
Alzheimer’s Disease and Inflammaging
 , , and
, , and 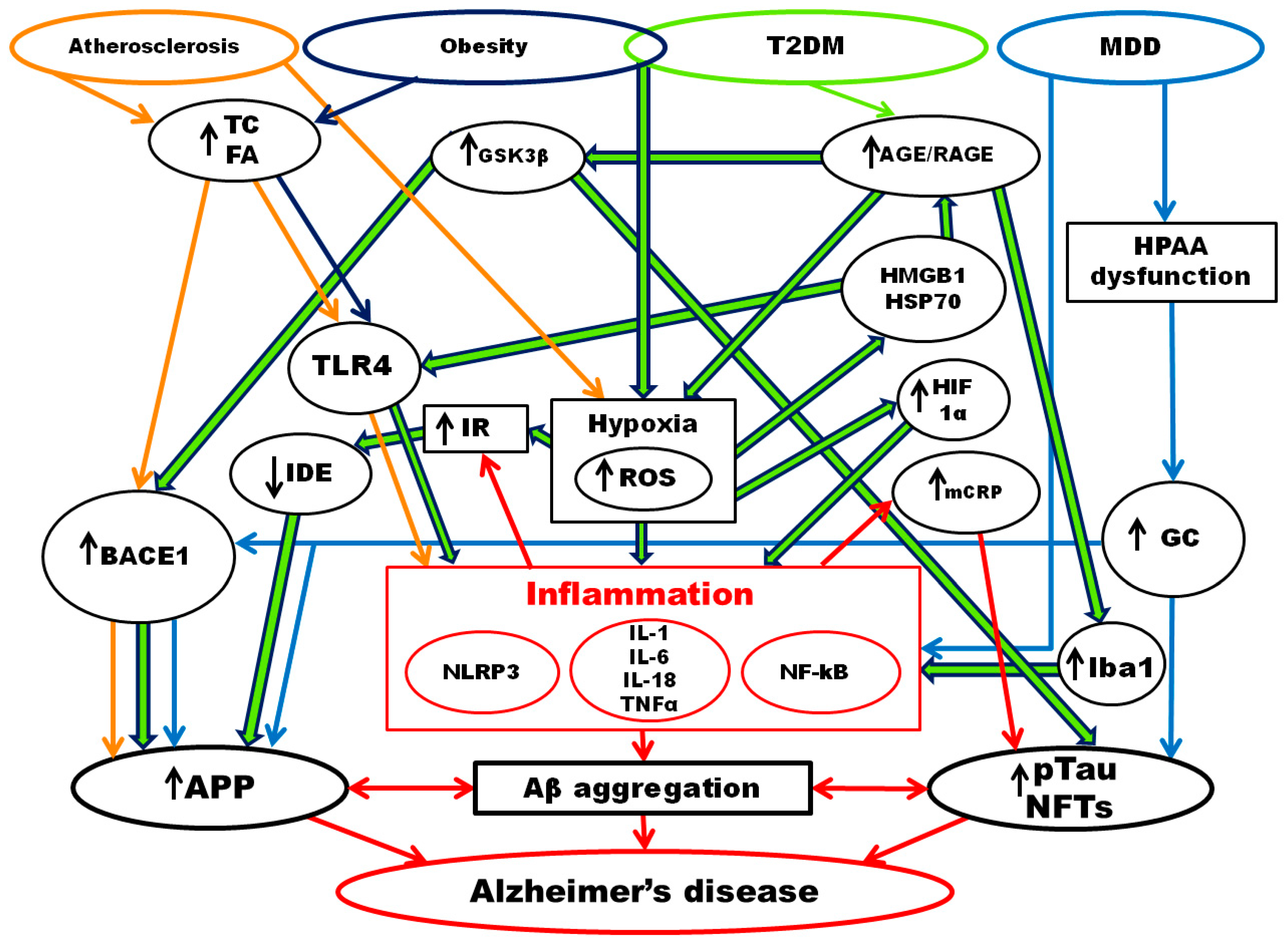{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Aging and Inflammation
3. Hypothesis of the Development of Alzheimer’s Disease
3.1. Genetic Hypothesis of AD Development
3.2. Hypothesis of the Amyloid Cascade in AD Development
3.3. Prions Hypothesis of AD Development
3.4. Oestrogen Hypothesis of AD Development
3.5. Inflammation and AD Development
3.6. Autoimmune Hypothesis of AD Development
3.7. Hypoxia and AD Development
4. Alzheimer’s Disease and Chronic Inflammatory Diseases
4.1. Atherosclerosis and Alzheimer’s Disease
4.2. Obesity and Alzheimer’s Disease
4.3. Type 2 Diabetes Mellitus and Alzheimer’s Disease
4.4. Major Depressive Disorder and Alzheimer’s Disease
5. Inflammaging and Alzheimer’s Disease
6. The Role of Adjuvant Therapy in AD Development
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AICD | APP IntraCellular Domain |
| APP | Amyloid-β Precursor Protein |
| Aβ | Amyloid-β |
| BACE-1 | B-site APP Cleavage Enzyme 1, β-secretase |
| DAMPs | Damage Associated Molecular Patterns |
| EGF | Epidermal Growth Factor |
| EOAD | Early Onset Alzheimer’s disease |
| GC | GlucoCorticoids |
| GSK3β | Glycogen Synthase Kinase 3 Beta |
| HIF1 | Hypoxia-inducible factor 1 |
| HMGB1 | High-Mobility Group Protein Box 1 |
| HSP | Heat Shock Proteins |
| Iba1 | Ionized calcium Binding Adaptor molecule 1 |
| IDE | Insulin Degrading Enzyme |
| IR | Insulin Resistance |
| LOAD | Late Onset Alzheimer’s disease |
| MDD | Major Depressive Disorder |
| MMP | Matrix MetalloProteinases |
| NFTs | NeuroFibrillary Tangles |
| NF-κB | Nuclear Factor Kappa-light-chain-enhancer of activated B cells |
| NLRP3 | NLR family Pyrin domain containing 3 |
| NPCs | NeuroProgenitor Cells |
| NSAIDs | nonsteroidal anti-inflammatory drugs |
| PRRs | Pattern-Recognition Receptor |
| RAGE | Receptor for Advanced Glycation End products |
| SASP | Senescence-Associated Secretory Phenotype |
| SA-β-Gal | Senescence-Associated β-galactosidase |
| SSRI | Serotonin Selective Reuptake Inhibitors |
| SYK | Non-receptor tyrosine kinase SYK |
| T2DM | Type 2 Diabetes Mellitus |
| TGF | Transforming Growth Factor |
| TLR4 | Toll-Like Receptors type 4 |
| TNFα | Tumor Necrosis Factor α |
| TREM2 | Triggering Receptor Expressed on Myeloid cells 2 |
| VEGF | Vascular Endothelial Growth Factor |
References
- World Health Organizatios (WHO). Ageing and Health. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/ageing-and-health (accessed on 20 July 2022).
