
Hydrogen Sulfide Attenuates Neuroinflammation by Inhibiting the NLRP3/Caspase-1/GSDMD Pathway in Retina or Brain Neuron following Rat Ischemia/Reperfusion
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. The Middle Cerebral Artery Occlusion/Reperfusion (I/R) Model
2.3. Evaluation of Neurological Deficits and Behavioral Analysis
2.4. TTC Staining
2.5. ELISA Assay for Inflammatory Cytokines
2.6. Western Blot Assay
2.7. Brain Frozen Section and Retina Paraffin Section
2.8. Hematoxylin and Eosin Staining (HE)
2.9. TUNEL Staining
2.10. Immunofluorescence
2.11. Statistical Analysis
3. Results
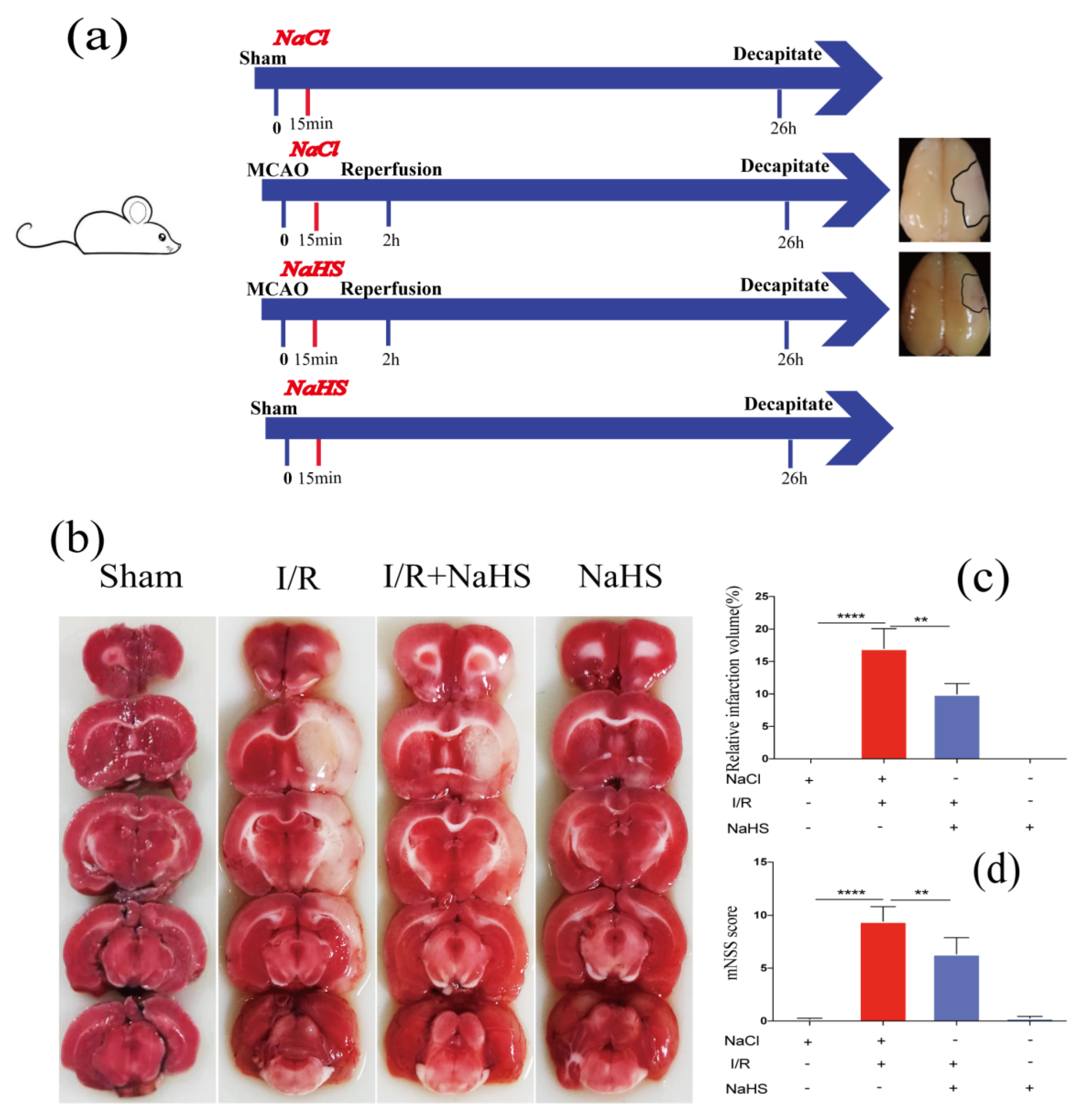
3.1. NaHS Rescued Post-Stroke Neurological Deficits and Inhibited Infarct Progression
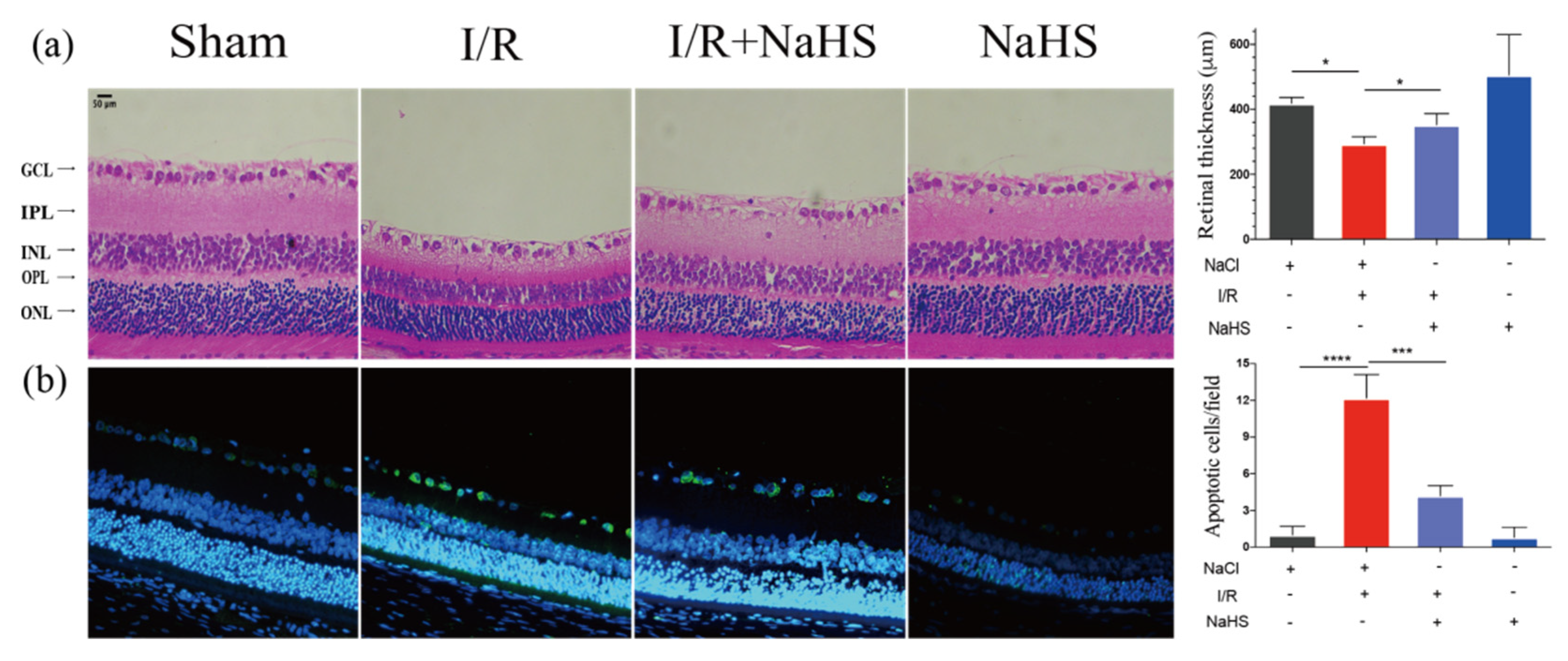
3.2. NaHS Improved Retinal Injury Induced by I/R
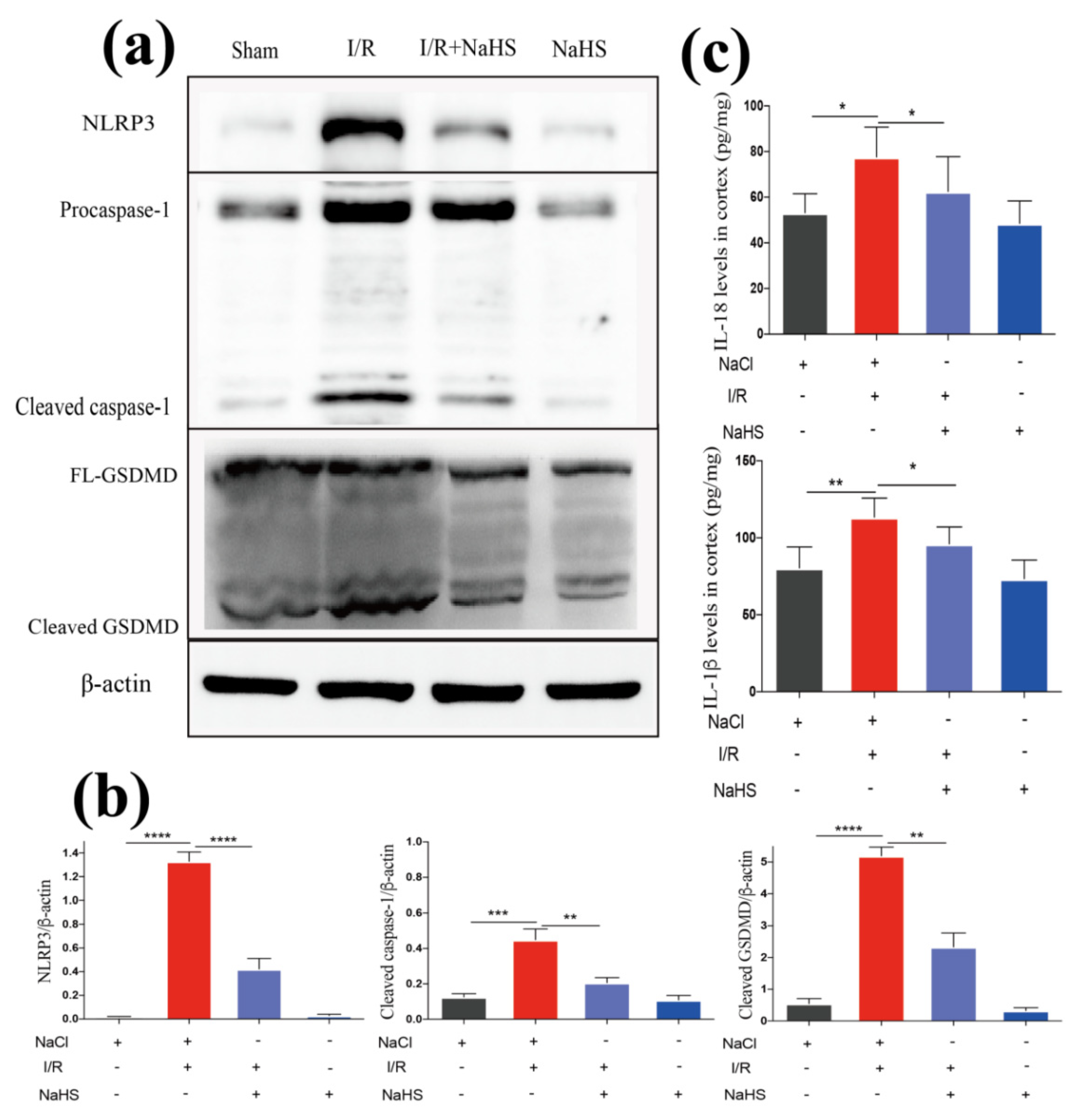
3.3. NaHS Improved Neuroinflammation by Suppressing I/R-Induced Inflammasome-Dependent Pyroptosis in Rat Brain Cortex
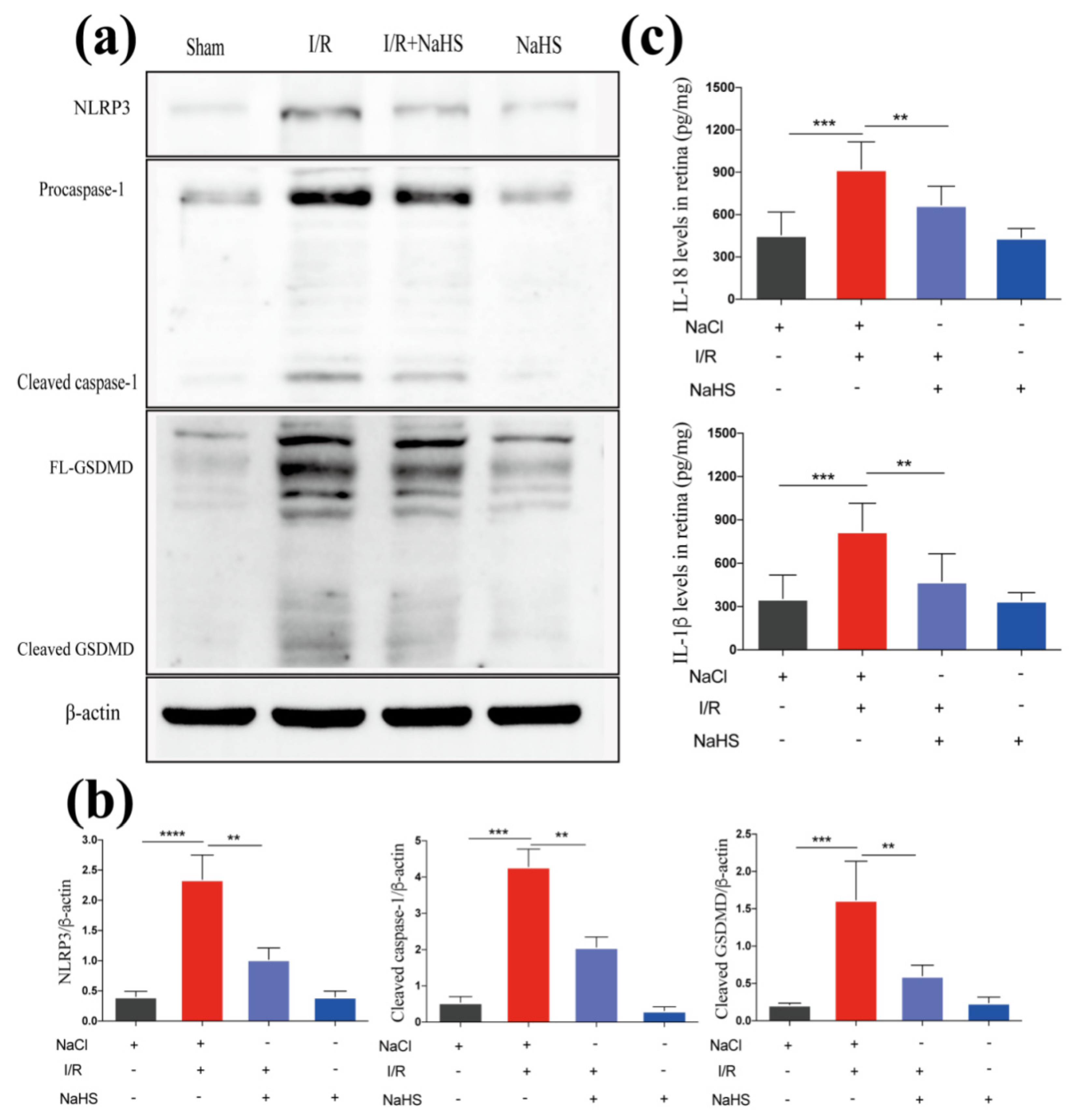
3.4. NaHS Improved Neuroinflammation by Suppressing I/R-Induced Inflammasome Mediated Pyroptosis in Rat Retina
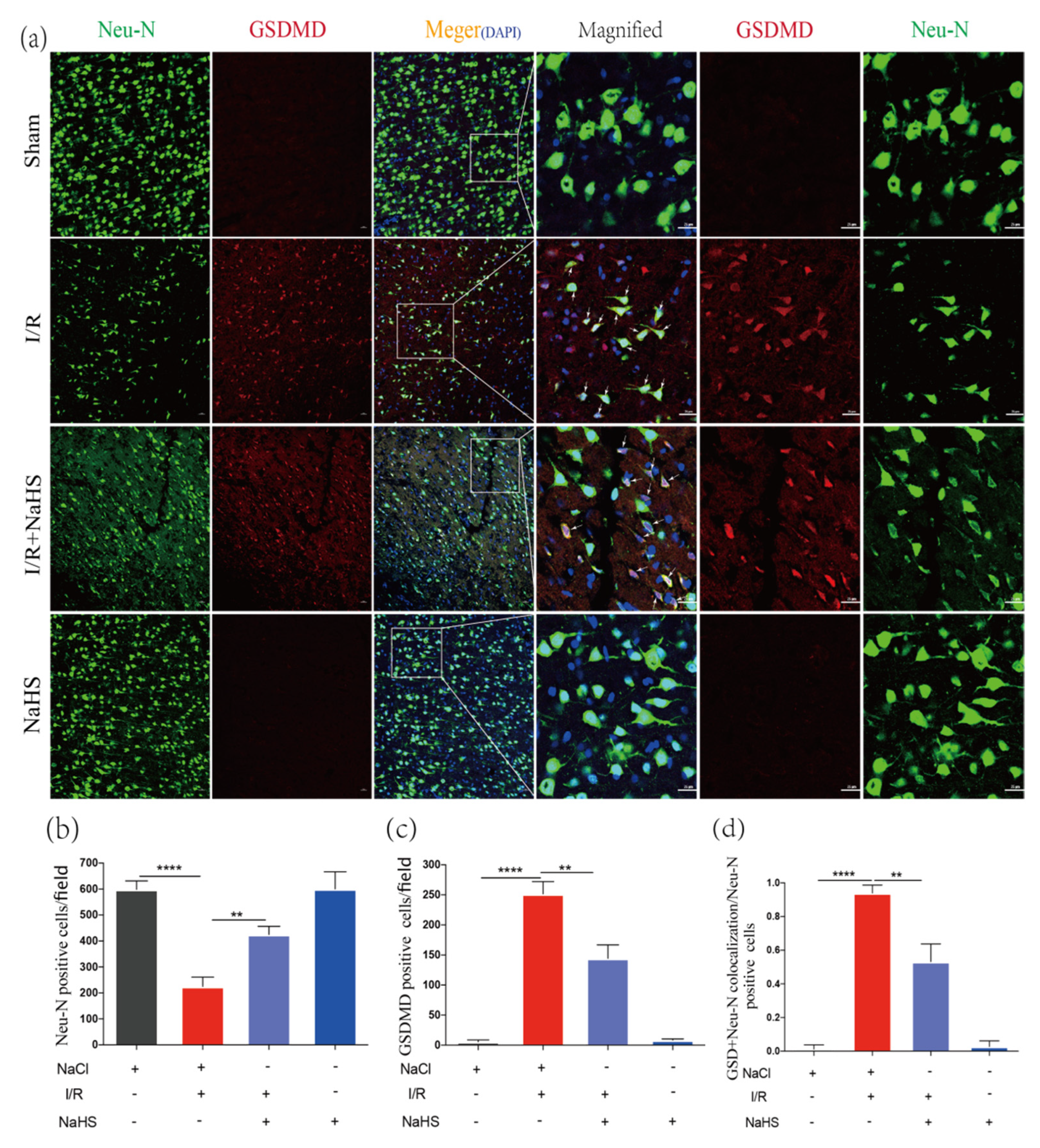
3.5. NaHS Prevented Neuron Pyroptosis in Rat Brain Cortex after Cerebral I/R Injury
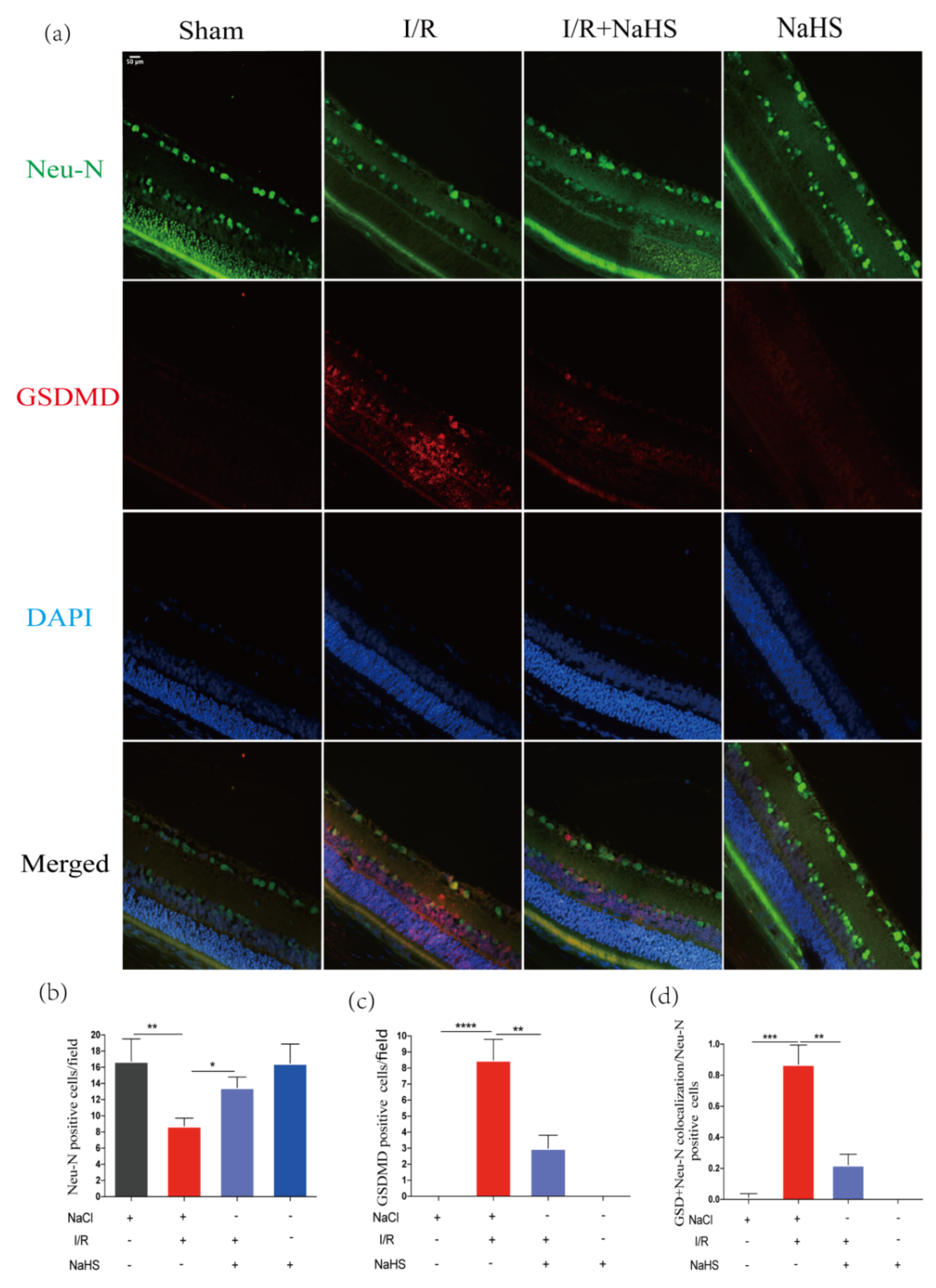
