
Tumor Microenvironment in Glioma Invasion

 , ,
, ,
Abstract
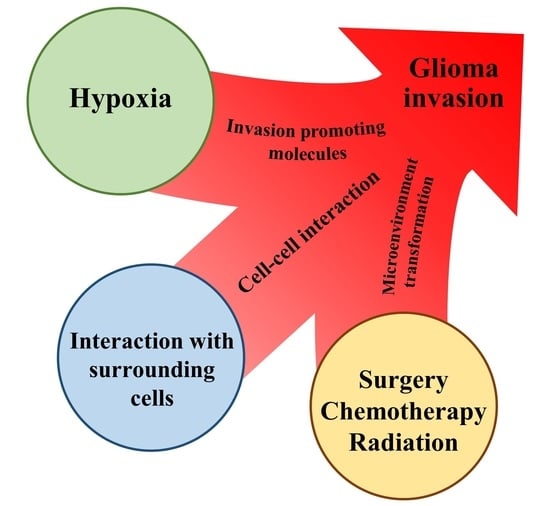
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Physical Environment
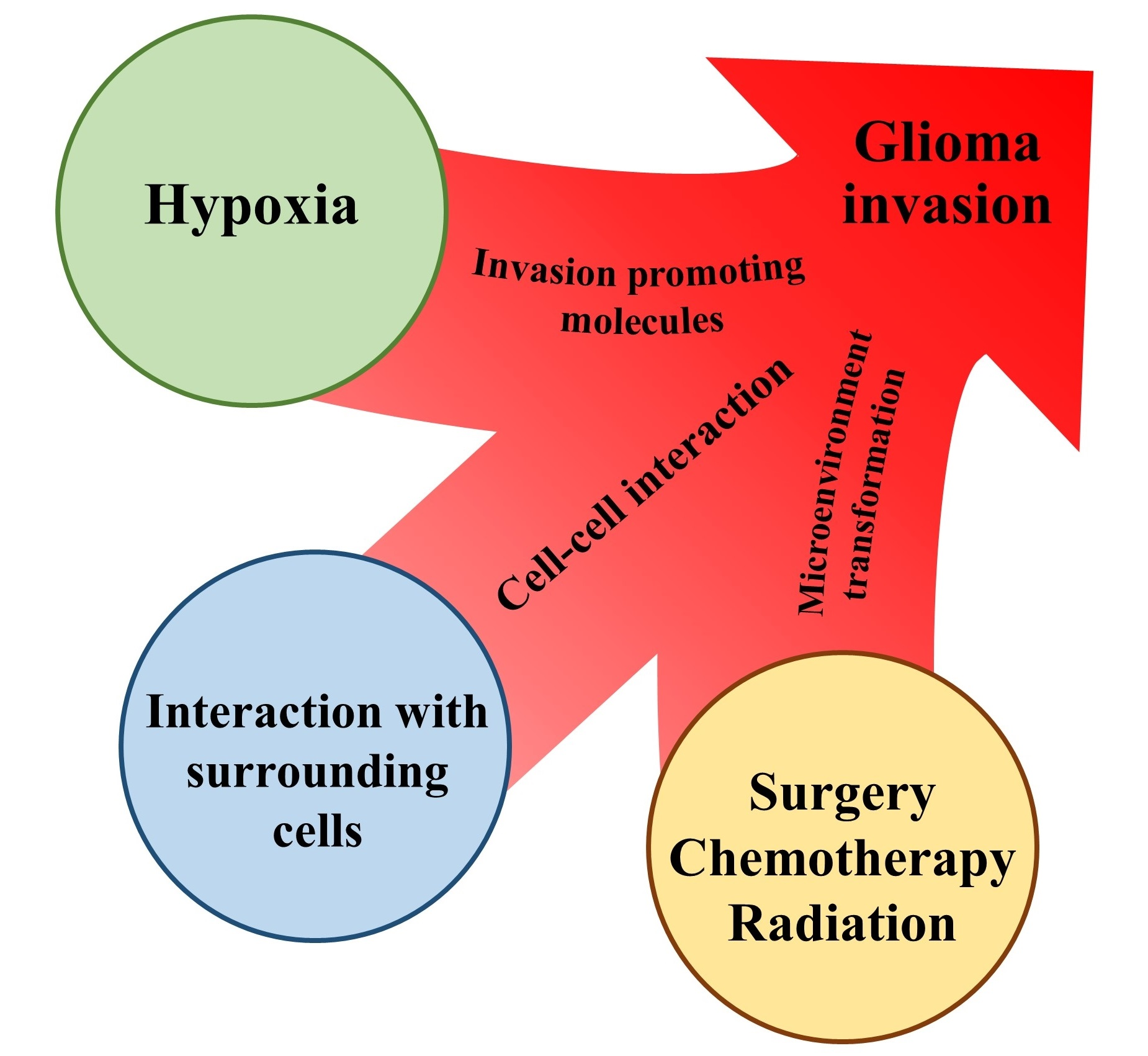
2.1. Hypoxia
2.1.1. Signaling Pathway Associated with Hypoxia-Inducible Factor (HIF)
2.1.2. GIC-Based Mechanism
2.1.3. Changing of Intracellular Metabolic Systems
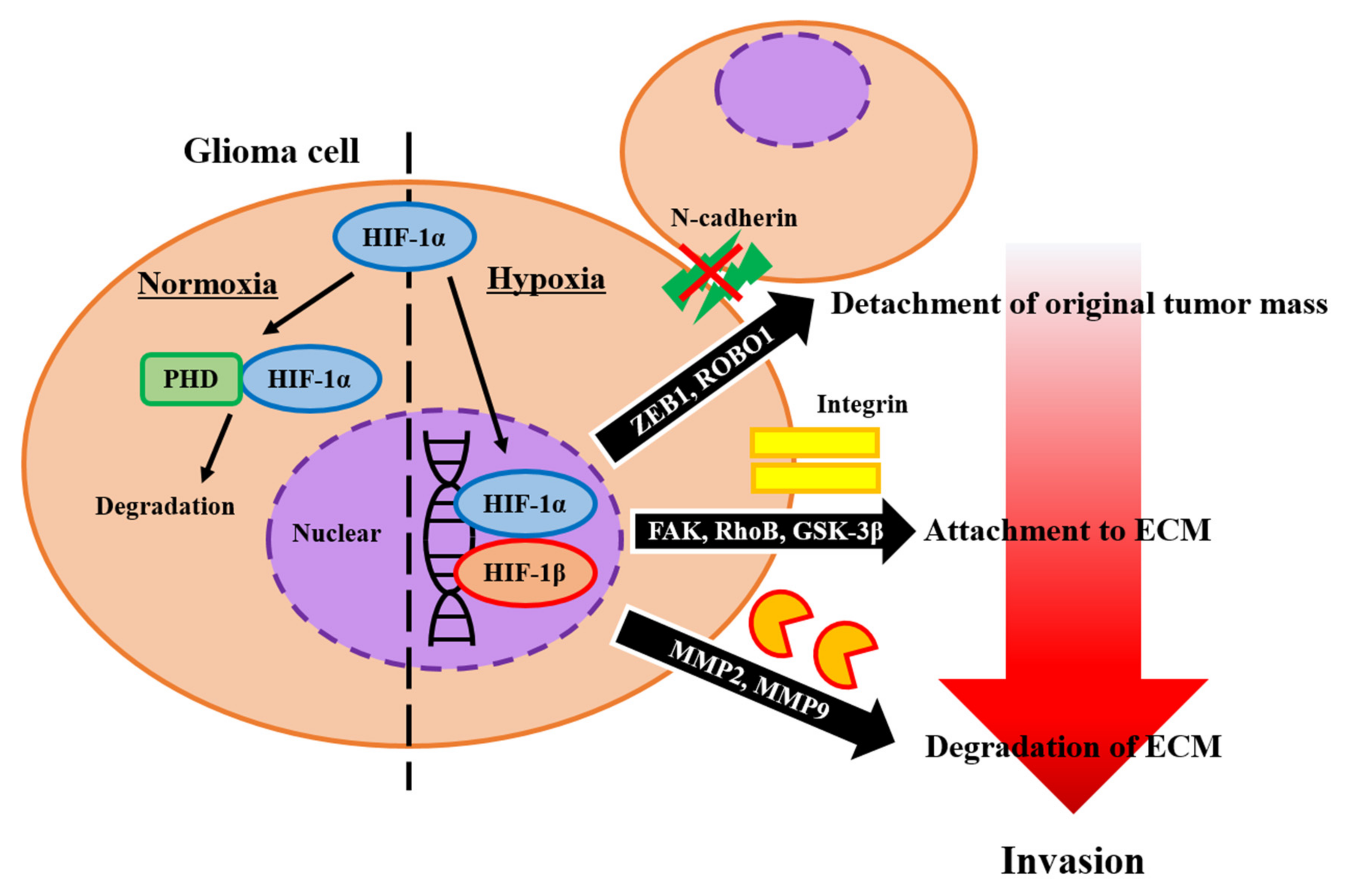
2.2. Extracellular Matrix
2.2.1. Normal Brain ECM
2.2.2. ECM Promoting Invasion
2.2.3. ECM Inhibiting Invasion
2.2.4. Enzymes Associated with ECM Foundation and Glioma Invasion
3. Interaction with Surrounding
3.1. Astrocyte and Oligodendrocyte
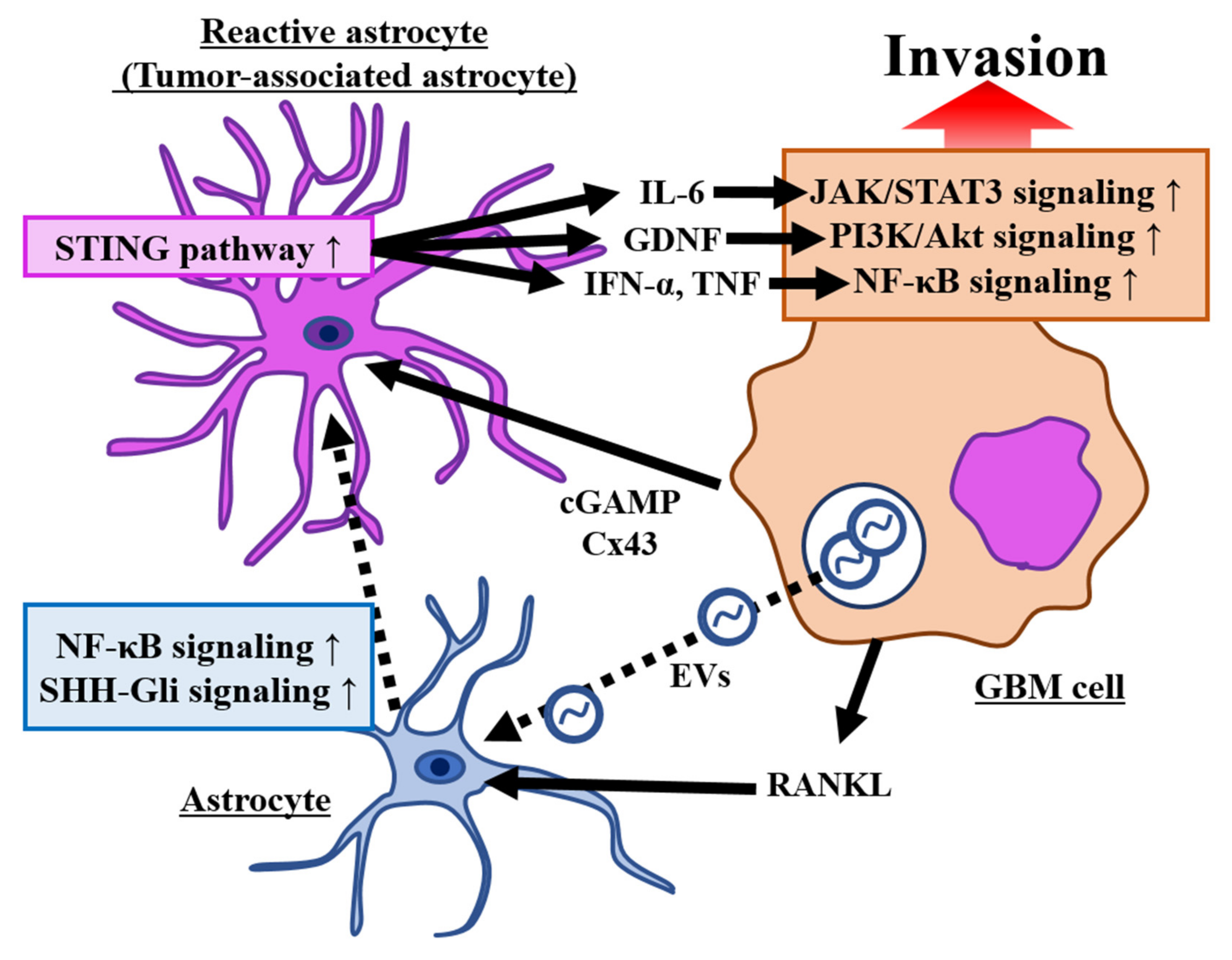
3.1.1. Role of Tumor-Associated Astrocytes (TAAs)
3.1.2. Activation of the Signaling Pathway Associated with Invasion by TAAs
3.1.3. The Crosstalk between Glioma Cells and Astrocytes via Extracellular Vesicles (EVs) Contributes to Glioma Invasion
3.2. Microglia
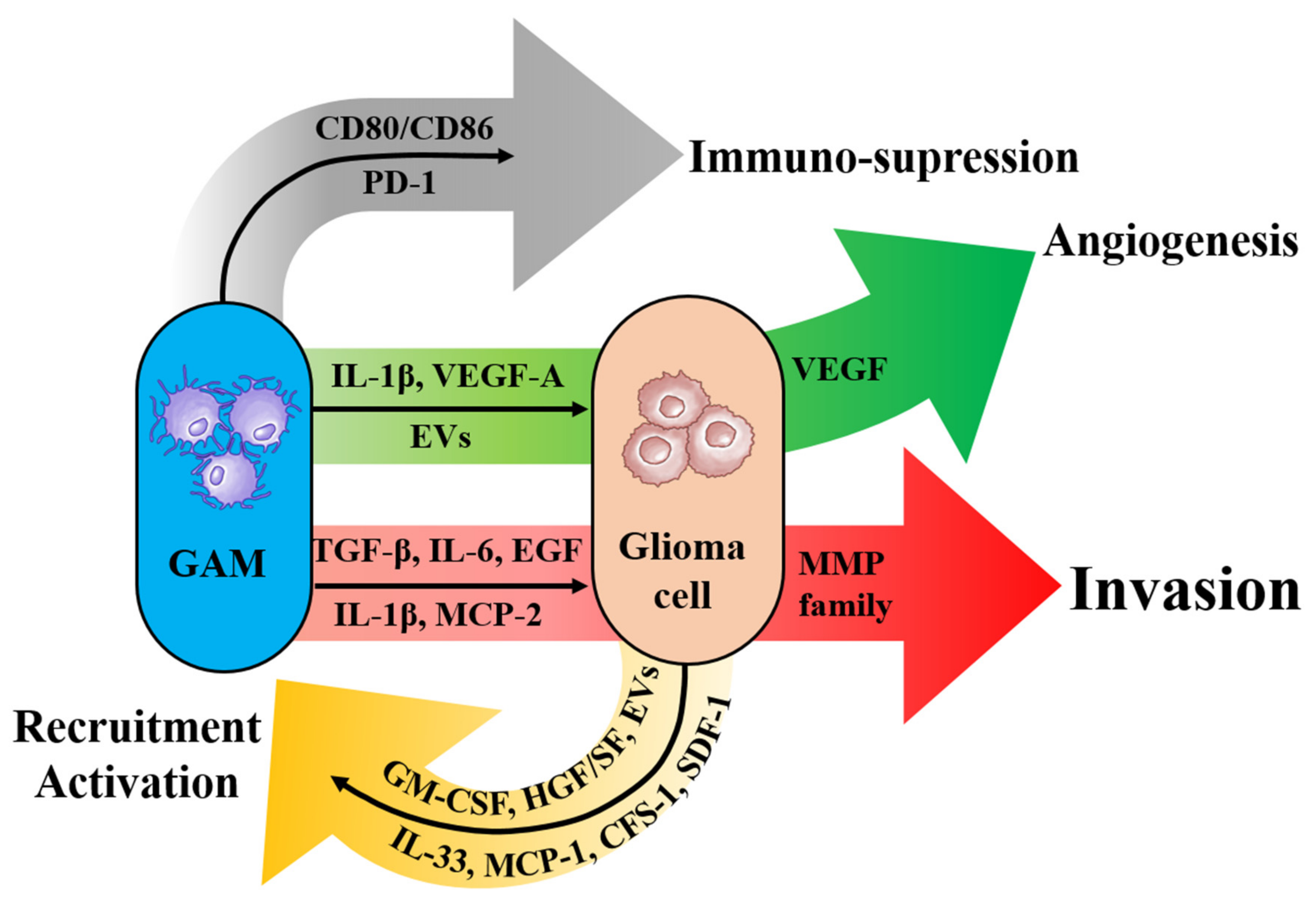
3.2.1. Introduction of Glioma-Associated Macrophages/Microglia (GAM)
3.2.2. Promotion of Tumor Invasion by GAMs
3.2.3. Roles of GAMs for Changing Microenvironments
3.3. Glioma Cell
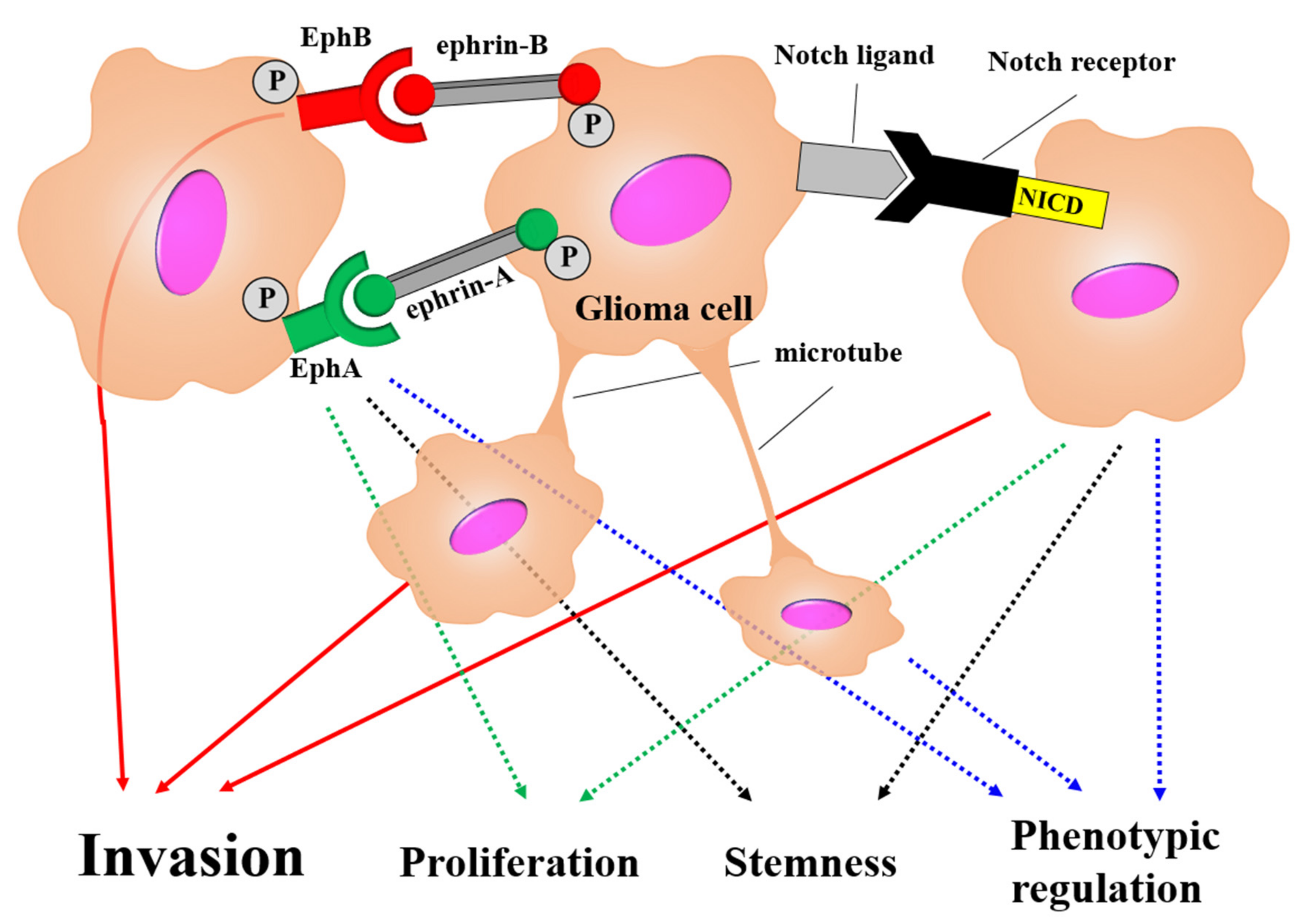
3.3.1. Tumor Cell Membrane Tube Network
3.3.2. Ephs–Ephrins Pathway
3.3.3. Notch Signal
4. Alteration of the Microenvironment Affected by Therapy
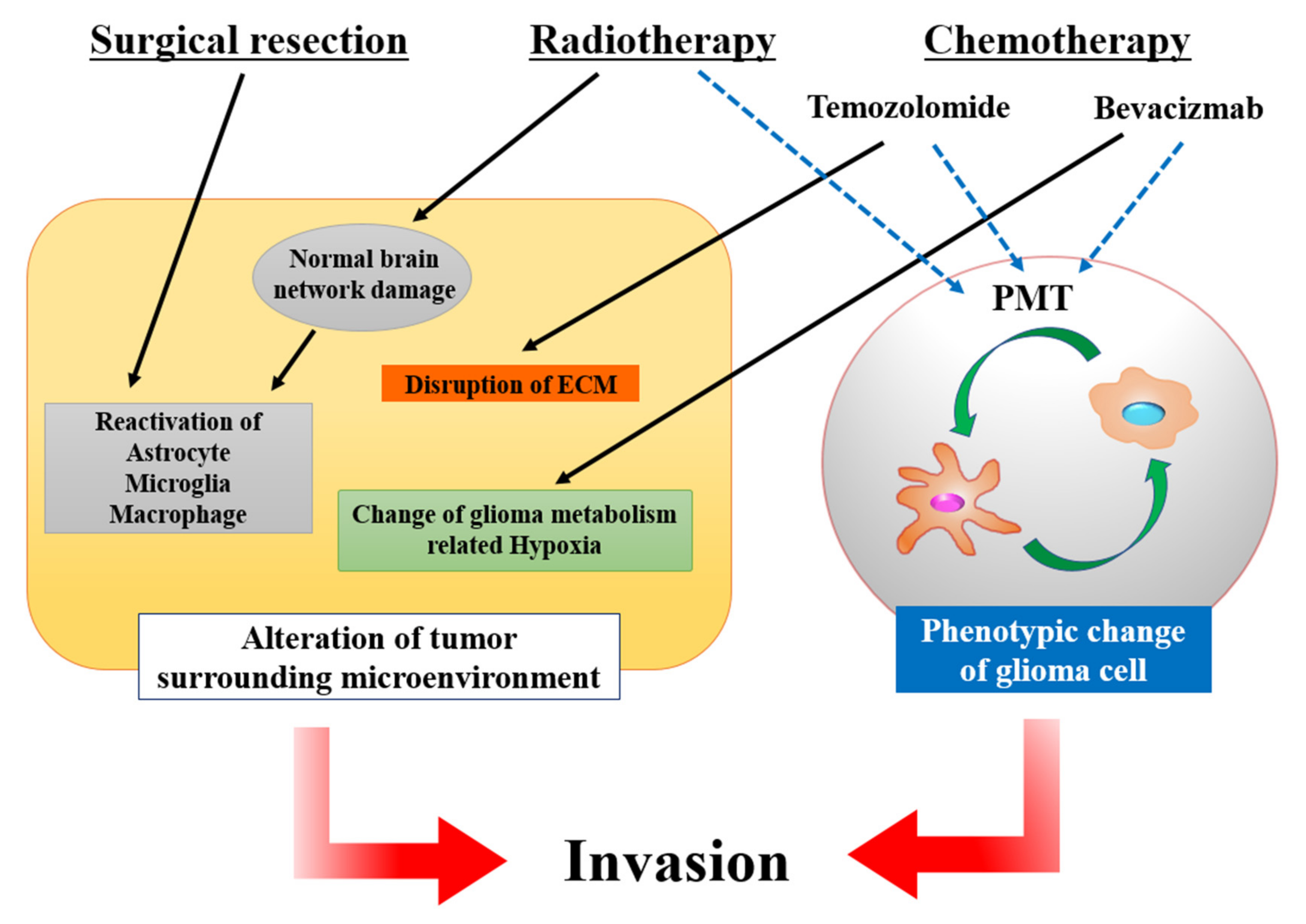
4.1. Operation
4.2. Chemotherapy
4.2.1. Temozolomide (TMZ)
4.2.2. Bevacizumab (BEV)
4.3. Radiation
5. Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro-Oncology 2021, 23, iii1–iii105. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Brain Tumor Registry of Japan (2005-2008). Neurol. Med. Chir. 2017, 57, 9–102. [CrossRef]
- Burger, P.C.; Heinz, E.R.; Shibata, T.; Kleihues, P. Topographic anatomy and CT correlations in the untreated glioblastoma multiforme. J. Neurosurg. 1988, 68, 698–704. [Google Scholar] [CrossRef]
- Nakada, M.; Nakada, S.; DeMuth, T.; Tran, N.L.; Hoelzinger, D.; Berens, M.E. Molecular targets of glioma invasion. Cell. Mol. Life Sci. 2007, 64, 458–478. [Google Scholar] [CrossRef]
- Nakada, M.; Kita, D.; Teng, L.; Pyko, I.V.; Watanabe, T.; Hayashi, Y.; Hamada, J.-I. Receptor Tyrosine Kinases: Principles and Functions in Glioma Invasion. Aldehyde Dehydrogenases 2020, 1202, 151–178. [Google Scholar] [CrossRef]
- Zhang, J.; Furuta, T.; Sabit, H.; Tamai, S.; Jiapaer, S.; Dong, Y.; Kinoshita, M.; Uchida, Y.; Ohtsuki, S.; Terasaki, T.; et al. Gelsolin inhibits malignant phenotype of glioblastoma and is regulated by miR-654-5p and miR-450b-5p. Cancer Sci. 2020, 111, 2413–2422. [Google Scholar] [CrossRef]
- Ikushima, H.; Todo, T.; Ino, Y.; Takahashi, M.; Miyazawa, K.; Miyazono, K. Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell 2009, 5, 504–514. [Google Scholar] [CrossRef] [Green Version]
- Tamase, A.; Muraguchi, T.; Naka, K.; Tanaka, S.; Kinoshita, M.; Hoshii, T.; Ohmura, M.; Shugo, H.; Ooshio, T.; Nakada, M.; et al. Identification of tumor-initiating cells in a highly aggressive brain tumor using promoter activity of nucleostemin. Proc. Natl. Acad. Sci. USA 2009, 106, 17163–17168. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Furuta, T.; Sabit, H.; Kitabayashi, T.; Jiapaer, S.; Kobayashi, M.; Ino, Y.; Todo, T.; Teng, L.; Hirao, A.; et al. Identi-fication of antipsychotic drug fluspirilene as a potential anti-glioma stem cell drug. Oncotarget 2017, 8, 111728–111741. [Google Scholar] [CrossRef]
- Kitabayashi, T.; Dong, Y.; Furuta, T.; Sabit, H.; Jiapaer, S.; Zhang, J.; Zhang, G.; Hayashi, Y.; Kobayashi, M.; Domoto, T.; et al. Identification of GSK3β inhibitor kenpaullone as a temozolomide enhancer against glioblastoma. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Zhang, G.; Tanaka, S.; Jiapaer, S.; Sabit, H.; Tamai, S.; Kinoshita, M.; Nakada, M. RBPJ contributes to the malignancy of glio-blastoma and induction of proneural-mesenchymal transition via IL-6-STAT3 pathway. Cancer Sci. 2020, 111, 4166–4176. [Google Scholar] [CrossRef]
- Evans, S.M.; Judy, K.D.; Dunphy, I.; Jenkins, W.T.; Hwang, W.-T.; Nelson, P.T.; Lustig, R.A.; Jenkins, K.; Magarelli, D.P.; Hahn, S.M.; et al. Hypoxia Is Important in the Biology and Aggression of Human Glial Brain Tumors. Clin. Cancer Res. 2004, 10, 8177–8184. [Google Scholar] [CrossRef] [Green Version]
- Evans, S.M.; Judy, K.D.; Dunphy, I.; Jenkins, W.T.; Nelson, P.T.; Collins, R.; Wileyto, E.P.; Jenkins, K.; Hahn, S.M.; Stevens, C.W.; et al. Comparative Measurements of Hypoxia in Human Brain Tumors Using Needle Electrodes and EF5 Binding. Cancer Res. 2004, 64, 1886–1892. [Google Scholar] [CrossRef] [Green Version]
- Evans, S.M.; Jenkins, K.W.; Jenkins, W.T.; Dilling, T.; Judy, K.D.; Schrlau, A.; Judkins, A.; Hahn, S.M.; Koch, C.J. Imaging and Analytical Methods as Applied to the Evaluation of Vasculature and Hypoxia in Human Brain Tumors. Radiat. Res. 2008, 170, 677–690. [Google Scholar] [CrossRef] [Green Version]
- Raza, S.M.; Lang, F.F.; Aggarwal, B.B.; Fuller, G.N.; Wildrick, D.M.; Sawaya, R. Necrosis and Glioblastoma: A Friend or a Foe? A Review and a Hypothesis. Neurosurgery 2002, 51, 2–13. [Google Scholar] [CrossRef]
- Brat, D.J.; Castellano-Sanchez, A.A.; Hunter, S.B.; Pecot, M.; Cohen, C.; Hammond, E.H.; Devi, S.N.; Kaur, B.; van Meir, E.G. Pseudopalisades in Glioblastoma Are Hypoxic, Express Extracellular Matrix Proteases, and Are Formed by an Actively Migrating Cell Population. Cancer Res. 2004, 64, 920–927. [Google Scholar] [CrossRef] [Green Version]
- Kaur, B.; Khwaja, F.W.; Severson, E.A.; Matheny, S.L.; Brat, D.J.; van Meir, E.G. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro-Oncology 2005, 7, 134–153. [Google Scholar] [CrossRef] [Green Version]
- Koritzinsky, M.; Seigneuric, R.; Magagnin, M.G.; Beucken, T.V.D.; Lambin, P.; Wouters, B.G. The hypoxic proteome is influenced by gene-specific changes in mRNA translation. Radiother. Oncol. 2005, 76, 177–186. [Google Scholar] [CrossRef]
