permission numbers 78/2018 and 70/2021. The Spna2R1098Q mice (R1098Q) were backcrossed for at least ten generations to C57Bl6/J (WT). The mice were divided into three age groups: young (8 wks old), adult (24 weeks old), and old (54 weeks old). All efforts were made to minimize the number of animals used for testing, and their distress, and suffering. Mice were housed in individually ventilated cages (Techniplast, Buguggiate (VA), Italy) (2–5 mice/cage) in air-conditioned rooms (temperature 22 ± 2 °C), with a controlled humidity level (55 ± 10%) and automatic regulation of the day/night cycle (12/12 h). Constant access to filtered and sterilized water and a standard sterilized rodent chow (Ssniff Spezialdiäten GmbH, Soest, Germany) was provided. Elements of environmental enrichment (wooden tunnels for abrasion of incisors, plastic domes, and materials to build lair and cardboard tubes to play) were provided in every cage. In 5–7 days before the planned experiments, the mice were tamed by handling. Handling activities were aimed at getting the animals used to picking up and taking into hands, and in the same sessions, the animals were getting used to the devices on which the experiments were conducted, as well as with the room where the tests were performed.
2.2. RotaRod
An in-house rotarod apparatus (designed and manufactured by Eng Robert Budziński) fitted with an automatic timer and falling sensor was used. Three constant rotational speeds were chosen for this study (4, 8, and 14 rpm). The cutoff time for a single trial was set to 120 s. Mice were given two practice trials, 48 and 24 h before the first session to habituate them to the test conditions. Then, each mouse had three trials per session, four sessions in total, with one session per day on four consecutive days. At least 3 min intervals between the trials were applied. The results of trials were averaged for analysis. If a mouse stopped walking (rotated passively) before reaching 120 s, it was counted as if it fell off the rod.
2.3. Parallel Rod Floor Test
The parallel rod floor test assesses unforced sensorimotor and balance deficits. The apparatus used for this test was constructed in-house (by Eng. Robert Budziński), according to published guidelines [