3.6. NaHS Prevented Neuron Pyroptosis in Rat Retina after Cerebral I/R Injury
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, M.; Wang, H.; Zhu, J.; Chen, W.; Wang, L.; Liu, S.; Li, Y.; Wang, L.; Liu, Y.; Yin, P.; et al. Cause-specific mortality for 240 causes in China during 1990–2013: A systematic subnational analysis for the Global Burden of Disease Study 2013. Lancet 2016, 387, 251–272. [Google Scholar] [CrossRef]
- Li, J.; Jin, X.; Liu, K.J.; Liu, W. Ischemia-reperfusion Injury in the Brain: Mechanisms and Potential Therapeutic Strategies. Biochem. Pharmacol. Open Access 2016, 5, 3044–3057. [Google Scholar] [CrossRef]
- London, A.; Benhar, I.; Schwartz, M. The retina as a window to the brain—From eye research to CNS disorders. Nat. Rev. Neurol. 2013, 9, 44–53. [Google Scholar] [CrossRef]
- Rodrigues-Neves, A.C.; Carecho, R.; Correia, S.C.; Carvalho, C.; Campos, E.J.; Baptista, F.I.; Moreira, P.I.; Ambrósio, A.F. Retina and Brain Display Early and Differential Molecular and Cellular Changes in the 3xTg-AD Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 3043–3060. [Google Scholar] [CrossRef] [PubMed]
- Hanus, J.; Zhang, H.; Wang, Z.; Liu, Q.; Zhou, Q.; Wang, S. Induction of necrotic cell death by oxidative stress in retinal pigment epithelial cells. Cell Death Dis. 2013, 4, e965. [Google Scholar] [CrossRef]