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The Link between Comorbidities, Genetics, and Alzheimer’s Disease. J. Neuroinflamm. 2018, 15, 276. [Google Scholar] [CrossRef] [PubMed]
- Libertini, G.; Graziamaria, C.; Valeria, C.; Olga, S.; Nicola, F. Introduction. In Evolutionary Gerontology and Geriatrics: Why and How We Age; Springer: Cham, Switzerland, 2021; pp. 1–31. [Google Scholar]
- Skulachev, V.P. New Data on Biochemical Mechanism of Programmed Senescence of Organisms and Antioxidant Defense of Mitochondria. Biochemistry 2009, 74, 1400–1403. [Google Scholar] [CrossRef] [PubMed]
- Ratushnyy, A.Y.; Rudimova, Y.V.; Buravkova, L.B. Replicative Senescence and Expression of Autophagy Genes in Mesenchymal Stromal Cells. Biochemistry 2020, 85, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- López-Olóriz, J.; López-Cancio, E.; Arenillas, J.F.; Hernández, M.; Jiménez, M.; Dorado, L.; Barrios, M.; Soriano-Raya, J.J.; Miralbell, J.; Cáceres, C.; et al. Asymptomatic Cervicocerebral Atherosclerosis, Intracranial Vascular Resistance and Cognition: The Asia-Neuropsychology Study. Atherosclerosis 2013, 230, 330–335. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-Aging. An Evolutionary Perspective on Immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Didier, E.S.; Sugimoto, C.; Bowers, L.C.; A Khan, I.; Kuroda, M.J. Immune Correlates of Aging in Outdoor-Housed Captive Rhesus Macaques (Macaca mulatta). Immun. Ageing 2012, 9, 25. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-Droplet-Accumulating Microglia Represent a Dysfunctional and Proinflammatory State in the Aging Brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, C. Giuliani, and A. Santoro. Inflammaging: A New Immune-Metabolic Viewpoint for Age-Related Diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Jura, M.; Kozak, L. Obesity and Related Consequences to Ageing. Age 2016, 38, 23. [Google Scholar] [CrossRef] [Green Version]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of P53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Vitale, G.; Capri, M.; Salvioli, S. Inflammaging and ‘Garb-Aging’. Trends Endocrinol. Metab. 2017, 28, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Dodig, S.; Čepelak, I.; Pavić, I. Hallmarks of Senescence and Aging. Biochem. Med. 2019, 29, 030501. [Google Scholar] [CrossRef] [PubMed]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. Nrf2 and Nf-κb Interplay in Cerebrovascular and Neurodegenerative Disorders: Molecular Mechanisms and Possible Therapeutic Approaches. Redox Biol. 2019, 21, 101059. [Google Scholar] [CrossRef] [PubMed]
- Tilstra, J.S.; Robinson, A.R.; Wang, J.; Gregg, S.Q.; Clauson, C.L.; Reay, D.P.; Nasto, L.A.; St Croix, C.M.; Usas, A.; Vo, N.; et al. Nf-Κb Inhibition Delays DNA Damage-Induced Senescence and Aging in Mice. J. Clin. Investig. 2012, 122, 2601–2612. [Google Scholar] [CrossRef]
- Kriete, A.; Mayo, K.L.; Yalamanchili, N.; Beggs, W.; Bender, P.; Kari, C.; Rodeck, U. Cell Autonomous Expression of Inflammatory Genes in Biologically Aged Fibroblasts Associated with Elevated Nf-Kappab Activity. Immun. Ageing 2008, 5, 5. [Google Scholar] [CrossRef]
- Carreno, G.; Guiho, R.; Martinez-Barbera, J.P. Cell Senescence in Neuropathology: A Focus on Neurodegeneration and Tumours. Neuropathol. Appl. Neurobiol. 2021, 47, 359–378. [Google Scholar] [CrossRef]
- Askarova, S.; Umbayev, B.; Masoud, A.R.; Kaiyrlykyzy, A.; Safarova, Y.; Tsoy, A.; Olzhayev, F.; Kushugulova, A. The Links between the Gut Microbiome, Aging, Modern Lifestyle and Alzheimer’s Disease. Front. Cell Infect. Microbiol. 2020, 10, 104. [Google Scholar] [CrossRef]
- World Health Organizations (WHO). Dementia. 2021. Available online: https://www.who.int/publications/i/item/9789241550543 (accessed on 20 July 2022).
- Möller, H.J.; Graeber, M.B. The Case Described by Alois Alzheimer in 1911. Historical and Conceptual Perspectives Based on the Clinical Record and Neurohistological Sections. Eur. Arch. Psychiatry Clin. Neurosci. 1998, 248, 111–122. [Google Scholar]
- Jack, C.R.; Bennett, D.A., Jr.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. Nia-Aa Research Framework: Toward a Biological Definition of Alzheimer’s Disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Nikolac Perkovic, M.; Pivac, N. Genetic Markers of Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019, 1192, 27–52. [Google Scholar] [PubMed]
- Grozeva, D.; Saad, S.; Menzies, G.E.; Sims, R. Benefits and Challenges of Rare Genetic Variation in Alzheimer’s Disease. Curr. Genet. Med. Rep. 2019, 7, 53–62. [Google Scholar] [CrossRef]
- Voskobiynyk, Y.; Roth, J.R.; Cochran, J.N.; Rush, T.; Carullo, N.V.; Mesina, J.S.; Waqas, M.; Vollmer, R.M.; Day, J.J.; McMahon, L.L.; et al. Alzheimer’s Disease Risk Gene Bin1 Induces Tau-Dependent Network Hyperexcitability. eLife 2020, 9, e57354. [Google Scholar] [CrossRef]
- Dafsari, F.S.; Jessen, F. Depression-an Underrecognized Target for Prevention of Dementia in Alzheimer’s Disease. Transl. Psychiatry 2020, 10, 160. [Google Scholar] [CrossRef]
- Grimaldi, L.M.; Casadei, V.M.; Ferri, C.; Veglia, F.; Licastro, F.; Annoni, G.; Biunno, I.; De Bellis, G.; Sorbi, S.; Mariani, C.; et al. Association of Early-Onset Alzheimer’s Disease with an Interleukin-1alpha Gene Polymorphism. Ann. Neurol. 2000, 47, 361–365. [Google Scholar] [CrossRef]
- Nicoll, J.A.; Mrak, R.E.; Graham, D.I.; Stewart, J.; Wilcock, G.; MacGowan, S.; Esiri, M.M.; Murray, L.S.; Dewar, D.; Love, S.; et al. Association of Interleukin-1 Gene Polymorphisms with Alzheimer’s Disease. Ann. Neurol. 2000, 47, 365–368. [Google Scholar] [CrossRef]
- Taipa, R.; Ferreira, V.; Brochado, P.; Robinson, A.; Reis, I.; Marques, F.; Mann, D.M.; Melo-Pires, M.; Sousa, N. Inflammatory Pathology Markers (Activated Microglia and Reactive Astrocytes) in Early and Late Onset Alzheimer Disease: A Post Mortem Study. Neuropathol. Appl. Neurobiol. 2018, 44, 298–313. [Google Scholar] [CrossRef]
- Canu, E.; Frisoni, G.B.; Agosta, F.; Pievani, M.; Bonetti, M.; Filippi, M. Early and Late Onset Alzheimer’s Disease Patients Have Distinct Patterns of White Matter Damage. Neurobiol. Aging 2012, 33, 1023–1033. [Google Scholar] [CrossRef]
- Elahi, F.M.; Casaletto, K.B.; La Joie, R.; Walters, S.M.; Harvey, D.; Wolf, A.; Edwards, L.; Rivera-Contreras, W.; Karydas, A.; Cobigo, Y.; et al. Plasma Biomarkers of Astrocytic and Neuronal Dysfunction in Early- and Late-Onset Alzheimer’s Disease. Alzheimer’s Dement. 2020, 16, 681–695. [Google Scholar] [CrossRef]