- Jögi, A.; Øra, I.; Nilsson, H.; Lindeheim, Å.; Makino, Y.; Poellinger, L.; Axelson, H.; Påhlman, S. Hypoxia alters gene expression in human neuroblastoma cells toward an immature and neural crest-like phenotype. Proc. Natl. Acad. Sci. USA 2002, 99, 7021–7026. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.-H.; Zheng, J.Z.; Leung, S.W.; Roe, R.; Semenza, G.L. Transactivation and inhibitory domains of hypoxia-inducible factor 1alpha. Modulation of transcriptional activity by oxygen tension. J. Biol. Chem. 1997, 272, 19253–19260. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Zagzag, D.; Zhong, H.; Scalzitti, J.M.; Laughner, E.; Simons, J.W.; Semenza, G.L. Expression of hypoxia-inducible factor 1alpha in brain tumors: Association with angiogenesis, invasion, and progression. Cancer 2000, 88, 2606–2618. [Google Scholar] [CrossRef]
- Krishnamachary, B.; Berg-Dixon, S.; Kelly, B.; Agani, F.; Feldser, D.; Ferreira, G.C.; Iyer, N.; LaRusch, J.; Pak, B.; Taghavi, P.; et al. Regulation of colon carcinoma cell invasion by hypoxia-inducible factor 1. Cancer Res. 2003, 63, 1138–1143. [Google Scholar]
- Bindra, R.S.; Crosby, M.E.; Glazer, P.M. Regulation of DNA repair in hypoxic cancer cells. Cancer Metastasis Rev. 2007, 26, 249–260. [Google Scholar] [CrossRef]
- Siebzehnrubl, F.A.; Silver, D.J.; Tugertimur, B.; Deleyrolle, L.P.; Siebzehnrubl, D.; Sarkisian, M.R.; Devers, K.G.; Yachnis, A.T.; Kupper, M.D.; Neal, D.; et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol. Med. 2013, 5, 1196–1212. [Google Scholar] [CrossRef]
- Joseph, J.V.; Conroy, S.; Pavlov, K.; Sontakke, P.; Tomar, T.; Eggens-Meijer, E.; Balasubramaniyan, V.; Wagemakers, M.; den Dunnen, W.F.; Kruyt, F.A. Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift me-diated by the HIF1α-ZEB1 axis. Cancer Lett. 2015, 359, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, W.; Li, Y.; Alvarez, A.; Li, Z.; Wang, Y.; Song, L.; Lv, D.; Nakano, I.; Hu, B.; et al. SHP-2-upregulated ZEB1 is important for PDGFRα-driven glioma epithelial–mesenchymal transition and invasion in mice and humans. Oncogene 2016, 35, 5641–5652. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Rhee, J.; Mahfooz, N.S.; Arregui, C.; Lilien, J.; Balsamo, J.; VanBerkum, M.F. Activation of the repulsive receptor Roundabout inhibits N-cadherin-mediated cell adhesion. Nat. Cell Biol. 2002, 4, 798–805. [Google Scholar] [CrossRef]
- Burridge, K.; Chrzanowska-Wodnicka, M. Focal adhesions, contractility, and signaling. Annu. Rev. Cell Dev. Biol. 1996, 12, 463–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, E.J. Integrin-associated proteins. Curr. Opin. Cell Biol. 2002, 14, 603–607. [Google Scholar] [CrossRef]
- Skuli, N.; Monferran, S.; Delmas, C.; Favre, G.; Bonnet, J.; Toulas, C.; Cohen-Jonathan Moyal, E. Alphavbeta3/alphavbeta5 integrins-FAK-RhoB: A novel pathway for hypoxia regulation in glioblastoma. Cancer Res. 2009, 69, 3308–3316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skuli, N.; Monferran, S.; Delmas, C.; Lajoie-Mazenc, I.; Favre, G.; Toulas, C.; Cohen-Jonathan-Moyal, E. Activation of RhoB by Hypoxia Controls Hypoxia-Inducible Factor-1α Stabilization through Glycogen Synthase Kinase-3 in U87 Glioblastoma Cells. Cancer Res. 2006, 66, 482–489. [Google Scholar] [CrossRef] [Green Version]
- Rao, J.S. Molecular mechanisms of glioma invasiveness: The role of proteases. Nat. Cancer 2003, 3, 489–501. [Google Scholar] [CrossRef]
- Fujiwara, S.; Nakagawa, K.; Harada, H.; Nagato, S.; Furukawa, K.; Teraoka, M.; Seno, T.; Oka, K.; Iwata, S.; Ohnishi, T. Silencing hypoxia-inducible factor-1alpha inhibits cell migration and invasion under hypoxic environment in malignant gliomas. Int. J. Oncol. 2007, 30, 793–802. [Google Scholar]
- Prasad, P.; Mittal, S.A.; Chongtham, J.; Mohanty, S.; Srivastava, T. Hypoxia-Mediated Epigenetic Regulation of Stemness in Brain Tumor Cells. Stem Cells 2017, 35, 1468–1478. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, T.; Otani, Y.; Kurozumi, K.; Date, I. Phenotypic Transition as a Survival Strategy of Glioma. Neurol. Medico-Chirurgica 2016, 56, 387–395. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Talasila, K.M.; Røsland, G.V.; Hagland, H.R.; Eskilsson, E.; Flønes, I.H.; Fritah, S.; Azuaje, F.; Atai, N.; Harter, P.N.; Mittelbronn, M.; et al. The angiogenic switch leads to a metabolic shift in human glioblastoma. Neuro-Oncology 2016, 19, 383–393. [Google Scholar] [CrossRef] [Green Version]
- Kathagen-Buhmann, A.; Schulte, A.; Weller, J.; Holz, M.; Herold-Mende, C.; Glass, R.; Lamszus, K. Glycolysis and the pentose phosphate pathway are differentially associated with the dichotomous regulation of glioblastoma cell migration versus pro-liferation. Neuro-Oncology 2016, 18, 1219–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda-Gonçalves, V.; Granja, S.; Martinho, O.; Honavar, M.; Pojo, M.; Costa, B.M.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; et al. Hypoxia-mediated upregulation of MCT1 expression supports the glycolytic phenotype of glioblastomas. Oncotarget 2016, 7, 46335–46353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kita, D.; Takino, T.; Nakada, M.; Takahashi, T.; Yamashita, J.; Sato, H. Expression of dominant-negative form of Ets-1 sup-presses fibronectin-stimulated cell adhesion and migration through down-regulation of integrin alpha5 expression in U251 glioma cell line. Cancer Res. 2001, 61, 7985–7991. [Google Scholar] [PubMed]
- Lau, L.W.; Cua, R.; Keough, M.B.; Haylock-Jacobs, S.; Yong, V.W. Pathophysiology of the brain extracellular matrix: A new target for remyelination. Nat. Rev. Neurosci. 2013, 14, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Franco, S.J.; Müller, U. Extracellular matrix functions during neuronal migration and lamination in the mammalian central nervous system. Dev. Neurobiol. 2011, 71, 889–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerrisk, M.E.; Cingolani, L.A.; Koleske, A.J. ECM receptors in neuronal structure, synaptic plasticity, and behavior. Progress in Brain Res. 2014, 214, 101–131. [Google Scholar] [CrossRef] [Green Version]
- Kalebic, N.; Gilardi, C.; Stepien, B.; Wilsch-Bräuninger, M.; Long, K.; Namba, T.; Florio, M.; Langen, B.; Lombardot, B.; Shevchenko, A.; et al. Neocortical Expansion Due to Increased Proliferation of Basal Progenitors Is Linked to Changes in Their Morphology. Cell Stem Cell 2019, 24, 535–550.e9. [Google Scholar] [CrossRef] [Green Version]