- Chien, H.; Dix, R.D. Evidence for Multiple Cell Death Pathways during Development of Experimental Cytomegalovirus Retinitis in Mice with Retrovirus-Induced Immunosuppression: Apoptosis, Necroptosis, and Pyroptosis. J. Virol. 2012, 86, 10961–10978. [Google Scholar] [CrossRef]
- Li, X.; Liu, Y.; Sun, M.; Gao, M.; Li, T.; Liang, J.; Zhai, Y.; Xu, M.; She, X.; Yang, S.; et al. Photoreceptors Degenerate Through Pyroptosis After Experimental Retinal Detachment. Investig. Opthalmology Vis. Sci. 2020, 61, 31. [Google Scholar] [CrossRef]
- D’Onofrio, P.M.; Koeberle, P.D. What can we learn about stroke from retinal ischemia models? Acta Pharmacol. Sin. 2013, 34, 91–103. [Google Scholar] [CrossRef]
- Liao, Y.; Cheng, J.; Kong, X.; Li, S.; Li, X.; Zhang, M.; Zhang, H.; Yang, T.; Dong, Y.; Li, J.; et al. HDAC3 inhibition ameliorates ischemia/reperfusion-induced brain injury by regulating the microglial cGAS-STING pathway. Theranostics 2020, 10, 9644–9662. [Google Scholar] [CrossRef]
- Madeira, M.H.; Boia, R.; Santos, P.F.; Ambrósio, A.F.; Santiago, A.R. Contribution of microglia-mediated neuroinflammation to retinal degenerative diseases. Mediat. Inflamm. 2015, 2015, 673090. [Google Scholar] [CrossRef] [Green Version]