- Castellani, R.J.; Plascencia-Villa, G.; Perry, G. The Amyloid Cascade and Alzheimer’s Disease Therapeutics: Theory Versus Observation. Lab. Investig. 2019, 99, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Hong, F.; Yang, S. Amyloidosis in Alzheimer’s Disease: Pathogeny, Etiology, and Related Therapeutic Directions. Molecules 2022, 27, 1210. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Nation, D.A.; Zlokovic, B.V. Apoe4 Accelerates Development of Dementia after Stroke: Is There a Role for Cerebrovascular Dysfunction? Stroke 2020, 51, 699–700. [Google Scholar] [CrossRef]
- Leite, D.M.; Seifi, M.; Ruiz-Perez, L.; Nguemo, F.; Plomann, M.; Swinny, J.D.; Battaglia, G. Syndapin-2 Mediated Transcytosis of Amyloid-Β across the Blood-Brain Barrier. Brain Commun. 2022, 4, fcac039. [Google Scholar] [CrossRef]
- Chai, A.B.; Leung, G.K.F.; Callaghan, R.; Gelissen, I.C. P-Glycoprotein: A Role in the Export of Amyloid-Β in Alzheimer’s Disease? FEBS J. 2020, 287, 612–625. [Google Scholar] [CrossRef]
- Vadukul, D.M.; Maina, M.; Franklin, H.; Nardecchia, A.; Serpell, L.C.; Marshall, K.E. Internalisation and Toxicity of Amyloid-Β 1–42 Are Influenced by Its Conformation and Assembly State Rather Than Size. FEBS Lett. 2020, 594, 3490–3503. [Google Scholar] [CrossRef]
- Brunnström, H.R.; Englund, E.M. Cause of Death in Patients with Dementia Disorders. Eur. J. Neurol. 2009, 16, 488–492. [Google Scholar] [CrossRef]
- Wegmann, S.; Biernat, J.; Mandelkow, E. A Current View on Tau Protein Phosphorylation in Alzheimer’s Disease. Curr. Opin. Neurobiol. 2021, 69, 131–138. [Google Scholar] [CrossRef]
- Bhatia, V.; Sharma, S. Role of Mitochondrial Dysfunction, Oxidative Stress and Autophagy in Progression of Alzheimer’s Disease. J. Neurol. Sci. 2021, 421, 117253. [Google Scholar] [CrossRef]
- Naseri, N.N.; Wang, H.; Guo, J.; Sharma, M.; Luo, W. The Complexity of Tau in Alzheimer’s Disease. Neurosci. Lett. 2019, 705, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Mesulam, M.M.; Mufson, E.J.; Levey, A.I.; Wainer, B.H. Cholinergic Innervation of Cortex by the Basal Forebrain: Cytochemistry and Cortical Connections of the Septal Area, Diagonal Band Nuclei, Nucleus Basalis (Substantia Innominata), and Hypothalamus in the Rhesus Monkey. J. Comp. Neurol. 1983, 214, 170–197. [Google Scholar] [CrossRef]
- Mesulam, M.M. Cholinergic Circuitry of the Human Nucleus Basalis and Its Fate in Alzheimer’s Disease. J. Comp. Neurol. 2013, 521, 4124–4144. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, J.; Ray, N.J.; A Hamilton, C.; Donaghy, P.C.; Firbank, M.; Roberts, G.; Allan, L.; Durcan, R.; Barnett, N.; O’Brien, J.T.; et al. Cholinergic White Matter Pathways in Dementia with Lewy Bodies and Alzheimer’s Disease. Brain 2022, 145, 1773–1784. [Google Scholar] [CrossRef]
- Guarnieri, G.; Sarchielli, E.; Comeglio, P.; Herrera-Puerta, E.; Piaceri, I.; Nacmias, B.; Benelli, M.; Kelsey, G.; Maggi, M.; Gallina, P.; et al. Tumor Necrosis Factor α Influences Phenotypic Plasticity and Promotes Epigenetic Changes in Human Basal Forebrain Cholinergic Neuroblasts. Int. J. Mol. Sci. 2020, 21, 6128. [Google Scholar] [CrossRef]
- Prusiner, S.B. Cell Biology. A Unifying Role for Prions in Neurodegenerative Diseases. Science 2012, 336, 1511–1513. [Google Scholar] [CrossRef]
- Gibbons, G.S.; Lee, V.M.Y.; Trojanowski, J.Q. Mechanisms of Cell-to-Cell Transmission of Pathological Tau: A Review. JAMA Neurol. 2019, 76, 101–108. [Google Scholar] [CrossRef]
- Shorter, J. Hsp104: A Weapon to Combat Diverse Neurodegenerative Disorders. Neurosignals 2008, 16, 63–74. [Google Scholar] [CrossRef]
- Low, K.J.Y.; Venkatraman, A.; Mehta, J.S.; Pervushin, K. Molecular Mechanisms of Amyloid Disaggregation. J. Adv. Res. 2022, 36, 113–132. [Google Scholar] [CrossRef]
- Meriin, A.B.; Sherman, M.Y. Role of Molecular Chaperones in Neurodegenerative Disorders. Int. J. Hyperth. 2005, 21, 403–419. [Google Scholar] [CrossRef]
- Campanella, C.; Pace, A.; Caruso Bavisotto, C.; Marzullo, P.; Marino Gammazza, A.; Buscemi, S.; Palumbo Piccionello, A. Heat Shock Proteins in Alzheimer’s Disease: Role and Targeting. Int. J. Mol. Sci. 2018, 19, 2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vendredy, L.; Adriaenssens, E.; Timmerman, V. Small Heat Shock Proteins in Neurodegenerative Diseases. Cell. Stress. Chaperones 2020, 25, 679–699. [Google Scholar] [CrossRef] [PubMed]
- Kuznik, B.I.; Chalisova, N.I.; Tzibikov, N.N.; Linkova, N.S.; Davidov, S.O. Stress, Aging and United Humoral Protective System of the Organism. Epigenetic Mechanisms of Regulation. Adv. Physiol. Sci. 2020, 51, 51–68. [Google Scholar]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; Van Duijn, C.M. Effects of Age, Sex, and Ethnicity on the Association between Apolipoprotein E Genotype and Alzheimer Disease. A Meta-Analysis. Apoe and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Jett, S.; Schelbaum, E.; Jang, G.; Yepez, C.B.; Dyke, J.P.; Pahlajani, S.; Brinton, R.D.; Mosconi, L. Ovarian Steroid Hormones: A Long Overlooked but Critical Contributor to Brain Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 948219. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mishra, A.; Brinton, R.D. Transitions in Metabolic and Immune Systems from Pre-Menopause to Post-Menopause: Implications for Age-Associated Neurodegenerative Diseases. F1000Research 2020, 9, 68. [Google Scholar] [CrossRef]
- Holmes, C. Review: Systemic Inflammation and Alzheimer’s Disease. Neuropathol. Appl. Neurobiol. 2013, 39, 51–68. [Google Scholar] [CrossRef]
- Fakhoury, M. Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol. 2018, 16, 508–518. [Google Scholar] [CrossRef]
- Agostinho, P.; Cunha, R.A.; Oliveira, C. Neuroinflammation, Oxidative Stress and the Pathogenesis of Alzheimer’s Disease. Curr. Pharm. Des. 2010, 16, 2766–2778. [Google Scholar] [CrossRef]
- Shao, Y.; Gearing, M.; Mirra, S.S. Astrocyte-Apolipoprotein E Associations in Senile Plaques in Alzheimer Disease and Vascular Lesions: A Regional Immunohistochemical Study. J. Neuropathol. Exp. Neurol. 1997, 56, 376–381. [Google Scholar] [CrossRef]
- Zotova, E.; Holmes, C.; Johnston, D.; Neal, J.W.; Nicoll, J.A.; Boche, D. Microglial Alterations in Human Alzheimer’s Disease Following Aβ42 Immunization. Neuropathol. Appl. Neurobiol. 2011, 37, 513–524. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. Polymorphisms in Inflammatory Genes and the Risk of Alzheimer Disease. Arch. Neurol. 2001, 58, 1790–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, G.R.; Jonas, L.A.; Li, Y.M. Friend, Foe or Both? Immune Activity in Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 337. [Google Scholar] [CrossRef] [PubMed]
- Labzin, L.I.; Heneka, M.T.; Latz, E. Innate Immunity and Neurodegeneration. Annu. Rev. Med. 2018, 69, 437–449. [Google Scholar] [CrossRef]
- Kamboh, M.I.; Demirci, F.Y.; Wang, X.; Minster, R.L.; Carrasquillo, M.M.; Pankratz, V.S.; Younkin, S.G.; Saykin, A.J.; Jun, G.; Baldwin, C.; et al. Genome-Wide Association Study of Alzheimer’s Disease. Transl. Psychiatry 2012, 2, e117. [Google Scholar] [CrossRef] [PubMed]