- Long, K.R.; Huttner, W.B. The Role of the Extracellular Matrix in Neural Progenitor Cell Proliferation and Cortical Folding During Human Neocortex Development. Front. Cell. Neurosci. 2022, 15, 804649. [Google Scholar] [CrossRef]
- Barnes, J.M.; Przybyla, L.; Weaver, V.M. Tissue mechanics regulate brain development, homeostasis and disease. J. Cell Sci. 2017, 130, 71–82. [Google Scholar] [CrossRef] [Green Version]
- Mohiuddin, E.; Wakimoto, H. Extracellular matrix in glioblastoma: Opportunities for emerging therapeutic approaches. Am. J. Cancer Res. 2021, 11, 3742–3754. [Google Scholar]
- Belousov, A.; Titov, S.; Shved, N.; Garbuz, M.; Malykin, G.; Gulaia, V.; Kagansky, A.; Kumeiko, V. The Extracellular Matrix and Biocompatible Materials in Glioblastoma Treatment. Front. Bioeng. Biotechnol. 2019, 7, 341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, M.; Nambu, E.; Furuyama, N.; Yoshida, Y.; Takino, T.; Hayashi, Y.; Sato, H.; Sai, Y.; Tsuji, T.; Miyamoto, K.-I.; et al. Integrin α3 is overexpressed in glioma stem-like cells and promotes invasion. Br. J. Cancer 2013, 108, 2516–2524. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Erfani, S.; Liu, Z.; Jia, C.; Chen, Y.; Xu, B.; Deng, X.; Alfáro, J.E.; Chen, L.; Napier, D.; et al. CD151-α3β1 integrin complexes are prognostic markers of glioblastoma and cooperate with EGFR to drive tumor cell motility and invasion. Oncotarget 2015, 6, 29675–29693. [Google Scholar] [CrossRef] [Green Version]
- Blandin, A.F.; Noulet, F.; Renner, G.; Mercier, M.C.; Choulier, L.; Vauchelles, R.; Ronde, P.; Carreiras, F.; Etienne-Selloum, N.; Vereb, G.; et al. Glioma cell dispersion is driven by α5 integrin-mediated cell-matrix and cell-cell interactions. Cancer Lett. 2016, 376, 328–338. [Google Scholar] [CrossRef] [Green Version]
- Nandhu, M.S.; Kwiatkowska, A.; Bhaskaran, V.; Hayes, J.; Hu, B.; Viapiano, M.S. Tumor-derived fibulin-3 activates pro-invasive NF-κB signaling in glioblastoma cells and their microenvironment. Oncogene 2017, 36, 4875–4886. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, T.A.; de Juan Pardo, E.M.; Kumar, S. The Mechanical Rigidity of the Extracellular Matrix Regulates the Structure, Motility, and Proliferation of Glioma Cells. Cancer Res. 2009, 69, 4167–4174. [Google Scholar] [CrossRef] [Green Version]
- Miroshnikova, Y.A.; Mouw, J.K.; Barnes, J.M.; Pickup, M.W.; Lakins, J.N.; Kim, Y.; Lobo, K.; Persson, A.; Reis, G.F.; McKnight, T.R.; et al. Tissue mechanics promote IDH1-dependent HIF1α–tenascin C feedback to regulate glioblastoma aggression. Nat. Cell Biol. 2016, 18, 1336–1345. [Google Scholar] [CrossRef]
- Nakada, M.; Yamada, A.; Takino, T.; Miyamori, H.; Takahashi, T.; Yamashita, J.; Sato, H. Suppression of membrane-type 1 matrix metalloproteinase (MMP)-mediated MMP-2 activation and tumor invasion by testican 3 and its splicing variant gene product, N-Tes. Cancer Res. 2001, 61, 8896–8902. [Google Scholar]
- Serres, E.; Debarbieux, F.; Stanchi, F.; Maggiorella, L.; Grall, D.; Turchi, L.; Burel-Vandenbos, F.; Figarella-Branger, D.; Virolle, T.; Rougon, G.; et al. Fibronectin expression in glioblastomas promotes cell cohesion, collective invasion of basement membrane in vitro and orthotopic tumor growth in mice. Oncogene 2013, 33, 3451–3462. [Google Scholar] [CrossRef]
- Shannon, S.; Vaca, C.; Jia, D.; Entersz, I.; Schaer, A.; Carcione, J.; Weaver, M.; Avidar, Y.; Pettit, R.; Nair, M.; et al. Dexame-thasone-Mediated Activation of Fibronectin Matrix Assembly Reduces Dispersal of Primary Human Glioblastoma Cells. PLoS ONE 2015, 10, e0135951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Han, L.; Dong, Y.; Tan, Y.; Li, Y.; Zhao, M.; Xie, H.; Ju, H.; Wang, H.; Zhao, Y.; et al. EGFRvIII/integrin β3 interaction in hypoxic and vitronectinenriching microenvironment promote GBM progression and metastasis. Oncotarget 2016, 7, 4680–4694. [Google Scholar] [CrossRef] [PubMed]
- Nakada, M.; Okada, Y.; Yamashita, J. The role of matrix metalloproteinases in glioma invasion. Front. Biosci. 2003, 8, e261–e269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, M.; Kita, D.; Futami, K.; Yamashita, J.; Fujimoto, N.; Sato, H.; Okada, Y. Roles of membrane type 1 matrix metallo-proteinase and tissue inhibitor of metalloproteinases 2 in invasion and dissemination of human malignant glioma. J. Neurosurg. 2001, 94, 464–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chintala, S.K.; Sawaya, R.; Gokaslan, Z.L.; Rao, J.S. Modulation of matrix metalloprotease-2 and invasion in human glioma cells by alpha 3 beta 1 integrin. Cancer Lett. 1996, 103, 201–208. [Google Scholar] [CrossRef]
- Vos, C.; Gartner, S.; Ransohoff, R.M.; McArthur, J.C.; Wahl, L.; Sjulson, L.; Hunter, E.; Conant, K. Matrix metalloprotease-9 release from monocytes increases as a function of differentiation: Implications for neuroinflammation and neurodegeneration. J. Neuroimmunol. 2000, 109, 221–227. [Google Scholar] [CrossRef]
- Eeckhout, Y.; Vaes, G. Further studies on the activation of procollagenase, the latent precursor of bone collagenase. Effects of lysosomal cathepsin B, plasmin and kallikrein, and spontaneous activation. Biochem. J. 1977, 166, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Choe, G.; Park, J.K.; Jouben-Steele, L.; Kremen, T.J.; Liau, L.; Vinters, H.V.; Cloughesy, T.F.; Mischel, P.S. Active matrix metalloproteinase 9 expression is associated with primary glioblastoma subtype. Clin. Cancer Res. 2002, 8, 2894–2901. [Google Scholar]
- Gomez, D.E.; Alonso, D.F.; Yoshiji, H.; Thorgeirsson, U.P. Tissue inhibitors of metalloproteinases: Structure, regulation and biological functions. Eur. J. Cell Biol. 1997, 74, 111–122. [Google Scholar]
- Fillmore, H.L.; VanMeter, T.E.; Broaddus, W.C. Membrane-type matrix metalloproteinases (MT-MMPs): Expression and func-tion during glioma invasion. J. Neurooncol. 2001, 53, 187–202. [Google Scholar] [CrossRef]