16]. The 150 mm × 150 mm arena surrounded by 200 mm high Plexiglass walls was made with parallel metal rods (1,2 mm diameter) with inter rod distance of 0.75 cm. The apparatus was fitted with an automatic misstep counter, an infrared movement detector, and electronic timer. Mice were individually placed at the center of the grid and allowed to freely walk a total distance of 10 m. A number of missteps and time needed to travel this distance was recorded. The test was performed on 4 consecutive days, one trial per day.
2.4. Footprint Test
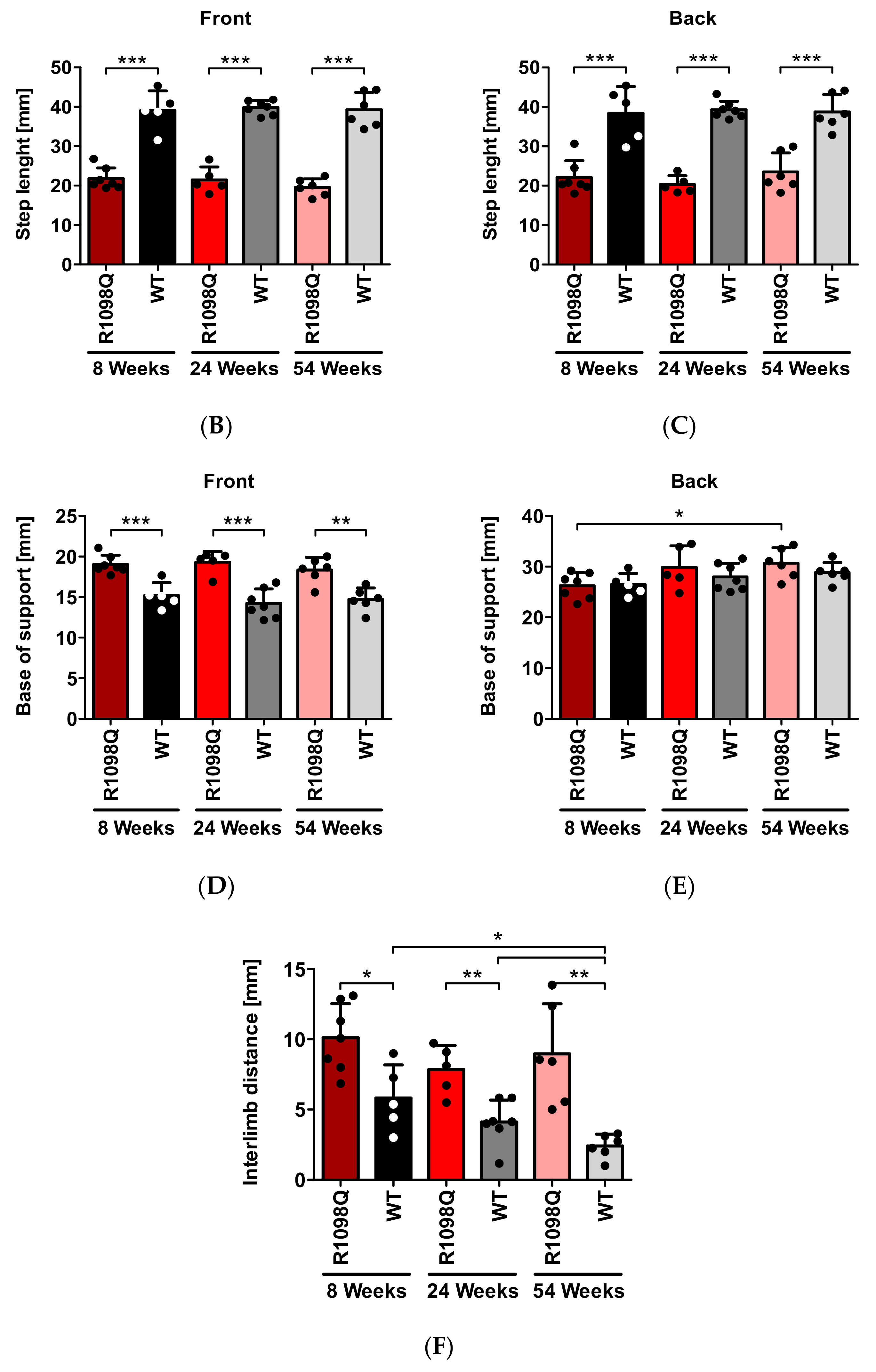
This test allows for a detailed analysis of motor coordination and gait during normal walking. Mice had their front and hind paws painted with non-toxic dyes (Winsor & Newton, London, UK) in green and black, respectively. The mice were then allowed to walk along a 300 × 100 mm runway into an enclosed box leaving their footprints on a sheet of Whatman filter paper (Whatman Ltd., Midestone, UK). The distances between the specific traces were measured (average values of at least 5 steps per mouse) to determine the following parameters: the stride length (the distance between successive contacts of the same paw), the step length (the distance between successive contacts of contralateral front or hind paws along the axis of the direction of motion), the base of support (BOS—a distance between successive contralateral front or hind paws perpendicular to the axis of direction of motion), and the interlimb distance (a distance between the ipsilateral front and hind paws).
2.5. Wire Hang Test
This test is best suited for measuring muscle coordination and endurance in rodents. The device used for this test consisted of a 2-mm thick, non-stretchable and non-vibrating, 350 mm long multistranded metallic wire (Inoxa Sp. z o.o., Jaworzno, Poland) stretched between two vertical stands of 300 mm high. On the bottom of the device, there was a layer of shock-absorbing bedding material to prevent injury of an animal when it fell. The mouse was hung at the center of the wire, and the time until it fell or until the test cutoff time (180 s) passed was measured.