- Sierksma, A.; Lu, A.; Mancuso, R.; Fattorelli, N.; Thrupp, N.; Salta, E.; Zoco, J.; Blum, D.; Buée, L.; De Strooper, B.; et al. Novel Alzheimer Risk Genes Determine the Microglia Response to Amyloid-Β but Not to Tau Pathology. EMBO Mol. Med. 2020, 12, e10606. [Google Scholar] [CrossRef]
- Hur, J.Y.; Frost, G.R.; Wu, X.; Crump, C.; Pan, S.J.; Wong, E.; Barros, M.; Li, T.; Nie, P.; Zhai, Y. The Innate Immunity Protein Ifitm3 Modulates Γ-Secretase in Alzheimer’s Disease. Nature 2020, 586, 735–740. [Google Scholar] [CrossRef]
- Baruch, K.; Deczkowska, A.; David, E.; Castellano, J.M.; Miller, O.; Kertser, A.; Berkutzki, T.; Barnett-Itzhaki, Z.; Bezalel, D.; Wyss-Coray, T.; et al. Aging. Aging-Induced Type I Interferon Response at the Choroid Plexus Negatively Affects Brain Function. Science 2014, 346, 89–93. [Google Scholar] [CrossRef]
- De Strooper, B. Aph-1, Pen-2, and Nicastrin with Presenilin Generate an Active Gamma-Secretase Complex. Neuron 2003, 38, 9–12. [Google Scholar] [CrossRef]
- Tumani, H.; Huss, A.; Bachhuber, F. The Cerebrospinal Fluid and Barriers—Anatomic and Physiologic Considerations. Handb. Clin. Neurol. 2017, 146, 21–32. [Google Scholar]
- Xie, J.; Van Hoecke, L.; Vandenbroucke, R.E. The Impact of Systemic Inflammation on Alzheimer’s Disease Pathology. Front. Immunol. 2021, 12, 796867. [Google Scholar] [CrossRef]
- Xie, J.; Gorlé, N.; Vandendriessche, C.; Van Imschoot, G.; Van Wonterghem, E.; Van Cauwenberghe, C.; Parthoens, E.; Van Hamme, E.; Lippens, S.; Van Hoecke, L. Low-Grade Peripheral Inflammation Affects Brain Pathology in the App(Nl-G-F)Mouse Model of Alzheimer’s Disease. Acta Neuropathol. Commun. 2021, 9, 163. [Google Scholar] [CrossRef]
- Kountouras, J.; Tsolaki, M.; Gavalas, E.; Boziki, M.; Zavos, C.; Karatzoglou, P.; Chatzopoulos, D.; Venizelos, I. Relationship between Helicobacter pylori Infection and Alzheimer Disease. Neurology 2006, 66, 938–940. [Google Scholar] [CrossRef]
- Dioguardi, M.; Crincoli, V.; Laino, L.; Alovisi, M.; Sovereto, D.; Mastrangelo, F.; Russo, L.L.; Muzio, L.L. The Role of Periodontitis and Periodontal Bacteria in the Onset and Progression of Alzheimer’s Disease: A Systematic Review. J. Clin. Med. 2020, 9, 495. [Google Scholar] [CrossRef]
- Carter, C.J.; France, J.; Crean, S.; Singhrao, S.K. The Porphyromonas Gingivalis/Host Interactome Shows Enrichment in Gwasdb Genes Related to Alzheimer’s Disease, Diabetes and Cardiovascular Diseases. Front. Aging Neurosci. 2017, 9, 408. [Google Scholar] [CrossRef]
- Kamer, A.R.; Craig, R.G.; Pirraglia, E.; Dasanayake, A.P.; Norman, R.G.; Boylan, R.J.; Nehorayoff, A.; Glodzik, L.; Brys, M.; de Leon, M.J. Tnf-Alpha and Antibodies to Periodontal Bacteria Discriminate between Alzheimer’s Disease Patients and Normal Subjects. J. Neuroimmunol. 2009, 216, 92–97. [Google Scholar] [CrossRef]
- Farhad, S.Z.; Amini, S.; Khalilian, A.; Barekatain, M.; Mafi, M.; Barekatain, M.; Rafei, E. The Effect of Chronic Periodontitis on Serum Levels of Tumor Necrosis Factor-Alpha in Alzheimer Disease. Dent. Res. J. 2014, 11, 549–552. [Google Scholar]
- Wozniak, M.A.; Mee, A.P.; Itzhaki, R.F. Herpes Simplex Virus Type 1 DNA Is Located within Alzheimer’s Disease Amyloid Plaques. J. Pathol. 2009, 217, 131–138. [Google Scholar] [CrossRef]
- Tsai, M.-C.; Cheng, W.-L.; Sheu, J.-J.; Huang, C.-C.; Shia, B.-C.; Kao, L.-T.; Lin, H.-C. Increased Risk of Dementia Following Herpes Zoster Ophthalmicus. PLoS ONE 2017, 12, e0188490. [Google Scholar] [CrossRef]
- Chen, V.C.; Wu, S.I.; Huang, K.Y.; Yang, Y.H.; Kuo, T.Y.; Liang, H.Y.; Huang, K.L.; Gossop, M. Herpes Zoster and Dementia: A Nationwide Population-Based Cohort Study. J. Clin. Psychiatry 2018, 79, 8164. [Google Scholar] [CrossRef]
- Tzeng, N.S.; Chung, C.H.; Lin, F.H.; Chiang, C.P.; Yeh, C.B.; Huang, S.Y.; Lu, R.B.; Chang, H.A.; Kao, Y.C.; Yeh, H.W. Anti-Herpetic Medications and Reduced Risk of Dementia in Patients with Herpes Simplex Virus Infections-a Nationwide, Population-Based Cohort Study in Taiwan. Neurotherapeutics 2018, 15, 417–429. [Google Scholar] [CrossRef]
- Lim, B.; Prassas, I.; Diamandis, E.P. Alzheimer Disease Pathogenesis: The Role of Autoimmunity. J. Appl. Lab. Med. 2021, 6, 756–764. [Google Scholar] [CrossRef]
- D’Andrea, M.R. Evidence Linking Neuronal Cell Death to Autoimmunity in Alzheimer’s Disease. Brain Res. 2003, 982, 19–30. [Google Scholar] [CrossRef]
- Nagele, R.G.; Clifford, P.M.; Siu, G.; Levin, E.C.; Acharya, N.K.; Han, M.; Kosciuk, M.C.; Venkataraman, V.; Zavareh, S.; Zarrabi, S.; et al. Brain-Reactive Autoantibodies Prevalent in Human Sera Increase Intraneuronal Amyloid-Β(1–42) Deposition. J. Alzheimers Dis. 2011, 25, 605–622. [Google Scholar] [CrossRef] [Green Version]
- Morgan, B.P. Complement in the Pathogenesis of Alzheimer’s Disease. Semin. Immunopathol. 2018, 40, 113–124. [Google Scholar] [CrossRef]
- Fernandez-Vizarra, P.; Lopez-Franco, O.; Mallavia, B.; Higuera-Matas, A.; Lopez-Parra, V.; Ortiz-Muñoz, G.; Ambrosio, E.; Egido, J.; Almeida, O.F.X.; Gomez-Guerrero, C. Immunoglobulin G Fc Receptor Deficiency Prevents Alzheimer-like Pathology and Cognitive Impairment in Mice. Brain 2012, 135, 2826–2837. [Google Scholar] [CrossRef]
- Loeffler, D.A.; Camp, D.M.; Bennett, D.A. Plaque Complement Activation and Cognitive Loss in Alzheimer’s Disease. J. Neuroinflamm. 2008, 5, 9. [Google Scholar] [CrossRef]
- Nath, A.; Hall, E.; Tuzova, M.; Dobbs, M.; Jons, M.; Anderson, C.; Woodward, J.; Guo, Z.; Fu, W.; Kryscio, R.; et al. Autoantibodies to Amyloid Beta-Peptide (Abeta) Are Increased in Alzheimer’s Disease Patients and Abeta Antibodies Can Enhance Abeta Neurotoxicity: Implications for Disease Pathogenesis and Vaccine Development. Neuromol. Med. 2003, 3, 29–39. [Google Scholar] [CrossRef]
- Kellner, A.; Matschke, J.; Bernreuther, C.; Moch, H.; Ferrer, I.; Glatzel, M. Autoantibodies against Beta-Amyloid Are Common in Alzheimer’s Disease and Help Control Plaque Burden. Ann. Neurol. 2009, 65, 24–31. [Google Scholar] [CrossRef]
- Mengel, D.; Röskam, S.; Neff, F.; Balakrishnan, K.; Deuster, O.; Gold, M.; Oertel, W.H.; Bacher, M.; Bach, J.-P.; Dodel, R. Naturally Occurring Autoantibodies Interfere with Β-Amyloid Metabolism and Improve Cognition in a Transgenic Mouse Model of Alzheimer’s Disease 24 H after Single Treatment. Transl. Psychiatry 2013, 3, e236. [Google Scholar] [CrossRef]
- Jha, N.K.; Jha, S.K.; Sharma, R.; Kumar, D.; Kumar, P. Hypoxia-Induced Signaling Activation in Neurodegenerative Diseases: Targets for New Therapeutic Strategies. J. Alzheimer’s Dis. 2018, 62, 15–38. [Google Scholar] [CrossRef] [PubMed]