- Nakada, M.; Nakamura, H.; Ikeda, E.; Fujimoto, N.; Yamashita, J.; Sato, H.; Seiki, M.; Okada, Y. Expression and Tissue Localization of Membrane-Type 1, 2, and 3 Matrix Metalloproteinases in Human Astrocytic Tumors. Am. J. Pathol. 1999, 154, 417–428. [Google Scholar] [CrossRef] [Green Version]
- Nagase, H.; Woessner, J.F., Jr. Matrix metalloproteinases. J. Biol. Chem. 1999, 274, 21491–21494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yana, I.; Weiss, S.J. Regulation of Membrane Type-1 Matrix Metalloproteinase Activation by Proprotein Convertases. Mol. Biol. Cell 2000, 11, 2387–2401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łukaszewicz-Zając, M.; Dulewicz, M.; Mroczko, B. A Disintegrin and Metalloproteinase (ADAM) Family: Their Significance in Malignant Tumors of the Central Nervous System (CNS). Int. J. Mol. Sci. 2021, 22, 10378. [Google Scholar] [CrossRef] [PubMed]
- Goddard, D.R.; Bunning, R.A.; Woodroofe, M.N. Astrocyte and endothelial cell expression of ADAM 17 (TACE) in adult human CNS. Glia 2001, 34, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Ikeda, E.; Okada, A.; Ohtsuka, T.; Shimoda, M.; Shiomi, T.; Yoshida, K.; Nakada, M.; Ohuchi, E.; Okada, Y. ADAM12 Is Selectively Overexpressed in Human Glioblastomas and Is Associated with Glioblastoma Cell Proliferation and Shedding of Heparin-Binding Epidermal Growth Factor. Am. J. Pathol. 2004, 165, 1743–1753. [Google Scholar] [CrossRef] [Green Version]
- Kanakis, D.; Lendeckel, U.; Theodosiou, P.; Dobrowolny, H.; Mawrin, C.; Keilhoff, G.; Bukowska, A.; Dietzmann, K.; Bogerts, B.; Bernstein, H.G. ADAM 12: A putative marker of oligodendrogliomas? Dis. Markers 2013, 34, 81–91. [Google Scholar] [CrossRef]
- Cesarini, V.; Silvestris, D.A.; Tassinari, V.; Tomaselli, S.; Alon, S.; Eisenberg, E.; Locatelli, F.; Gallo, A. ADAR2/miR-589-3p axis controls glioblastoma cell migration/invasion. Nucleic Acid Res. 2018, 46, 2045–2059. [Google Scholar] [CrossRef]
- Held-Feindt, J.; Paredes, E.B.; Blömer, U.; Seidenbecher, C.; Stark, A.M.; Mehdorn, H.M.; Mentlein, R. Matrix-degrading pro-teases ADAMTS4 and ADAMTS5 (disintegrins and metalloproteinases with thrombospondin motifs 4 and 5) are expressed in human glioblastomas. Int. J. Cancer 2006, 118, 55–61. [Google Scholar] [CrossRef]
- Held-Feindt, J.; Mentlein, R. CD70/CD27 ligand, a member of the TNF family, is expressed in human brain tumors. Int. J. Cancer 2002, 98, 352–356. [Google Scholar] [CrossRef]
- Nakada, M.; Miyamori, H.; Kita, D.; Takahashi, T.; Yamashita, J.; Sato, H.; Miura, R.; Yamaguchi, Y.; Okada, Y. Human gli-oblastomas overexpress ADAMTS-5 that degrades brevican. Acta Neuropathol. 2005, 110, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix Metalloproteinase Inhibitors and Cancer—Trials and Tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Levin, V.A.; Phuphanich, S.; Yung, W.K.A.; Forsyth, P.A.; del Maestro, R.; Perry, J.R.; Fuller, G.N.; Baillet, M. Randomized, double-blind, placebo-controlled trial of marimastat in glioblastoma multiforme patients following surgery and irradiation. J. Neuro-Oncol. 2006, 78, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Ohi, H.; Sugimura, M.; Shinohara, H.; Fujii, T.; Terao, T. Inhibition of in vitro ovarian cancer cell invasion by modulation of urokinase-type plasminogen activator and cathepsin B. Cancer Res. 1992, 52, 3610–3614. [Google Scholar] [PubMed]
- Kostoulas, G.; Lang, A.; Nagase, H.; Baici, A. Stimulation of angiogenesis through cathepsin B inactivation of the tissue in-hibitors of matrix metalloproteinases. FEBS Lett. 1999, 455, 286–290. [Google Scholar] [CrossRef] [Green Version]
- Sivaparvathi, M.; Sawaya, R.; Wang, S.W.; Rayford, A.; Yamamoto, M.; Liottat, L.A.; Nicolson, G.L.; Rao, J.S. Overexpression and localization of cathepsin B during the progression of human gliomas. Clin. Exp. Metastasis 1995, 13, 49–56. [Google Scholar] [CrossRef]
- Mikkelsen, T.; Yan, P.-S.; Ho, K.-L.; Sameni, M.; Sloane, B.F.; Rosenblum, M.L. Immunolocalization of cathepsin B in human glioma: Implications for tumor invasion and angiogenesis. J. Neurosurg. 1995, 83, 285–290. [Google Scholar] [CrossRef]
- Reith, A.; Rucklidge, G.J. Invasion of brain tissue by primary glioma: Evidence for the involvement of urokinase-type plas-minogen activator as an activator of type IV collagenase. Biochem. Biophys. Res. Commun. 1992, 186, 348–354. [Google Scholar] [CrossRef]
- Koshelnick, Y.; Ehart, M.; Hufnagl, P.; Heinrich, P.C.; Binder, B.R. Urokinase receptor is associated with the components of the JAK1/STAT1 signaling pathway and leads to activation of this pathway upon receptor clustering in the human kidney epi-thelial tumor cell line TCL-598. J. Biol. Chem. 1997, 272, 28563–28567. [Google Scholar] [CrossRef] [Green Version]
- Dumler, I.; Weis, A.; Mayboroda, O.; Maasch, C.; Jerke, U.; Haller, H.; Gulba, D.C. The Jak/Stat Pathway and Urokinase Receptor Signaling in Human Aortic Vascular Smooth Muscle Cells. J. Biol. Chem. 1998, 273, 315–321. [Google Scholar] [CrossRef] [Green Version]
- Aguirre-Ghiso, J.A.; Liu, D.; Mignatti, A.; Kovalski, K.; Ossowski, L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol. Biol. Cell 2001, 12, 863–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasi, F.; Carmeliet, P. uPAR: A versatile signalling orchestrator. Nat. Rev. Mol. Cell Biol. 2002, 3, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sawaya, R.; Mohanam, S.; Bindal, A.K.; Bruner, J.M.; Oka, K.; Rao, V.H.; Tomonaga, M.; Nicolson, G.L.; Rao, J.S. Expression and localization of urokinase-type plasminogen activator in human astrocytomas in vivo. Cancer Res. 1994, 54, 3656–3661. [Google Scholar] [PubMed]
- Gladson, C.L.; Pijuan-Thompson, V.; Olman, M.A.; Gillespie, G.Y.; Yacoub, I.Z. Up-regulation of urokinase and urokinase receptor genes in malignant astrocytoma. Am. J. Pathol. 1995, 146, 1150–1160. [Google Scholar]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2012, 60, 502–514. [Google Scholar] [CrossRef]
- Vasile, F.; Dossi, E.; Rouach, N. Human astrocytes: Structure and functions in the healthy brain. Anat. Embryol. 2017, 222, 2017–2029. [Google Scholar] [CrossRef] [Green Version]