2.6. Novel Object Recognition Test (NOR)
The NOR test was used for evaluating cognitive functions, in particular, recognition memory in mice. Each mouse was habituated to an empty arena (500 × 300 × 500 mm) one day before the test. At the day of the test (training session) each mouse was exposed to two identical objects placed at the center of arena, 150 mm apart, for 10 min. On the next day (test day) the mice were allowed to explore one unchanged and one new object for 10 min. The sessions were recorded using a camera (Xiaomi, Beijing, China) mounted above the arena. After each session, the arena and the objects were cleaned with distilled water and 70% ethanol to remove any residual scent. Exploration was defined as time spent pointing the head toward an object at a distance of 15 mm from the object. Climbing and sitting on the objects were not considered exploratory behaviors.
2.7. Behavioral Assessment of Seizure
The duration of handling-induced seizures was recorded with a digital timer after briefly turning the animal’s head down. The severity of rotarod-induced seizure episodes in R1098Q mice was assessed using an adapted Racine behavioral scale [
17]. Seizure episodes were recorded with a digital camera (Xiaomi, Beijing, China) and analyzed for the presence of characteristic behavioral features. The prevalence and duration of observed behavioral features were shown in
Supplementary Videos S1–S10 and summarized in
Supplementary Table S1.
2.8. Statistical Analysis and Data Presentation
Data are presented as means ±SD. The Shapiro–Wilk test was performed to assess data normality. On this basis, either Student’s t-test, for pairwise comparisons, or one-way ANOVA for multiple comparisons of normally distributed values was used followed by Bonferroni post hoc test. For non-normally distributed values either the Mann–Whitney or the Kruskal–Wallis ANOVA test followed by Dunn’s post hoc test for multiple non-parametric comparisons was used. GraphPad Prism software v7 (Dotmatics, Boston, MA, USA) was used for the graphical representation of data.
4. Discussion
In this study, we further extended our initial observations regarding the phenotype of R1098Q mice to draw parallels to reported clinical presentations of pathogenic missense
SPTAN1 and
SPTBN variants in human patients. It is worth noting that although