- Merelli, A.; Rodríguez, J.C.G.; Folch, J.; Regueiro, M.R.; Camins, A.; Lazarowski, A. Understanding the Role of Hypoxia Inducible Factor During Neurodegeneration for New Therapeutics Opportunities. Curr. Neuropharmacol. 2018, 16, 1484–1498. [Google Scholar] [CrossRef]
- Zhang, F.; Niu, L.; Li, S.; Le, W. Pathological Impacts of Chronic Hypoxia on Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 902–909. [Google Scholar] [CrossRef]
- Ruas, J.L.; Poellinger, L. Hypoxia-Dependent Activation of Hif into a Transcriptional Regulator. Semin. Cell. Dev. Biol. 2005, 16, 514–522. [Google Scholar] [CrossRef]
- Lall, R.; Mohammed, R.; Ojha, U. What Are the Links between Hypoxia and Alzheimer’s Disease? Neuropsychiatr. Dis. Treat. 2019, 15, 1343–1354. [Google Scholar] [CrossRef]
- Li, L.; Zhang, X.; Yang, D.; Luo, G.; Chen, S.; Le, W. Hypoxia Increases Abeta Generation by Altering Beta- and Gamma-Cleavage of App. Neurobiol. Aging 2009, 30, 1091–1098. [Google Scholar] [CrossRef]
- Jakubauskienė, E.; Kanopka, A. Alternative Splicing and Hypoxia Puzzle in Alzheimer’s and Parkinson’s Diseases. Genes 2021, 12, 1272. [Google Scholar] [CrossRef]
- Li, W.; He, P.; Huang, Y.; Li, Y.-F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective Autophagy of Intracellular Organelles: Recent Research Advances. Theranostics 2021, 11, 222–256. [Google Scholar] [CrossRef]
- Shen, Y.; Hua, L.; Yeh, C.K.; Shen, L.; Ying, M.; Zhang, Z.; Liu, G.; Li, S.; Chen, S.; Chen, X.; et al. Ultrasound with Microbubbles Improves Memory, Ameliorates Pathology and Modulates Hippocampal Proteomic Changes in a Triple Transgenic Mouse Model of Alzheimer’s Disease. Theranostics 2020, 10, 11794–11819. [Google Scholar] [CrossRef]
- Andrews, S.J.; Fulton-Howard, B.; O’Reilly, P.; Marcora, E.; Goate, A.M. Causal Associations between Modifiable Risk Factors and the Alzheimer’s Phenome. Ann. Neurol. 2021, 89, 54–65. [Google Scholar] [CrossRef]
- Roher, A.E.; Tyas, S.L.; Maarouf, C.L.; Daugs, I.D.; Kokjohn, T.A.; Emmerling, M.R.; Garami, Z.; Belohlavek, M.; Sabbagh, M.N.; Sue, L.I.; et al. Intracranial Atherosclerosis as a Contributing Factor to Alzheimer’s Disease Dementia. Alzheimer’s Dement. 2011, 7, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Urbanova, B.; Tomek, A.; Mikulik, R.; Magerova, H.; Horinek, D.; Hort, J. Neurosonological Examination: A Non-Invasive Approach for the Detection of Cerebrovascular Impairment in Ad. Front. Behav. Neurosci. 2014, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Paffen, E.; DeMaat, M.P. C-Reactive Protein in Atherosclerosis: A Causal Factor? Cardiovasc. Res. 2006, 71, 30–39. [Google Scholar] [CrossRef]
- Slevin, M.; Matou, S.; Zeinolabediny, Y.; Corpas, R.; Weston, R.; Liu, D.; Boras, E.; Di Napoli, M.; Petcu, E.; Sarroca, S.; et al. Monomeric C-Reactive Protein--a Key Molecule Driving Development of Alzheimer’s Disease Associated with Brain Ischaemia? Sci. Rep. 2015, 5, 13281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of Action, Role in Disease, and Therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. Nlrp3 Is Activated in Alzheimer’s Disease and Contributes to Pathology in App/Ps1 Mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Westerterp, M.; Fotakis, P.; Ouimet, M.; Bochem, A.E.; Zhang, H.; Molusky, M.M.; Wang, W.; Abramowicz, S.; la Bastide-van Gemert, S.; Wang, N.; et al. Cholesterol Efflux Pathways Suppress Inflammasome Activation, Netosis, and Atherogenesis. Circulation 2018, 138, 898–912. [Google Scholar] [CrossRef]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; E Mullick, A.; Golenbock, D.T.; et al. Cd36 Coordinates Nlrp3 Inflammasome Activation by Facilitating Intracellular Nucleation of Soluble Ligands into Particulate Ligands in Sterile Inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef]
- Tan, H.-W.; Liu, X.; Bi, X.-P.; Xing, S.-S.; Li, L.; Gong, H.-P.; Zhong, M.; Wang, Z.-H.; Zhang, Y.; Zhang, W. Il-18 Overexpression Promotes Vascular Inflammation and Remodeling in a Rat Model of Metabolic Syndrome. Atherosclerosis 2010, 208, 350–357. [Google Scholar] [CrossRef]
- Valerio, A.; Boroni, F.; Benarese, M.; Sarnico, I.; Ghisi, V.; Bresciani, L.G.; Ferrario, M.; Borsani, G.; Spano, P.; Pizzi, M. Nf-Kappab Pathway: A Target for Preventing Beta-Amyloid (Abeta)-Induced Neuronal Damage and Abeta42 Production. Eur. J. Neurosci. 2006, 23, 1711–1720. [Google Scholar] [CrossRef]
- Aslam, F. Obesity and Crp. Ann. Rheum. Dis. 2018, 77, e52. [Google Scholar] [CrossRef]
- Mavri, A.; Alessi, M.C.; Geel-Georgelin, O.; Fina, F.; Sentocnik, J.T.; Bastelica, D.; Stegnar, M.; Juhan-Vague, I. Subcutaneous Abdominal, but Not Femoral Fat Expression of Plasminogen Activator Inhibitor-1 (Pai-1) Is Related to Plasma Pai-1 Levels and Insulin Resistance and Decreases after Weight Loss. Diabetologia 2001, 44, 2025–2031. [Google Scholar] [CrossRef]
- Marques-Vidal, P.; Bochud, M.; Bastardot, F.; Lüscher, T.; Ferrero, F.; Gaspoz, J.M.; Paccaud, F.; Urwyler, A.; von Känel, R.; Hock, C.; et al. Association between Inflammatory and Obesity Markers in a Swiss Population-Based Sample (Colaus Study). Obes. Facts. 2012, 5, 734–744. [Google Scholar] [CrossRef]
- Christiansen, T.; Richelsen, B.; Bruun, J.M. Monocyte Chemoattractant Protein-1 Is Produced in Isolated Adipocytes, Associated with Adiposity and Reduced after Weight Loss in Morbid Obese Subjects. Int. J. Obes. 2005, 29, 146–150. [Google Scholar] [CrossRef] [Green Version]
- Hartigh, L.J.D.; Wang, S.; Goodspeed, L.; Ding, Y.; Averill, M.; Subramanian, S.; Wietecha, T.; O’Brien, K.D.; Chait, A. Deletion of Serum Amyloid A3 Improves High Fat High Sucrose Diet-Induced Adipose Tissue Inflammation and Hyperlipidemia in Female Mice. PLoS ONE 2014, 9, e108564. [Google Scholar] [CrossRef]
- Piché, M.E.; Tchernof, A.; Després, J.P. Obesity Phenotypes, Diabetes, and Cardiovascular Diseases. Circ. Res. 2020, 126, 1477–1500. [Google Scholar] [CrossRef]
- Gudala, K.; Bansal, D.; Schifano, F.; Bhansali, A. Diabetes Mellitus and Risk of Dementia: A Meta-Analysis of Prospective Observational Studies. J. Diabetes Investig. 2013, 4, 640–650. [Google Scholar] [CrossRef]
- Anstey, K.J.; Peters, R.; Zheng, L.; Barnes, D.E.; Brayne, C.; Brodaty, H.; Chalmers, J.; Clare, L.; Dixon, R.A.; Dodge, H.; et al. Future Directions for Dementia Risk Reduction and Prevention Research: An International Research Network on Dementia Prevention Consensus. J. Alzheimer’s Dis. 2020, 78, 3–12. [Google Scholar] [CrossRef]
- Gomez, G.; Beason-Held, L.L.; Bilgel, M.; An, Y.; Wong, D.F.; Studenski, S.; Ferrucci, L.; Resnick, S.M. Metabolic Syndrome and Amyloid Accumulation in the Aging Brain. J. Alzheimer’s Dis. 2018, 65, 629–639. [Google Scholar] [CrossRef]
- Palta, P.; Rippon, B.; Tahmi, M.; Sherwood, G.; Soto, L.; Ceballos, F.; Laing, K.; He, H.; Reitz, C.; Razlighi, Q.; et al. Metabolic Syndrome and Its Components in Relation to In vivo Brain Amyloid and Neurodegeneration in Late Middle Age. Neurobiol. Aging 2021, 97, 89–96. [Google Scholar] [CrossRef]