- Malik, A.; Kanneganti, T.-D. Inflammasome activation and assembly at a glance. J. Cell Sci. 2017, 130, 3955–3963. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Qian, J.; Zhang, P.; Li, H.; Shen, H.; Li, X.; Chen, G. Gasdermin D serves as a key executioner of pyroptosis in experimental cerebral ischemia and reperfusion model both in vivo and in vitro. J. Neurosci. Res. 2019, 97, 645–660. [Google Scholar] [CrossRef]
- Poh, L.; Kang, S.-W.; Baik, S.-H.; Ng, G.Y.Q.; She, D.T.; Balaganapathy, P.; Dheen, S.T.; Magnus, T.; Gelderblom, M.; Sobey, C.G.; et al. Evidence that NLRC4 inflammasome mediates apoptotic and pyroptotic microglial death following ischemic stroke. Brain Behav. Immun. 2019, 75, 34–47. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Liu, H.-D.; Li, W.; Chen, Z.-R.; Hu, Y.-C.; Zhang, D.-D.; Shen, W.; Zhou, M.-L.; Zhu, L.; Hang, C.-H. Expression of the NLRP3 Inflammasome in Cerebral Cortex After Traumatic Brain Injury in a Rat Model. Neurochem. Res. 2013, 38, 2072–2083. [Google Scholar] [CrossRef]
- Yang, F.; Wang, Z.; Wei, X.; Han, H.; Meng, X.; Zhang, Y.; Shi, W.; Li, F.; Xin, T.; Pang, Q.; et al. NLRP3 Deficiency Ameliorates Neurovascular Damage in Experimental Ischemic Stroke. J. Cereb. Blood Flow Metab. 2014, 34, 660–667. [Google Scholar] [CrossRef] [PubMed]