SPTAN1 variants affecting intrahelical stabilizing interactions within spectrin repeats share a similar proposed pathogenic mechanism, they present a complex spectrum of spastic and ataxia phenotypes. Van de Vondel et al. described a patient bearing the pArg1624Cys variant who presented early-onset cerebellar ataxia, dystonic head movements, and distal muscular atrophy without intellectual disability while other patient carrying pArg1098Gln variant displayed cerebellar ataxia with severe intellectual disability and seizures [
1]. Even the same heterozygous
SPTAN1 pAsp1616Asn mutation in three members of the same family, reported by Terrone et al. have a wide spectrum of epilepsy manifestations ranging from benign non-recurrent seizures during mild acute gastroenteritis to early onset generalized epilepsy [
19]. Therefore, at present, it seems impossible to a priori predict the severity of phenotype caused by a
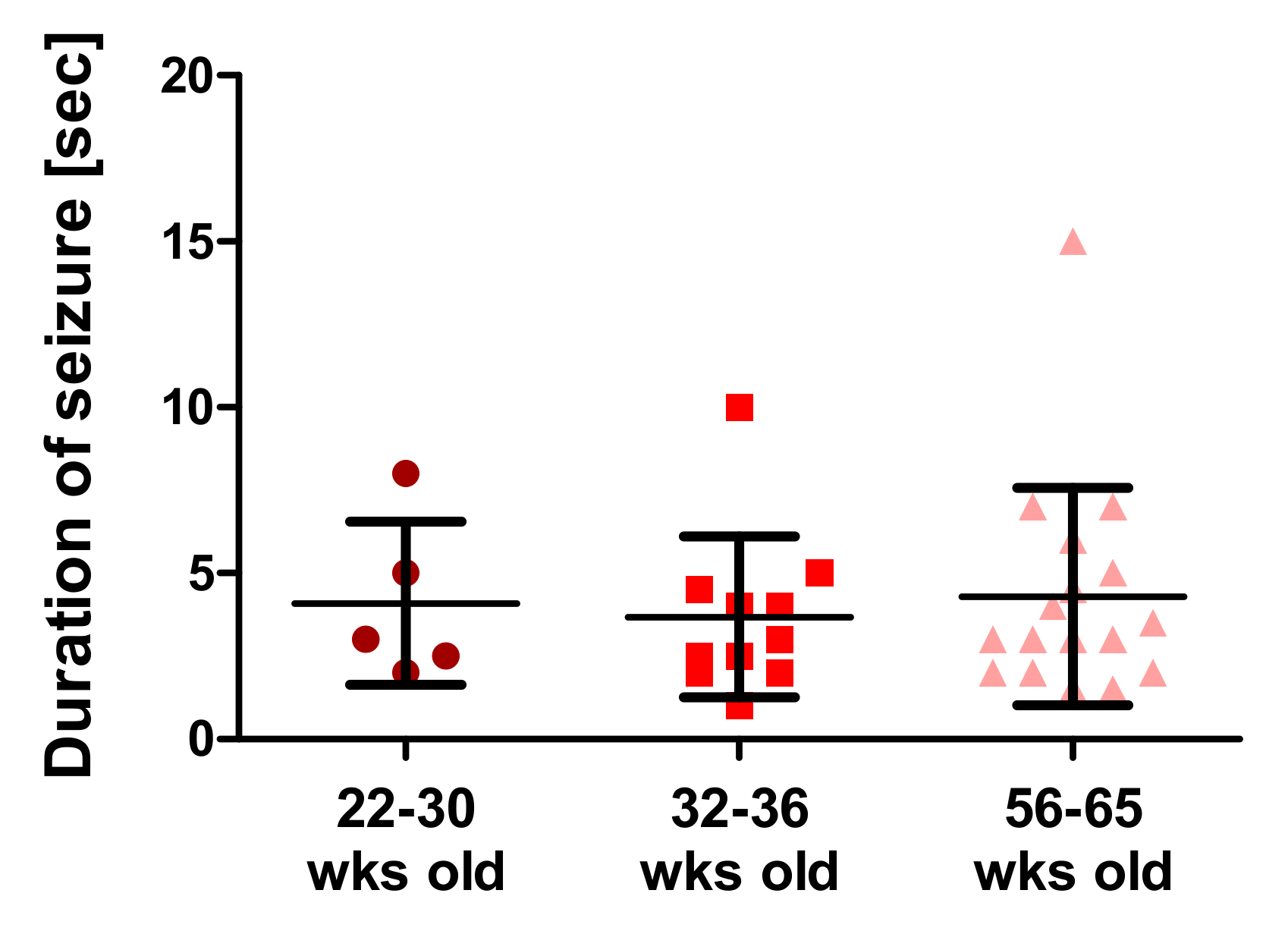
SPTAN1 mutation affecting the stability of spectrin repeats. Even within a well-genetically defined R1098Q mouse model, the severity of motor incoordination and lengths of rotarod-induced seizures may differ between littermates (
Figure 1,
Table S1). This observation can also be extended to other spectrin mutations. For example in the case of three patients carrying the same Arg1003Trp mutation of
SPTBN1 (βII spectrin), only one suffers from seizures and only one has an abnormal brain MRI, while all of them share developmental delay features [
9]. In the above example, genetic background, sex, and age differences were proposed to contribute to various disease presentations. However, in the case of inbred R1098Q mice, the variable penetrance of phenotype might also suggest other mechanisms resulting in either uneven expression of
SPTAN1 alleles and/or random pairing of mutated/healthy αII chains with β-spectrins. Another intriguing possibility assumes local instability of mutant αII protein affecting its mechanical properties and interactions with binding partners. Indeed, using thermal circular dichroism we had previously shown that R1098Q-mutant αII peptides had a higher thermal propensity to unfold over a wide range of temperatures [
20]. We speculate that physical stressors acting on neurons during synaptic remodeling may cause the premature unfolding of mutant spectrin heterodimers, contributing to neuropathology.