- Schenk, S.; Saberi, M.; Olefsky, J.M. Insulin Sensitivity: Modulation by Nutrients and Inflammation. J. Clin. Investig. 2008, 118, 2992–3002. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A Central Role for Jnk in Obesity and Insulin Resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and Systemic Insulin Resistance Resulting from Hepatic Activation of Ikk-Beta and Nf-Kappab. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef]
- Arkan, M.C.; Hevener, A.L.; Greten, F.R.; Maeda, S.; Li, Z.W.; Long, J.M.; Wynshaw-Boris, A.; Poli, G.; Olefsky, J.; Karin, M. Ikk-Beta Links Inflammation to Obesity-Induced Insulin Resistance. Nat. Med. 2005, 11, 191–198. [Google Scholar] [CrossRef]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef]
- Willette, A.A.; Bendlin, B.B.; Starks, E.J.; Birdsill, A.C.; Johnson, S.C.; Christian, B.T.; Okonkwo, O.C.; La Rue, A.; Hermann, B.P.; Koscik, R.L.; et al. Association of Insulin Resistance with Cerebral Glucose Uptake in Late Middle-Aged Adults at Risk for Alzheimer Disease. JAMA Neurol. 2015, 72, 1013–1020. [Google Scholar] [CrossRef]
- Nuzzo, D.; Baldassano, S.; Amato, A.; Picone, P.; Galizzi, G.; Caldara, G.F.; Di Carlo, M.; Mulè, F. Glucagon-Like Peptide-2 Reduces the Obesity-Associated Inflammation in the Brain. Neurobiol. Dis. 2019, 121, 296–304. [Google Scholar] [CrossRef]
- Rao, R.V.; Descamps, O.; John, V.; Bredesen, D.E. Ayurvedic Medicinal Plants for Alzheimer’s Disease: A Review. Alzheimer’s Res. Ther. 2012, 4, 22. [Google Scholar] [CrossRef]
- Grimm, M.O.; Grimm, H.S.; Hartmann, T. Amyloid Beta as a Regulator of Lipid Homeostasis. Trends Mol. Med. 2007, 13, 337–344. [Google Scholar] [CrossRef]
- Arvanitakis, Z.; Wilson, R.S.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Diabetes Mellitus and Risk of Alzheimer Disease and Decline in Cognitive Function. Arch. Neurol. 2004, 61, 661–666. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, C.; Hua, S.; Liao, H.; Wang, M.; Xiong, Y.; Cao, F. An Updated Meta-Analysis of Cohort Studies: Diabetes and Risk of Alzheimer’s Disease. Diabetes Res. Clin. Pract. 2017, 124, 41–47. [Google Scholar] [CrossRef]
- Li, J.; Tang, Y.; Cai, D. Ikkβ/Nf-Κb Disrupts Adult Hypothalamic Neural Stem Cells to Mediate a Neurodegenerative Mechanism of Dietary Obesity and Pre-Diabetes. Nat. Cell. Biol. 2012, 14, 999–1012. [Google Scholar] [CrossRef]
- Khan, M.I.; Rath, S.; Adhami, V.M.; Mukhtar, H. Hypoxia Driven Glycation: Mechanisms and Therapeutic Opportunities. Semin. Cancer Biol. 2018, 49, 75–82. [Google Scholar] [CrossRef]
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common Neurodegenerative Pathways in Obesity, Diabetes, and Alzheimer’s Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1037–1045. [Google Scholar] [CrossRef]
- Choi, B.-R.; Cho, W.-H.; Kim, J.; Lee, H.J.; Chung, C.; Jeon, W.K.; Han, J.-S. Increased Expression of the Receptor for Advanced Glycation End Products in Neurons and Astrocytes in a Triple Transgenic Mouse Model of Alzheimer’s Disease. Exp. Mol. Med. 2014, 46, e75. [Google Scholar] [CrossRef]
- Arancio, O.; Zhang, H.P.; Chen, X.; Lin, C.; Trinchese, F.; Puzzo, D.; Liu, S.; Hegde, A.; Yan, S.F.; Stern, A.; et al. Rage Potentiates Abeta-Induced Perturbation of Neuronal Function in Transgenic Mice. EMBO J. 2014, 23, 4096–4105. [Google Scholar] [CrossRef]
- Senatus, L.M.; Schmidt, A.M. The Age-Rage Axis: Implications for Age-Associated Arterial Diseases. Front. Genet. 2017, 8, 187. [Google Scholar] [CrossRef]
- Liang, Z.; Wu, L.; Gong, S.; Liu, X. The Cognitive Dysfunction Related to Alzheimer Disease or Cerebral Small Vessel Disease: What’s the Differences. Medicine 2021, 100, e26967. [Google Scholar] [CrossRef]
- Kehmeier, M.N.; Walker, A.E. Sex Differences in Large Artery Stiffness: Implications for Cerebrovascular Dysfunction and Alzheimer’s Disease. Front. Aging 2021, 2, 791208. [Google Scholar] [CrossRef]
- Du, X.L.; Song, L.; Schulz, P.E.; Xu, H.; Chan, H. Risk of Developing Alzheimer’s Disease and Related Dementias in Association with Cardiovascular Disease, Stroke, Hypertension, and Diabetes in a Large Cohort of Women with Breast Cancer and with up to 26 Years of Follow-Up. J. Alzheimer’s Dis. 2022, 87, 415–432. [Google Scholar] [CrossRef]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-Degrading Enzyme Regulates the Levels of Insulin, Amyloid Beta-Protein, and the Beta-Amyloid Precursor Protein Intracellular Domain In Vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef]
- Kim, M.; Hersh, L.B.; Leissring, M.A.; Ingelsson, M.; Matsui, T.; Farris, W.; Lu, A.; Hyman, B.T.; Selkoe, D.J.; Bertram, L.; et al. Decreased Catalytic Activity of the Insulin-Degrading Enzyme in Chromosome 10-Linked Alzheimer Disease Families. J. Biol. Chem. 2007, 282, 7825–7832. [Google Scholar] [CrossRef]
- Cook, D.G.; Leverenz, J.B.; McMillan, P.J.; Kulstad, J.J.; Ericksen, S.; Roth, R.A.; Schellenberg, G.D.; Jin, L.W.; Kovacina, K.S.; Craft, S. Reduced Hippocampal Insulin-Degrading Enzyme in Late-Onset Alzheimer’s Disease Is Associated with the Apolipoprotein E-Epsilon4 Allele. Am. J. Pathol. 2003, 162, 313–319. [Google Scholar] [CrossRef]
- Bahniwal, M.; Little, J.P.; Klegeris, A. High Glucose Enhances Neurotoxicity and Inflammatory Cytokine Secretion by Stimulated Human Astrocytes. Curr. Alzheimer Res. 2017, 14, 731–741. [Google Scholar] [CrossRef]
- Flores-Dorantes, M.T.; Díaz-López, Y.E.; Gutiérrez-Aguilar, R. Environment and Gene Association with Obesity and Their Impact on Neurodegenerative and Neurodevelopmental Diseases. Front. Neurosci. 2020, 14, 863. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ta, Q.T.H.; Nguyen, T.K.O.; Nguyen, T.T.D.; Giau, V.V. Type 3 Diabetes and Its Role Implications in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 3165. [Google Scholar] [CrossRef]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s Disease a Type 3 Diabetes? A Critical Appraisal. Biochim. Biophys. Acta Mol. Basis. Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef]
- Snowden, M.B.; Atkins, D.C.; Steinman, L.E.; Bell, J.F.; Bryant, L.L.; Copeland, C.; Fitzpatrick, A.L. Longitudinal Association of Dementia and Depression. Am. J. Geriatr. Psychiatry 2015, 23, 897–905. [Google Scholar] [CrossRef]
- Chi, S.; Wang, C.; Jiang, T.; Zhu, X.-C.; Yu, J.-T.; Tan, L. The Prevalence of Depression in Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Curr. Alzheimer Res. 2015, 12, 189–198. [Google Scholar] [CrossRef]
- Ismail, Z.; Elbayoumi, H.; Fischer, C.E.; Hogan, D.B.; Millikin, C.P.; Schweizer, T.; Mortby, M.E.; Smith, E.E.; Patten, S.B.; Fiest, K.M. Prevalence of Depression in Patients with Mild Cognitive Impairment: A Systematic Review and Meta-Analysis. JAMA Psychiatry 2017, 74, 58–67. [Google Scholar] [CrossRef]