- Galvao, R.P.; Kasina, A.; McNeill, R.S.; Harbin, J.E.; Foreman, O.; Verhaak, R.G.W.; Nishiyama, A.; Miller, C.R.; Zong, H. Transformation of quiescent adult oligodendrocyte precursor cells into malignant glioma through a multistep reactivation process. Proc. Natl. Acad. Sci. USA 2014, 111, E4214–E4223. [Google Scholar] [CrossRef] [Green Version]
- Sugiarto, S.; Persson, A.I.; Munoz, E.G.; Waldhuber, M.; Lamagna, C.; Andor, N.; Hanecker, P.; Ayers-Ringler, J.; Phillips, J.; Siu, J.; et al. Asymmetry-Defective Oligodendrocyte Progenitors Are Glioma Precursors. Cancer Cell 2011, 20, 328–340. [Google Scholar] [CrossRef] [Green Version]
- Hide, T.; Shibahara, I.; Kumabe, T. Novel concept of the border niche: Glioblastoma cells use oligodendrocytes progenitor cells (GAOs) and microglia to acquire stem cell-like features. Brain Tumor Pathol. 2019, 36, 63–73. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonnière, L.; Bernard, M.; et al. The Hedgehog Pathway Promotes Blood-Brain Barrier Integrity and CNS Immune Quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef] [Green Version]
- Horng, S.; Therattil, A.; Moyon, S.; Gordon, A.; Kim, K.; Argaw, A.T.; Hara, Y.; Mariani, J.N.; Sawai, S.; Flodby, P.; et al. Astrocytic tight junctions control inflammatory CNS lesion pathogenesis. J. Clin. Investig. 2017, 127, 3136–3151. [Google Scholar] [CrossRef] [PubMed]
- John Lin, C.-C.; Yu, K.; Hatcher, A.; Huang, T.-W.; Lee, H.K.; Carlson, J.; Weston, M.C.; Chen, F.; Zhang, Y.; Zhu, W.; et al. Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci. 2017, 20, 396–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, R.C.D.; Wang, C.-Y.; Zheng, H.; Deneen, B.; Wu, J.Q. Brain Region-Specific Gene Signatures Revealed by Distinct Astrocyte Subpopulations Unveil Links to Glioma and Neurodegenerative Diseases. Eneuro 2019, 6, 6. [Google Scholar] [CrossRef]
- Irvin, D.M.; McNeill, R.S.; Bash, R.E.; Miller, C.R. Intrinsic Astrocyte Heterogeneity Influences Tumor Growth in Glioma Mouse Models. Brain Pathol. 2016, 27, 36–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Hu, X.; Qian, L.; O’Callaghan, J.P.; Hong, J.-S. Astrogliosis in CNS Pathologies: Is There A Role for Microglia? Mol. Neurobiol. 2010, 41, 232–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boccazzi, M.; Ceruti, S. Where do you come from and what are you going to become, reactive astrocyte? Stem Cell Investig. 2016, 3, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiland, D.H.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.W.C.; Strähle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat. Commun. 2019, 10, 2541. [Google Scholar] [CrossRef] [Green Version]
- Biasoli, D.; Sobrinho, M.F.; da Fonseca, A.C.C.; de Matos, D.G.; Romao, L.; de Moraes Maciel, R.; Rehen, S.K.; Moura-Neto, V.; Borges, H.L.; Lima, F.R.S. Glioblastoma cells inhibit astrocytic p53-expression favoring cancer malignancy. Oncogenesis 2014, 3, e123. [Google Scholar] [CrossRef] [Green Version]
- Placone, A.L.; Quiñones-Hinojosa, A.; Searson, P.C. The role of astrocytes in the progression of brain cancer: Complicating the picture of the tumor microenvironment. Tumor Biol. 2015, 37, 61–69. [Google Scholar] [CrossRef]
- Kim, J.-K.; Jin, X.; Sohn, Y.-W.; Jin, X.; Jeon, H.-Y.; Kim, E.-J.; Ham, S.W.; Jeon, H.-M.; Chang, S.-Y.; Oh, S.-Y.; et al. Tumoral RANKL activates astrocytes that promote glioma cell invasion through cytokine signaling. Cancer Lett. 2014, 353, 194–200. [Google Scholar] [CrossRef]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcino-ma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sin, W.C.; Aftab, Q.; Bechberger, J.F.; Leung, J.H.; Chen, H.; Naus, C.C. Astrocytes promote glioma invasion via the gap junction protein connexin43. Oncogene 2016, 35, 1504–1516. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, S.; Spray, D.C. Glioblastoma–Astrocyte Connexin 43 Gap Junctions Promote Tumor Invasion. Mol. Cancer Res. 2021, 20, 319–331. [Google Scholar] [CrossRef]
- Jin, P.; Shin, S.H.; Chun, Y.S.; Shin, H.W.; Shin, Y.J.; Lee, Y.; Kim, D.; Nam, D.H.; Park, J.W. Astrocyte-derived CCL20 rein-forces HIF-1-mediated hypoxic responses in glioblastoma by stimulating the CCR6-NF-κB signaling pathway. Oncogene 2018, 37, 3070–3087. [Google Scholar] [CrossRef] [PubMed]
- Ugbode, C.; Smith, I.; Whalley, B.J.; Hirst, W.; Rattray, M. Sonic hedgehog signalling mediates astrocyte crosstalk with neurons to confer neuroprotection. J. Neurochem. 2017, 142, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Pan, L.; Che, X.; Cui, D.; Li, C. Sonic Hedgehog/GLI₁ signaling pathway inhibition restricts cell migration and in-vasion in human gliomas. Neurol. Res. 2010, 32, 975–980. [Google Scholar] [CrossRef]
- Chen, W.; Xia, T.; Wang, D.; Huang, B.; Zhao, P.; Wang, J.; Qu, X.; Li, X. Human astrocytes secrete IL-6 to promote glioma migration and invasion through upregulation of cytomembrane MMP14. Oncotarget 2016, 7, 62425–62438. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef]
- Liu, Q.; Li, G.; Li, R.; Shen, J.; He, Q.; Deng, L.; Zhang, C.; Zhang, J. IL-6 promotion of glioblastoma cell invasion and angio-genesis in U251 and T98G cell lines. J. Neuroncol. 2010, 100, 165–176. [Google Scholar] [CrossRef]
- Chen, Z.; Yuan, S.J.; Li, K.; Zhang, Q.; Li, T.F.; An, H.C.; Xu, H.Z.; Yue, Y.; Han, M.; Xu, Y.H.; et al. Doxorubi-cin-polyglycerol-nanodiamond conjugates disrupt STAT3/IL-6-mediated reciprocal activation loop between glioblastoma cells and astrocytes. J. Control. Release 2020, 320, 469–483. [Google Scholar] [CrossRef]
- Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Na’Ara, S.; Gil, Z. Paracrine regulation of glioma cells invasion by astrocytes is mediated by glial-derived neurotrophic factor. Int. J. Cancer 2015, 137, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Valdebenito, S.; Malik, S.; Luu, R.; Loudig, O.; Mitchell, M.; Okafo, G.; Bhat, K.; Prideaux, B.; Eugenin, E.A. Tunneling nanotubes, TNT, communicate glioblastoma with surrounding non-tumor astrocytes to adapt them to hypoxic and metabolic tumor conditions. Sci. Rep. 2021, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ciccocioppo, F.; Lanuti, P.; Marchisio, M.; Miscia, S. Extracellular Vesicles Involvement in the Modulation of the Glioblastoma Environment. J. Oncol. 2020, 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [Green Version]