- An, P.; Xie, J.; Qiu, S.; Liu, Y.; Wang, J.; Xiu, X.; Li, L.; Tang, M. Hispidulin exhibits neuroprotective activities against cerebral ischemia reperfusion injury through suppressing NLRP3-mediated pyroptosis. Life Sci. 2019, 232, 116599. [Google Scholar] [CrossRef]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef]
- Kaneko, Y.; Kimura, Y.; Kimura, H.; Niki, I. L-cysteine inhibits insulin release from the pancreatic beta-cell: Possible involvement of metabolic production of hy-drogen sulfide, a novel gasotransmitter. Diabetes 2006, 55, 1391–1397. [Google Scholar] [CrossRef] [Green Version]
- Hosoki, R.; Matsuki, N.; Kimura, H. The Possible Role of Hydrogen Sulfide as an Endogenous Smooth Muscle Relaxant in Synergy with Nitric Oxide. Biochem. Biophys. Res. Commun. 1997, 237, 527–531. [Google Scholar] [CrossRef]
- va Sikura, K.; Combi, Z.; Potor, L.; Szerafin, T.; Hendrik, Z.; Méhes, G.; Gergely, P.; Whiteman, M.; Beke, L.; Fürtös, I.; et al. Hydrogen sulfide inhibits aortic valve calcification in heart via regulating RUNX2 by NF-κB, a link between inflammation and mineralization. J. Adv. Res. 2021, 27, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jin, S.; Teng, X.; Hu, Z.; Zhang, Z.; Qiu, X.; Tian, D.; Wu, Y. Hydrogen Sulfide Attenuates LPS-Induced Acute Kidney Injury by Inhibiting Inflammation and Oxidative Stress. Oxid. Med. Cell. Longev. 