- Kessing, L.V.; Andersen, P.K. Does the Risk of Developing Dementia Increase with the Number of Episodes in Patients with Depressive Disorder and in Patients with Bipolar Disorder? J. Neurol Neurosurg. Psychiatry 2004, 75, 1662–1666. [Google Scholar] [CrossRef]
- Byers, A.L.; Yaffe, K. Depression and Risk of Developing Dementia. Nat. Rev. Neurol. 2011, 7, 323–331. [Google Scholar] [CrossRef]
- Köhler, C.A.; Freitas, T.H.; Maes, M.; De Andrade, N.Q.; Liu, C.S.; Fernandes, B.S.; Stubbs, B.; Solmi, M.; Veronese, N.; Herrmann, N.; et al. Peripheral Cytokine and Chemokine Alterations in Depression: A Meta-Analysis of 82 Studies. Acta Psychiatr. Scand. 2017, 135, 373–387. [Google Scholar] [CrossRef]
- Cheng, Y.; Pardo, M.; Armini, R.D.S.; Martinez, A.; Mouhsine, H.; Zagury, J.-F.; Jope, R.S.; Beurel, E. Stress-Induced Neuroinflammation Is Mediated by Gsk3-Dependent Tlr4 Signaling That Promotes Susceptibility to Depression-Like Behavior. Brain Behav. Immun. 2016, 53, 207–222. [Google Scholar] [CrossRef] [Green Version]
- Ushakova, V.M.; Gorlova, A.V.; Zubkov, E.A.; Morozova, A.Y.; Zorkina, Y.A.; Pavlov, D.A.; Inozemtsev, A.N.; Chekhonin, V.P. Experimental Models of Depressive Disorder. Neurosci. Behav. Physiol. 2019, 69, 230–247. [Google Scholar]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the Distribution and Morphology of Microglia in the Normal Adult Mouse Brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Simon, M.S.; Schiweck, C.; Arteaga-Henríquez, G.; Poletti, S.; Haarman, B.C.M.; Dik, W.A.; Schwarz, M.; Vrieze, E.; Mikova, O.; Joergens, S.; et al. Monocyte Mitochondrial Dysfunction, Inflammaging, and Inflammatory Pyroptosis in Major Depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 111, 110391. [Google Scholar] [CrossRef]
- Hermida, A.P.; McDonald, W.M.; Steenland, K.; Levey, A. The Association between Late-Life Depression, Mild Cognitive Impairment and Dementia: Is Inflammation the Missing Link? Expert Rev. Neurother. 2012, 12, 1339–1350. [Google Scholar] [CrossRef]
- Sudheimer, K.D.; O’Hara, R.; Spiegel, D.; Powers, B.; Kraemer, H.C.; Neri, E.; Weiner, M.; Hardan, A.; Hallmayer, J.; Dhabhar, F.S. Cortisol, Cytokines, and Hippocampal Volume Interactions in the Elderly. Front. Aging Neurosci. 2014, 6, 153. [Google Scholar] [CrossRef]
- Green, K.N.; Billings, L.M.; Roozendaal, B.; McGaugh, J.L.; LaFerla, F.M. Glucocorticoids Increase Amyloid-Beta and Tau Pathology in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2006, 26, 9047–9056. [Google Scholar] [CrossRef]
- Yang, C.; Guo, X.; Wang, G.; Wang, H.; Liu, Z.; Liu, H.; Zhu, Z.; Li, Y. Changes in Tau Phosphorylation Levels in the Hippocampus and Frontal Cortex Following Chronic Stress. Braz. J. Med. Biol. Res. 2014, 47, 237–244. [Google Scholar] [CrossRef]
- Walker, F.R. A Critical Review of the Mechanism of Action for the Selective Serotonin Reuptake Inhibitors: Do These Drugs Possess Anti-Inflammatory Properties and How Relevant Is This in the Treatment of Depression? Neuropharmacology 2013, 67, 304–317. [Google Scholar] [CrossRef]
- Trovato, A.; Siracusa, R.; Di Paola, R.; Scuto, M.; Ontario, M.L.; Bua, O.; Di Mauro, P.; Toscano, M.A.; Petralia, C.C.T.; Maiolino, L.; et al. Redox Modulation of Cellular Stress Response and Lipoxin A4 Expression by Hericium Erinaceus in Rat Brain: Relevance to Alzheimer’s Disease Pathogenesis. Immun. Ageing 2016, 13, 23. [Google Scholar] [CrossRef]
- Pang, Y.; Fan, L.-W. Dysregulation of Neurogenesis by Neuroinflammation: Key Differences in Neurodevelopmental and Neurological Disorders. Neural Regen. Res. 2017, 12, 366–371. [Google Scholar] [CrossRef]
- Thevaranjan, N.; Puchta, A.; Schulz, C.; Naidoo, A.; Szamosi, J.C.; Verschoor, C.P.; Loukov, D.; Schenck, L.P.; Jury, J.; Foley, K.P.; et al. Age-Associated Microbial Dysbiosis Promotes Intestinal Permeability, Systemic Inflammation, and Macrophage Dysfunction. Cell Host Microbe 2017, 21, 455–466. [Google Scholar] [CrossRef] [Green Version]
- Gombar, S.; Jung, H.J.; Dong, F.; Calder, B.; Atzmon, G.; Barzilai, N.; Tian, X.-L.; Pothof, J.; Hoeijmakers, J.H.; Campisi, J.; et al. Comprehensive Microrna Profiling in B-Cells of Human Centenarians by Massively Parallel Sequencing. BMC Genom. 2012, 13, 353. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting Edge: Nf-Kappab Activating Pattern Recognition and Cytokine Receptors License Nlrp3 Inflammasome Activation by Regulating Nlrp3 Expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Gritsenko, A.; Yu, S.; Martin-Sanchez, F.; Diaz-Del-Olmo, I.; Nichols, E.-M.; Davis, D.; Brough, D.; Lopez-Castejon, G. Priming Is Dispensable for Nlrp3 Inflammasome Activation in Human Monocytes in Vitro. Front. Immunol. 2020, 11, 565924. [Google Scholar] [CrossRef]
- Royce, G.H.; Brown-Borg, H.M.; Deepa, S.S. The Potential Role of Necroptosis in Inflammaging and Aging. Geroscience 2019, 41, 795–811. [Google Scholar] [CrossRef]
- Caccamo, A.; Branca, C.; Piras, I.S.; Ferreira, E.; Huentelman, M.J.; Liang, W.S.; Readhead, B.; Dudley, J.T.; Spangenberg, E.E.; Green, K.N.; et al. Necroptosis Activation in Alzheimer’s Disease. Nat. Neurosci. 2017, 20, 1236–1246. [Google Scholar] [CrossRef]
- Ofengeim, D.; Mazzitelli, S.; Ito, Y.; DeWitt, J.P.; Mifflin, L.; Zou, C.; Das, S.; Adiconis, X.; Chen, H.; Zhu, H.; et al. Ripk1 Mediates a Disease-Associated Microglial Response in Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2017, 114, E8788–E8797. [Google Scholar] [CrossRef]
- Green, D.R.; Levine, B. To Be or Not to Be? How Selective Autophagy and Cell Death Govern Cell Fate. Cell 2014, 157, 65–75. [Google Scholar] [CrossRef]
- Madruga, E.; Maestro, I.; Martínez, A. Mitophagy Modulation, a New Player in the Race against Als. Int. J. Mol. Sci. 2022, 22, 740. [Google Scholar] [CrossRef]
- Feldman, N.; Rotter-Maskowitz, A.; Okun, E. Damps as Mediators of Sterile Inflammation in Aging-Related Pathologies. Ageing Res. Rev. 2015, 24, 29–39. [Google Scholar] [CrossRef]
- Fang, C.; Wei, X.; Wei, Y. Mitochondrial DNA in the Regulation of Innate Immune Responses. Protein Cell 2016, 7, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Downs, K.P.; Nguyen, H.; Dorfleutner, A.; Stehlik, C. An Overview of the Non-Canonical Inflammasome. Mol. Asp. Med. 2020, 76, 100924. [Google Scholar] [CrossRef]
- Biber, K.; Neumann, H.; Inoue, K.; Boddeke, H.W. Neuronal ‘on’ and ‘Off’ Signals Control Microglia. Trends Neurosci. 2007, 30, 596–602. [Google Scholar] [CrossRef]
- Gui, Y.; Marks, J.D.; Das, S.; Hyman, B.T.; Serrano-Pozo, A. Characterization of the 18 Kda Translocator Protein (Tspo) Expression in Post-Mortem Normal and Alzheimer’s Disease Brains. Brain Pathol. 2020, 30, 151–164. [Google Scholar] [CrossRef]