- Maas, S.L.N.; Breakefield, X.O.; Weaver, A.M. Extracellular Vesicles: Unique Intercellular Delivery Vehicles. Trends Cell Biol. 2017, 27, 172–188. [Google Scholar] [CrossRef] [Green Version]
- Oushy, S.; Hellwinkel, J.E.; Wang, M.; Nguyen, G.J.; Gunaydin, D.; Harland, T.A.; Anchordoquy, T.J.; Graner, M.W. Glio-blastoma multiforme-derived extracellular vesicles drive normal astrocytes towards a tumour-enhancing phenotype. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160477. [Google Scholar] [CrossRef] [Green Version]
- Taheri, B.; Soleimani, M.; Aval, S.F.; Memari, F.; Zarghami, N. C6 glioma-derived microvesicles stimulate the proliferative and metastatic gene expression of normal astrocytes. Neurosci. Lett. 2018, 685, 173–178. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, S.; Yao, J.; Lowery, F.J.; Zhang, Q.; Huang, W.C.; Li, P.; Li, M.; Wang, X.; Zhang, C.; et al. Microenviron-ment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 2015, 527, 100–104. [Google Scholar] [CrossRef]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [Green Version]
- Antunes, A.R.P.; Scheyltjens, I.; Lodi, F.; Messiaen, J.; Antoranz, A.; Duerinck, J.; Kancheva, D.; Martens, L.; de Vlaminck, K.; van Hove, H.; et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 2021, 24, 595–610. [Google Scholar] [CrossRef]
- Annovazzi, L.; Mellai, M.; Bovio, E.; Mazzetti, S.; Pollo, B.; Schiffer, D. Microglia immunophenotyping in gliomas. Oncol. Lett. 2017, 15, 998–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016, 1, e85841. [Google Scholar] [CrossRef] [PubMed]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.-P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17. [Google Scholar] [CrossRef] [PubMed]
- Fecci, P.E.; Ochiai, H.; Mitchell, D.A.; Grossi, P.M.; Sweeney, A.E.; Archer, G.E.; Cummings, T.; Allison, J.P.; Bigner, D.D.; Sampson, J.H. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without af-fecting regulatory T-cell function. Clin. Cancer Res. 2007, 13, 2158–2167. [Google Scholar] [CrossRef] [Green Version]
- Landry, A.P.; Balas, M.; Alli, S.; Spears, J.; Zador, Z. Distinct regional ontogeny and activation of tumor associated macrophages in human glioblastoma. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Szulzewsky, F.; Yerevanian, A.; Chen, Z.; Heinzmann, D.; Rasmussen, R.D.; Alvarez-Garcia, V.; Kim, Y.; Wang, B.; Tamagno, I.; et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget 2015, 6, 15077–15094. [Google Scholar] [CrossRef] [Green Version]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef]
- Da Fonseca, A.C.C.; Wang, H.; Fan, H.; Chen, X.; Zhang, I.; Zhang, L.; Lima, F.R.S.; Badie, B. Increased expression of stress inducible protein 1 in glioma-associated microglia/macrophages. J. Neuroimmunol. 2014, 274, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Wallmann, T.; Zhang, X.-M.; Wallerius, M.; Bolin, S.; Joly, A.-L.; Sobocki, C.; Leiss, L.; Jiang, Y.; Bergh, J.; Holland, E.C.; et al. Microglia Induce PDGFRB Expression in Glioma Cells to Enhance Their Migratory Capacity. iScience 2018, 9, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szulzewsky, F.; Schwendinger, N.; Guneykaya, D.; Cimino, P.; Hambardzumyan, D.; Synowitz, M.; Holland, E.C.; Kettenmann, H. Loss of host-derived osteopontin creates a glioblastoma-promoting microenvironment. Neuro-Oncology 2018, 20, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Platten, M.; Weller, M. Glioma cell invasion: Regulation of metalloproteinase activity by TGF-beta. J. Neurooncol. 2001, 53, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, L.; Dang, W.Q.; Cao, M.F.; Xiao, J.F.; Lv, S.Q.; Jiang, W.J.; Yao, X.H.; Lu, H.M.; Miao, J.Y.; et al. CCL8 secreted by tumor-associated macrophages promotes invasion and stemness of glioblastoma cells via ERK1/2 signaling. Lab. Investig. 2020, 100, 619–629. [Google Scholar] [CrossRef]
- Hu, F.; Dzaye, O.D.A.; Hahn, A.; Yu, Y.; Scavetta, R.J.; Dittmar, G.; Kaczmarek, A.K.; Dunning, K.; Ricciardelli, C.; Rinnenthal, J.L.; et al. Glioma-derived versican promotes tumor expansion via glioma-associated microglial/macrophages Toll-like receptor 2 signaling. Neuro-Oncology 2014, 17, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Markovic, D.S.; Vinnakota, K.; Chirasani, S.; Synowitz, M.; Raguet, H.; Stock, K.; Sliwa, M.; Lehmann, S.; Kälin, R.; van Rooijen, N.; et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc. Natl. Acad. Sci. USA 2009, 106, 12530–12535. [Google Scholar] [CrossRef] [Green Version]
- Vinnakota, K.; Hu, F.; Ku, M.-C.; Georgieva, P.B.; Szulzewsky, F.; Pohlmann, A.; Waiczies, H.; Niendorf, T.; Lehnardt, S.; Hanisch, U.-K.; et al. Toll-like receptor 2 mediates microglia/brain macrophage MT1-MMP expression and glioma expansion. Neuro-Oncology 2013, 15, 1457–1468. [Google Scholar] [CrossRef] [Green Version]
- Kaffes, I.; Szulzewsky, F.; Chen, Z.; Herting, C.J.; Gabanic, B.; Vega, J.E.V.; Shelton, J.; Switchenko, J.M.; Ross, J.L.; McSwain, L.F.; et al. Human Mesenchymal glioblastomas are characterized by an increased immune cell presence compared to Proneural and Classical tumors. OncoImmunology 2019, 8, e1655360. [Google Scholar] [CrossRef]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in gli-oblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Xu, J.; Chen, Z.; Wang, H.; Xue, H.; Yang, C.; Guo, Q.; Qi, Y.; Guo, X.; Qian, M.; et al. Transfer of MicroRNA via Macrophage-Derived Extracellular Vesicles Promotes Proneural-to-Mesenchymal Transition in Glioma Stem Cells. Cancer Immunol. Res. 2020, 8, 966–981. [Google Scholar] [CrossRef]