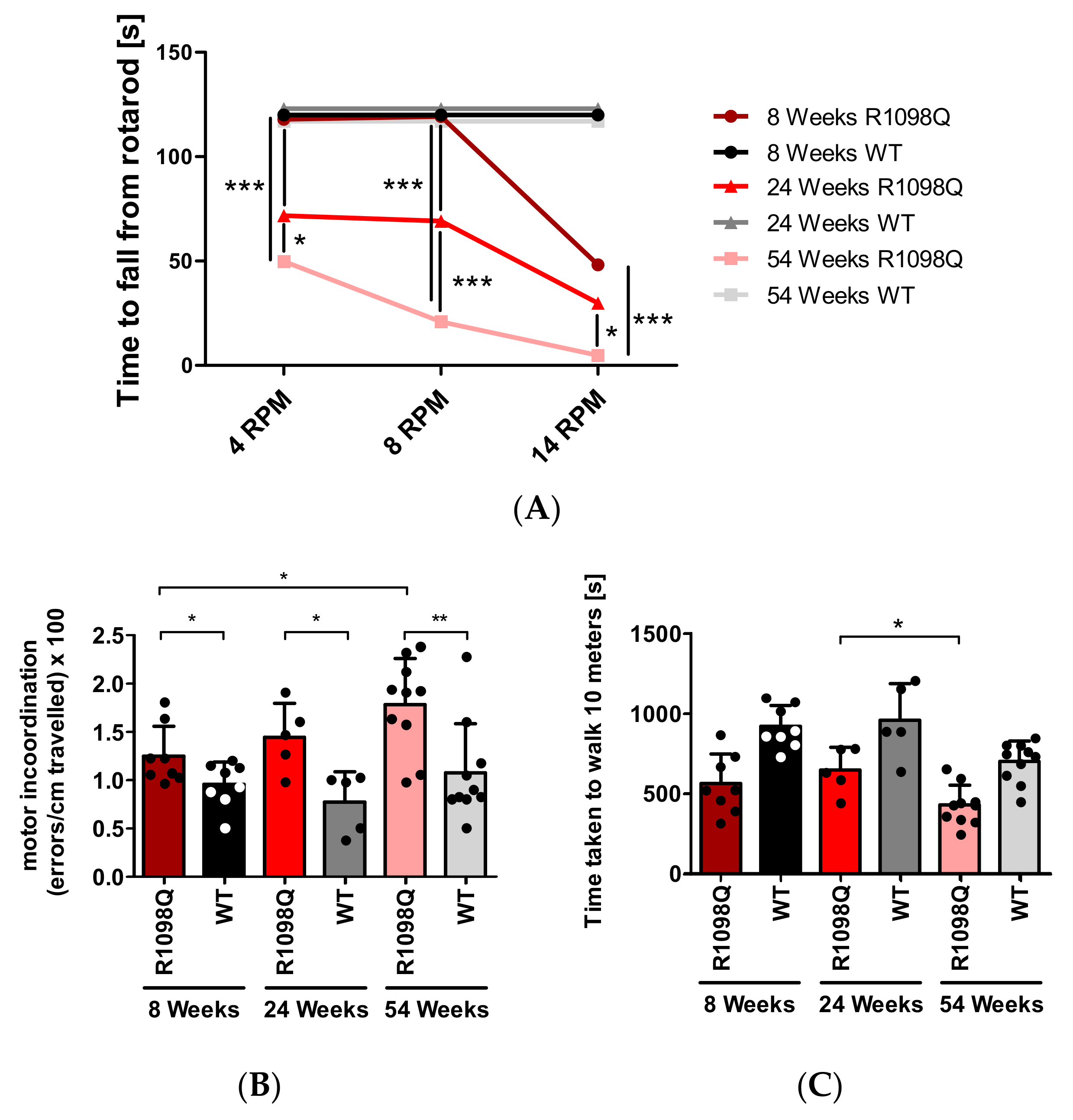
In this study, we also confirmed progressive motor incoordination of R1098Q mice using forced (rotarod) and unforced (grid walking, footprint) walking tests. Progressive ataxia developing in mice older than 3 wks was also reported in βIII spectrin knockout mice (
SPTBN2) [
10]. In contrast to β III knockout mice, however, the grid walking and wire hang tests revealed marked hyperactivity and muscle weakness of R1098Q mice, respectively (
Figure 1C and
Figure 3). Additionally, animal care operators reported voluntary biting behavior and aggressive behavior of R1098Q mice (data not shown). This compares well to a recent case report by Luongo-Zink et al., who analyzed a 9-year-old patient carrying de novo
SPTAN1 Ser889Cys heterozygous variant [
21]. This patient was diagnosed with attention deficit hyperactivity disorder (ADHD) and an autism spectrum disorder. It had self-regulation difficulty that interfered with daily life, including low frustration tolerance, aggression, and impulsivity (kicking, slapping, biting, and