- Onyango, I.G.; Bennett, J.P.; Stokin, G.B. Mitochondrially-Targeted Therapeutic Strategies for Alzheimer’s Disease. Curr. Alzheimer Res. 2021, 18, 753–771. [Google Scholar] [CrossRef]
- Holthoff, V.A.; Ferris, S.; Ihl, R.; Robert, P.; Winblad, B.; Gauthier, S.; Sternberg, K.; Tennigkeit, F. Validation of the Relevant Outcome Scale for Alzheimer’s Disease: A Novel Multidomain Assessment for Daily Medical Practice. Alzheimers Res. Ther. 2011, 3, 27. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s Disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Fairley, L.H.; Wong, J.H.; Barron, A.M. Mitochondrial Regulation of Microglial Immunometabolism in Alzheimer’s Disease. Front. Immunol. 2021, 12, 624538. [Google Scholar] [CrossRef]
- O’Bryant, S.E.; Waring, S.C.; Hobson, V.; Hall, J.R.; Moore, C.B.; Bottiglieri, T.; Massman, P.; Diaz-Arrastia, R. Decreased C-Reactive Protein Levels in Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 49–53. [Google Scholar] [CrossRef]
- O’Bryant, S.E.; Zhang, F.; Johnson, L.A.; Hall, J.; Edwards, M.; Grammas, P.; Oh, E.; Lyketsos, C.G.; Rissman, R.A. A Precision Medicine Model for Targeted Nsaid Therapy in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 66, 97–104. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Y.; Wang, D.; Zhang, J.; Zhang, F. Nsaid Exposure and Risk of Alzheimer’s Disease: An Updated Meta-Analysis from Cohort Studies. Front. Aging Neurosci. 2018, 10, 83. [Google Scholar] [CrossRef]
- Green, R.C.; Schneider, L.S.; Amato, D.A.; Beelen, A.P.; Wilcock, G.; Swabb, E.A.; Zavitz, K.H. Effect of Tarenflurbil on Cognitive Decline and Activities of Daily Living in Patients with Mild Alzheimer Disease: A Randomized Controlled Trial. JAMA 2009, 302, 2557–2564. [Google Scholar] [CrossRef]
- Aisen, P.S.; Davis, K.L.; Berg, J.D.; Schafer, K.; Campbell, K.; Thomas, R.G.; Weiner, M.F.; Farlow, M.R.; Sano, M.; Grundman, M.; et al. A Randomized Controlled Trial of Prednisone in Alzheimer’s Disease. Alzheimer’s Disease Cooperative Study. Neurology 2000, 54, 588–593. [Google Scholar] [CrossRef]
- Soininen, H.; West, C.; Robbins, J.; Niculescu, L. Long-Term Efficacy and Safety of Celecoxib in Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2007, 23, 8–21. [Google Scholar] [CrossRef]
- Hampel, H.; Caraci, F.; Cuello, A.C.; Caruso, G.; Nisticò, R.; Corbo, M.; Baldacci, F.; Toschi, N.; Garaci, F.; Chiesa, P.A.; et al. A Path toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Front. Immunol. 2020, 11, 456. [Google Scholar] [CrossRef]
- Hölscher, C. New Drug Treatments Show Neuroprotective Effects in Alzheimer’s and Parkinson’s Diseases. Neural Regen. Res. 2014, 9, 1870–1873. [Google Scholar] [CrossRef]
- Tumminia, A.; Vinciguerra, F.; Parisi, M.; Frittitta, L. Type 2 Diabetes Mellitus and Alzheimer’s Disease: Role of Insulin Signalling and Therapeutic Implications. Int. J. Mol. Sci. 2018, 19, 3306. [Google Scholar] [CrossRef]
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J.; et al. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimer’s Dis. 2017, 57, 1325–1334. [Google Scholar] [CrossRef]
- Chiang, M.-C.; Cheng, Y.-C.; Chen, S.-J.; Yen, C.-H.; Huang, R.-N. Metformin Activation of Ampk-Dependent Pathways Is Neuroprotective in Human Neural Stem Cells against Amyloid-Beta-Induced Mitochondrial Dysfunction. Exp. Cell Res. 2016, 347, 322–331. [Google Scholar] [CrossRef]
- Chung, M.-M.; Chen, Y.-L.; Pei, D.; Cheng, Y.-C.; Sun, B.; Nicol, C.J.; Yen, C.-H.; Chen, H.-M.; Liang, Y.-J.; Chiang, M.-C. The Neuroprotective Role of Metformin in Advanced Glycation End Product Treated Human Neural Stem Cells Is Ampk-Dependent. Biochim. Biophys. Acta 2015, 1852, 720–731. [Google Scholar] [CrossRef]
- American Diabetes Association. Introduction: Standards of Medical Care in Diabetes-2022. Diabetes Care 2022, 45, S1–S2. [Google Scholar] [CrossRef]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, D.E.; Hansen, T.B.; et al. 2019 Esc Guidelines on Diabetes, Pre-Diabetes, and Cardiovascular Diseases Developed in Collaboration with the Easd. Eur. Heart J. 2020, 41, 255–323. [Google Scholar] [CrossRef]
- Ng, T.P.; Feng, L.; Yap, K.B.; Lee, T.S.; Tan, C.H.; Winblad, B. Long-Term Metformin Usage and Cognitive Function among Older Adults with Diabetes. J. Alzheimer’s Dis. 2014, 41, 61–68. [Google Scholar] [CrossRef]
- Guo, M.; Mi, J.; Jiang, Q.-M.; Xu, J.-M.; Tang, Y.-Y.; Tian, G.; Wang, B. Metformin May Produce Antidepressant Effects through Improvement of Cognitive Function among Depressed Patients with Diabetes Mellitus. Clin. Exp. Pharmacol. Physiol. 2014, 41, 650–656. [Google Scholar] [CrossRef]
- Hsu, C.-C.; Wahlqvist, M.L.; Lee, M.-S.; Tsai, H.-N. Incidence of Dementia Is Increased in Type 2 Diabetes and Reduced by the Use of Sulfonylureas and Metformin. J. Alzheimer’s Dis. 2011, 24, 485–493. [Google Scholar] [CrossRef]
- Cheng, C.; Lin, C.-H.; Tsai, Y.-W.; Tsai, C.-J.; Chou, P.-H.; Lan, T.-H. Type 2 Diabetes and Antidiabetic Medications in Relation to Dementia Diagnosis. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1299–1305. [Google Scholar] [CrossRef]
- Landreth, G. Therapeutic Use of Agonists of the Nuclear Receptor Ppargamma in Alzheimer’s Disease. Curr. Alzheimer Res. 2007, 4, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Shang, Y.; Jiang, L.; Shi, T.L.; Wang, L. The Peroxisome Proliferators Activated Receptor-Gamma Agonists as Therapeutics for the Treatment of Alzheimer’s Disease and Mild-to-Moderate Alzheimer’s Disease: A Meta-Analysis. Int. J. Neurosci. 2016, 126, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Femminella, G.D.; Bencivenga, L.; Petraglia, L.; Visaggi, L.; Gioia, L.; Grieco, F.V.; De Lucia, C.; Komici, K.; Corbi, G.; Edison, P.; et al. Antidiabetic Drugs in Alzheimer’s Disease: Mechanisms of Action and Future Perspectives. J. Diabetes Res. 2017, 2017, 7420796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hölscher, C. The Role of Glp-1 in Neuronal Activity and Neurodegeneration. Vitam. Horm. 2010, 84, 331–354. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosyreva, A.M.; Sentyabreva, A.V.; Tsvetkov, I.S.; Makarova, O.V. Alzheimer’s Disease and Inflammaging. Brain Sci. 2022, 12, 1237. https://doi.org/10.3390/brainsci12091237
Kosyreva AM, Sentyabreva AV, Tsvetkov IS, Makarova OV. Alzheimer’s Disease and Inflammaging. Brain Sciences. 2022; 12(9):1237. https://doi.org/10.3390/brainsci12091237
Chicago/Turabian StyleKosyreva, Anna Mikhailovna, Alexandra Vladislavovna Sentyabreva, Ivan Sergeevich Tsvetkov, and Olga Vasilievna Makarova. 2022. "Alzheimer’s Disease and Inflammaging" Brain Sciences 12, no. 9: 1237. https://doi.org/10.3390/brainsci12091237