2018, 2018, 6717212. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Du, A.; Li, D.; Sui, J.; Mayhan, W.G.; Zhao, H. Dynamic change of hydrogen sulfide during global cerebral ischemia–reperfusion and its effect in rats. Brain Res. 2010, 1345, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Markand, S.; Tawfik, A.; Ha, Y.; Gnana-Prakasam, J.; Sonne, S.; Ganapathy, V.; Sen, N.; Xian, M.; Smith, S.B. Cystathionine Beta Synthase Expression in Mouse Retina. Curr. Eye Res. 2013, 38, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Pong, W.W.; Eldred, W.D. Interactions of the gaseous neuromodulators nitric oxide, carbon monoxide, and hydrogen sulfide in the salamander retina. J. Neurosci. Res. 2009, 87, 2356–2364. [Google Scholar] [CrossRef]
- Minamishima, S.; Bougaki, M.; Sips, P.; De Yu, J.; Minamishima, Y.A.; Elrod, J.; Lefer, D.J.; Bloch, K.D.; Ichinose, F. Hydrogen Sulfide Improves Survival After Cardiac Arrest and Cardiopulmonary Resuscitation via a Nitric Oxide Synthase 3–Dependent Mechanism in Mice. Circulation 2009, 120, 888–896. [Google Scholar] [CrossRef]