pushing). Of interest, ADHD patients carrying autosomal dominant βII variants were recently reported and a similar hyperactivity phenotype in an open field test was also observed in heterozygous βII deficient mice [
9]. These data underline common features of a certain heterozygous missense mutation of
SPTAN1 and
SPTBN1 that are well reflected by murine models.
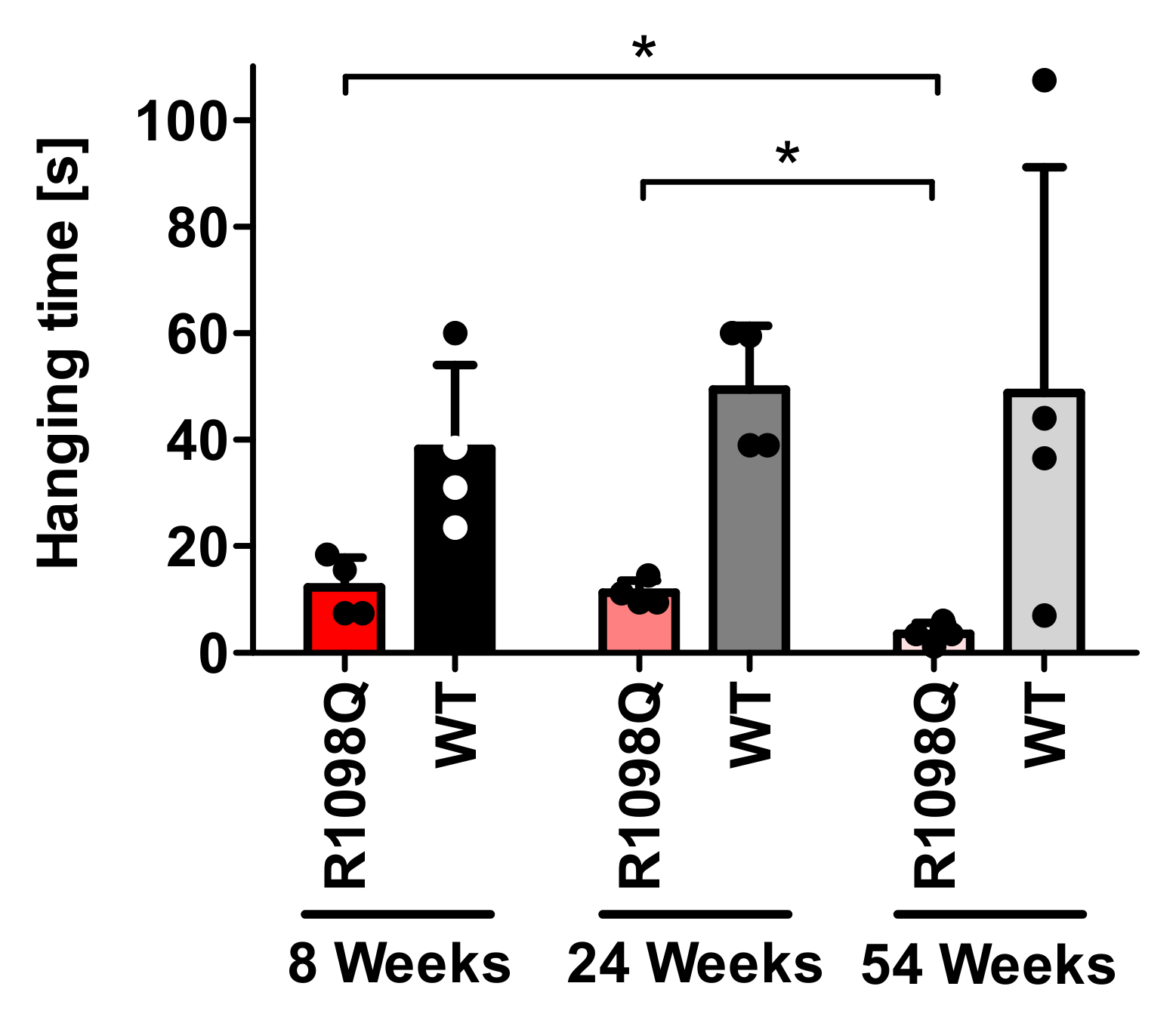
Muscle weakness of R1098Q mice inferred from the poor performance in the wire hang test is reminiscent of the symptoms presented by patients carrying heterozygous
SPTAN1 nonsense mutations reported by Beiyer et al. [
3]. Similar to human patients for whom the phenotype is a slowly progressive, juvenile onset, the R1098Q mice show a slow decline of hanging latencies reaching the statistical significance between the groups of adult and old and between young and old mice (
Figure 3). As to mechanism behind the peripheral neuropathy caused by dominant negative
SPTAN1 variants, haploinsufficiency caused by nonsense-mediated decay of mRNA encoding mutant alleles has been previously proposed [