- Saio, M.; Okada, M.; Kito, Y.; Ohe, N.; Yano, H.; Yoshimura, S.; Iwama, T.; Takami, T. Tumor-associated macrophage/microglia infiltration in human gliomas is correlated with MCP-3, but not MCP-1. Int. J. Oncol. 2009, 34, 1621–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boeck, A.; Ahn, B.Y.; D’Mello, C.; Lun, X.; Menon, S.V.; Alshehri, M.M.; Szulzewsky, F.; Shen, Y.; Khan, L.; Dang, N.H.; et al. Glioma-derived IL-33 orchestrates an inflammatory brain tumor microenvironment that accelerates glioma progression. Nat. Commun. 2020, 11, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Kros, J.M.; Cheng, C.; Mustafa, D. The contribution of tumor-associated macrophages in glioma neo-angiogenesis and implications for anti-angiogenic strategies. Neuro-Oncology 2017, 19, 1435–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutsui, T.; Kawahara, H.; Kimura, R.; Dong, Y.; Jiapaer, S.; Sabit, H.; Zhang, J.; Yoshida, T.; Nakada, M.; Hanayama, R. Glioma-derived extracellular vesicles promote tumor progression by conveying WT1. Carcinogenesis 2020, 41, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef]
- Wang, S.-C.; Yu, C.-F.; Hong, J.-H.; Tsai, C.-S.; Chiang, C.-S. Radiation Therapy-Induced Tumor Invasiveness Is Associated with SDF-1-Regulated Macrophage Mobilization and Vasculogenesis. PLoS ONE 2013, 8, e69182. [Google Scholar] [CrossRef] [Green Version]
- Stafford, J.H.; Hirai, T.; Deng, L.; Chernikova, S.B.; Urata, K.; West, B.L.; Brown, J.M. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro-Oncology 2016, 18, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Wang, J.; Yang, Z.-Y.; Guo, Y.-F.; Kuang, J.-Y.; Bian, X.-W.; Yu, S.-C. Targeting different domains of gap junction protein to control malignant glioma. Neuro-Oncology 2017, 20, 885–896. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.-C.; Xiao, H.-L.; Jiang, X.-F.; Wang, Q.-L.; Li, Y.; Yang, X.-J.; Ping, Y.-F.; Duan, J.J.; Jiang, J.-Y.; Ye, X.-Z.; et al. Connexin 43 Reverses Malignant Phenotypes of Glioma Stem Cells by Modulating E-Cadherin. Stem Cells 2012, 30, 108–120. [Google Scholar] [CrossRef]
- Hitomi, M.; Deleyrolle, L.P.; Mulkearns-Hubert, E.E.; Jarrar, A.; Li, M.; Sinyuk, M.; Otvos, B.; Brunet, S.; Flavahan, W.A.; Hubert, C.G.; et al. Differential Connexin Function Enhances Self-Renewal in Glioblastoma. Cell Rep. 2015, 11, 1031–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forster, T.; Rausch, V.; Zhang, Y.; Isayev, O.; Heilmann, K.; Schoensiegel, F.; Liu, L.; Nessling, M.; Richter, K.; Labsch, S.; et al. Sulforaphane counteracts aggressiveness of pancreatic cancer driven by dysregulated Cx43-mediated gap junctional inter-cellular communication. Oncotarget 2014, 5, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Kochanowski, P.; Catapano, J.; Pudełek, M.; Wróbel, T.; Madeja, Z.; Ryszawy, D.; Czyż, J. Temozolomide Induces the Acqui-sition of Invasive Phenotype by O6-Methylguanine-DNA Methyltransferase (MGMT)(+) Glioblastoma Cells in a Snail-1/Cx43-Dependent Manner. Int. J. Mol. Sci. 2021, 22, 4150. [Google Scholar] [CrossRef] [PubMed]
- Day, B.W.; Stringer, B.W.; Boyd, A.W. Eph receptors as therapeutic targets in glioblastoma. Br. J. Cancer 2014, 111, 1255–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferluga, S.; Debinski, W. Ephs and Ephrins in malignant gliomas. Growth Factors 2014, 32, 190–201. [Google Scholar] [CrossRef] [Green Version]
- Nakada, M.; Hayashi, Y.; Hamada, J.-I. Role of Eph/ephrin tyrosine kinase in malignant glioma. Neuro-Oncology 2011, 13, 1163–1170. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Li, Y.; Mao, X. A review on the role of different ephrins in glioma. Eur. J. Pharmacol. 2021, 917, 174588. [Google Scholar] [CrossRef]
- Rossmeisl, J.H.; Herpai, D.; Quigley, M.; Cecere, T.E.; Robertson, J.L.; D’Agostino, R.B.; Hinckley, J.; Tatter, S.B.; Dickinson, P.J.; Debinski, W. Phase I trial of convection-enhanced delivery of IL13RA2 and EPHA2 receptor targeted cytotoxins in dogs with spontaneous intracranial gliomas. Neuro-Oncology 2021, 23, 422–434. [Google Scholar] [CrossRef]
- Liu, D.-P.; Wang, Y.; Koeffler, H.P.; Xie, N. Ephrin-A1 is a negative regulator in glioma through down-reguation of EphA2 and FAK. Int. J. Oncol. 2007, 30, 865–871. [Google Scholar] [CrossRef]
- Wykosky, J.; Debinski, W. The EphA2 Receptor and EphrinA1 Ligand in Solid Tumors: Function and Therapeutic Targeting. Mol. Cancer Res. 2008, 6, 1795–1806. [Google Scholar] [CrossRef] [Green Version]
- Nakada, M.; Niska, J.A.; Miyamori, H.; McDonough, W.S.; Wu, J.; Sato, H.; Berens, M.E. The phosphorylation of EphB2 re-ceptor regulates migration and invasion of human glioma cells. Cancer Res. 2004, 64, 3179–3185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, M.; Drake, K.L.; Nakada, S.; Niska, J.; Berens, M.E. Ephrin-B3 Ligand Promotes Glioma Invasion through Activation of Rac1. Cancer Res. 2006, 66, 8492–8500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, M.; Anderson, E.M.; Demuth, T.; Nakada, S.; Reavie, L.B.; Drake, K.L.; Hoelzinger, D.B.; Berens, M.E. The phosphorylation of ephrin-B2 ligand promotes glioma cell migration and invasion. Int. J. Cancer 2010, 126, 1155–1165. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-Q.; Gong, L.-L.; Wen, Z.-H.; Wang, J.; Xu, C.-S.; Huang, X.-D. Delta-Like Ligand 4 Correlates with Endothelial Proliferation and Vessel Maturation in Human Malignant Glioma. Onkologie 2012, 35, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-L.; Sainson, R.C.; Oon, C.E.; Turley, H.; Leek, R.; Sheldon, H.; Bridges, E.; Shi, W.; Snell, C.; Bowden, E.T.; et al. DLL4-Notch Signaling Mediates Tumor Resistance to Anti-VEGF Therapy In Vivo. Cancer Res. 2011, 71, 6073–6083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, Y.; Furuta, T.; Sabit, H.; Tamai, S.; Dong, Y.; Jiapaer, S.; Zhang, J.; Zhang, G.; Oishi, M.; Miyashita, K.; et al. Ligand-dependent EphB4 activation serves as an anchoring signal in glioma cells. Cancer Lett. 2019, 449, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.; Mancini, A.; Colapietro, A.; Monache, S.D.; Sferra, R.; Vitale, F.; Cristiano, L.; Martellucci, S.; Marampon, F.; Mattei, V.; et al. The Small Molecule Ephrin Receptor Inhibitor, GLPG1790, Reduces Renewal Capabilities of Cancer Stem Cells, Showing Anti-Tumour Efficacy on Preclinical Glioblastoma Models. Cancers 2019, 11, 359. [Google Scholar] [CrossRef] [Green Version]