- Yin, J.; Tu, C.; Zhao, J.; Ou, D.; Chen, G.; Liu, Y.; Xiao, X. Exogenous hydrogen sulfide protects against global cerebral ischemia/reperfusion injury via its anti-oxidative, anti-inflammatory and anti-apoptotic effects in rats. Brain Res. 2012, 1491, 188–196. [Google Scholar] [CrossRef]
- Tao, L.; Yu, Q.; Zhao, P.; Yang, Q.; Wang, B.; Yang, Y.; Kuai, J.; Ding, Q. Preconditioning with hydrogen sulfide ameliorates cerebral ischemia/reperfusion injury in a mouse model of transient middle cerebral artery occlusion. Chem. Interact. 2019, 310, 108738. [Google Scholar] [CrossRef]
- Franke, M.; Bieber, M.; Kraft, P.; Weber, A.N.; Stoll, G.; Schuhmann, M.K. The NLRP3 inflammasome drives inflammation in ischemia/reperfusion injury after transient middle cerebral artery occlusion in mice. Brain Behav. Immun. 2021, 92, 221–231. [Google Scholar] [CrossRef]
- Wang, L.; Ren, W.; Wu, Q.; Liu, T.; Wei, Y.; Ding, J.; Zhou, C.; Xu, H.; Yang, S. NLRP3 Inflammasome Activation: A Therapeutic Target for Cerebral Ischemia–Reperfusion Injury. Front. Mol. Neurosci. 2022, 15, 847440. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Tuipulotu, D.E.; Tan, W.H.; Kay, C.; Man, S.M. Emerging Activators and Regulators of Inflammasomes and Pyroptosis. Trends Immunol. 2019, 40, 1035–1052. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Peng, M.; Xu, P.; Huang, F.; Xie, Y.; Li, J.; Hong, Y.; Guo, H.; Liu, Q.; Zhu, W. Low-density lipoprotein receptor (LDLR) regulates NLRP3-mediated neuronal pyroptosis following cerebral ische-mia/reperfusion injury. J. Neuroinflamm. 2020, 17, 330. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, K.-L.; Li, W.-H.; Liu, Y.-J.; Wei, Y.-J.; Ren, Y.-K.; Mai, C.-D.; Zhang, S.-Y.; Zuo, Y.; Sun, Z.-Z.; Li, D.-L.; et al. Hydrogen Sulfide Attenuates Neuroinflammation by Inhibiting the NLRP3/Caspase-1/GSDMD Pathway in Retina or Brain Neuron following Rat Ischemia/Reperfusion. Brain Sci. 2022, 12, 1245. https://doi.org/10.3390/brainsci12091245
Yang K-L, Li W-H, Liu Y-J, Wei Y-J, Ren Y-K, Mai C-D, Zhang S-Y, Zuo Y, Sun Z-Z, Li D-L, et al. Hydrogen Sulfide Attenuates Neuroinflammation by Inhibiting the NLRP3/Caspase-1/GSDMD Pathway in Retina or Brain Neuron following Rat Ischemia/Reperfusion. Brain Sciences. 2022; 12(9):1245. https://doi.org/10.3390/brainsci12091245
Chicago/Turabian StyleYang, Kun-Li, Wen-Hong Li, Ya-Jie Liu, Ying-Juan Wei, Yan-Kai Ren, Chen-Di Mai, Si-Yu Zhang, Yue Zuo, Zhen-Zhou Sun, Dong-Liang Li, and et al. 2022. "Hydrogen Sulfide Attenuates Neuroinflammation by Inhibiting the NLRP3/Caspase-1/GSDMD Pathway in Retina or Brain Neuron following Rat Ischemia/Reperfusion" Brain Sciences 12, no. 9: 1245. https://doi.org/10.3390/brainsci12091245