3]. In the case of R1098Q mice, the allelic imbalance has not been assessed experimentally, nor a possibility of missense-mediated decay reported to affect the splicing efficiency was tested [
22]. We only observed that the total expression level of αII in the brains of heterozygous mice did not significantly differ from the wildtype littermates [
15]. This result has to be treated with caution because of possible species-specific differences in the degree of transcriptional compensation of the murine
Spna2 (
SPTAN1) gene [
14].
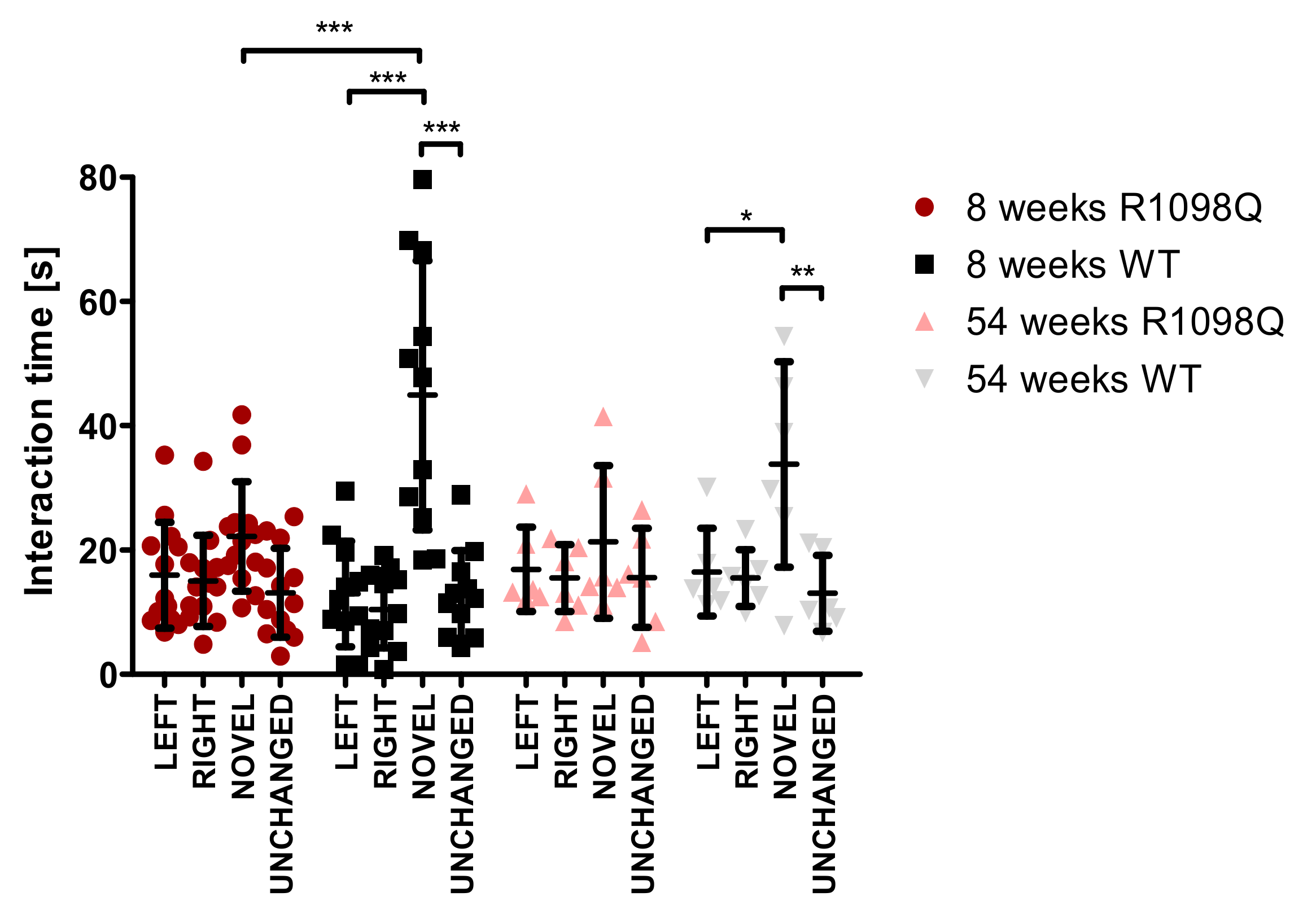
Spatial learning and memory loss are reportedly associated with recurrent seizure episodes in mice [
23,
24]. Since the R1098Q mice exhibited spontaneous and stress-induced seizures (
Figure 5, and
Supplementary Table S1), we assessed the spontaneous visual–spatial memory of young and old R1098Q mice using the novel object recognition test. We could find a highly significant difference between the performance of 8-wk-old R1098Q mice in comparison to their WT littermates. This result is consistent with neuronal loss in the neocortex and CA1-CA2 regions of the hippocampus [
15]. It also compares well to the reported poor immediate auditory-verbal and visual–spatial memory of the aforementioned 9-year-old SPTAN1 Ser889Cys patient [
21].
Taking together, the R1098Q mice present complex phenotype reflecting neuropathology of the central and peripheral nervous systems. For the first time, we document stress-induced seizure episodes and spatial memory impairments in R1098Q mice that compare well to clinical presentations of missense SPTAN1 variants in humans. Despite genetic homogeneity of R1098Q mice, we still observe an important variability in motor and memory performance, as well as in the severity and duration of seizure episodes between littermates. This observation is consistent with available clinical data regarding variable presentations of the same human missense SPTAN1 mutation among family members. It could also be extended to other variants of SPTBN genes.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