- Teng, L.; Nakada, M.; Furuyama, N.; Sabit, H.; Furuta, T.; Hayashi, Y.; Takino, T.; Dong, Y.; Sato, H.; Sai, Y.; et al. Ligand-dependent EphB1 signaling suppresses glioma invasion and correlates with patient survival. Neurooncology 2013, 15, 1710–1720. [Google Scholar] [CrossRef] [Green Version]
- Ables, J.L.; Breunig, J.; Eisch, A.J.; Rakic, P. Not(ch) just development: Notch signalling in the adult brain. Nat. Rev. Neurosci. 2011, 12, 269–283. [Google Scholar] [CrossRef] [Green Version]
- Stockhausen, M.-T.; Kristoffersen, K.; Poulsen, H.S. The functional role of Notch signaling in human gliomas. Neuro-Oncology 2009, 12, 199–211. [Google Scholar] [CrossRef] [Green Version]
- Bolós, V.; Blanco, M.; Medina, V.; Aparicio, G.; Díaz-Prado, S.; Grande, E. Notch signalling in cancer stem cells. Clin. Transl. Oncol. 2009, 11, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranganathan, P.; Weaver, K.L.; Capobianco, A.J. Notch signalling in solid tumours: A little bit of everything but not all the time. Nat. Cancer 2011, 11, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Hurlbut, G.D.; Kankel, M.W.; Lake, R.J.; Artavanis-Tsakonas, S. Crossing paths with Notch in the hyper-network. Curr. Opin. Cell Biol. 2007, 19, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Fu, J.; Zheng, S.; Yao, J.; Wang, S.; Liu, D.D.; Yuan, Y.; Sulman, E.P.; Lang, F.F.; Colman, H.; et al. A High Notch Pathway Activation Predicts Response to γ Secretase Inhibitors in Proneural Subtype of Glioma Tumor-Initiating Cells. Stem Cells 2013, 32, 301–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Chen, T.; Zhang, J.; Mao, Q.; Li, S.; Xiong, W.; Qiu, Y.; Xie, Q.; Ge, J. Notch1 promotes glioma cell migration and invasion by stimulating β-catenin and NF-κB signaling via AKT activation. Cancer Sci. 2012, 103, 181–190. [Google Scholar] [CrossRef]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.-M.; Maciaczyk, J.; et al. NOTCH Pathway Blockade Depletes CD133-Positive Glioblastoma Cells and Inhibits Growth of Tumor Neurospheres and Xenografts. Stem Cells 2010, 28, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Moore, G.; Annett, S.; McClements, L.; Robson, T. Top Notch Targeting Strategies in Cancer: A Detailed Overview of Recent Insights and Current Perspectives. Cells 2020, 9, 1503. [Google Scholar] [CrossRef]
- Peereboom, D.M.; Ye, X.; Mikkelsen, T.; Lesser, G.J.; Lieberman, F.S.; Robins, H.I.; Ahluwalia, M.S.; Sloan, A.E.; Grossman, S.A. A Phase II and Pharmacodynamic Trial of RO4929097 for Patients With Recurrent/Progressive Glioblastoma. Neurosurgery 2020, 88, 246–251. [Google Scholar] [CrossRef]
- McGirt, M.J.; Chaichana, K.L.; Attenello, F.J.; Weingart, J.D.; Than, K.; Burger, P.C.; Olivi, A.; Brem, H.; Quinoñes-Hinojosa, A. Extent of surgical resection is independently associated with survival in patients with hemispheric infiltrating low-grade gli-omas. Neurosurgery 2008, 63, 700–707, Author Reply 707–708. [Google Scholar] [CrossRef] [Green Version]
- Sanai, N.; Polley, M.Y.; McDermott, M.W.; Parsa, A.T.; Berger, M.S. An extent of resection threshold for newly diagnosed glioblastomas. J. Neurosurg. 2011, 115, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Fujii, Y.; Muragaki, Y.; Maruyama, T.; Nitta, M.; Saito, T.; Ikuta, S.; Iseki, H.; Hongo, K.; Kawamata, T. Threshold of the extent of resection for WHO Grade III gliomas: Retrospective volumetric analysis of 122 cases using intraoperative MRI. J. Neurosurg. 2018, 129, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunduz, N.; Fisher, B.; Saffer, E.A. Effect of surgical removal on the growth and kinetics of residual tumor. Cancer Res. 1979, 39, 3861–3865. [Google Scholar] [PubMed]
- Hingtgen, S.; Figueiredo, J.-L.; Farrar, C.; Duebgen, M.; Martinez-Quintanilla, J.; Bhere, D.; Shah, K. Real-time multi-modality imaging of glioblastoma tumor resection and recurrence. J. Neuro-Oncol. 2012, 111, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okolie, O.; Bago, J.R.; Schmid, R.; Irvin, D.M.; Bash, R.E.; Miller, C.R.; Hingtgen, S.D. Reactive astrocytes potentiate tumor aggressiveness in a murine glioma resection and recurrence model. Neuro-Oncology 2016, 18, 1622–1633. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Jeon, H.; Othmer, H. The role of the tumor microenvironment in glioblastoma: A mathematical model. IEEE Trans. Biomed. Eng. 2016, 64, 1. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Lee, D.; Lawler, S. Collective invasion of glioma cells through OCT1 signalling and interaction with reactive astrocytes after surgery. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190390. [Google Scholar] [CrossRef]
- Naus, C.C.; Aftab, Q.; Sin, W.C. Common mechanisms linking connexin43 to neural progenitor cell migration and glioma invasion. Semin. Cell Dev. Biol. 2016, 50, 59–66. [Google Scholar] [CrossRef]
- Tomar, M.S.; Kumar, A.; Srivastava, C.; Shrivastava, A. Elucidating the mechanisms of Temozolomide resistance in gliomas and the strategies to overcome the resistance. Biochim. Biophys. Acta 2021, 1876, 188616. [Google Scholar] [CrossRef]
- Omar, A.I.; Mason, W.P. Temozolomide: The evidence for its therapeutic efficacy in malignant astrocytomas. Core Evid. 2010, 4, 93–111. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Yang, X.; Mei, S.; Sun, Y.; Li, J. Acquisition of temozolomide resistance by the rat C6 glioma cell line increases cell mi-gration and side population phenotype. Oncol. Rep. 2019, 42, 2355–2362. [Google Scholar] [CrossRef]
- Stepanenko, A.A.; Andreieva, S.V.; Korets, K.V.; Mykytenko, D.O.; Baklaushev, V.P.; Huleyuk, N.L.; Kovalova, O.A.; Kotsarenko, K.V.; Chekhonin, V.P.; Vassetzky, Y.S.; et al. Temozolomide promotes genomic and phenotypic changes in glioblastoma cells. Cancer Cell Int. 2016, 16, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, C.; Zaiden, N.; Chong, K.H.; See, S.J.; Wong, M.C.; Ang, B.T.; Tang, C. Characterization of a side population of astro-cytoma cells in response to temozolomide. J. Neurosurg. 2008, 109, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.P.; Miao, S.; Wu, Y.; Zhang, W.; Zhang, X.F.; Ma, H.Z.; Xin, H.L.; Feng, J.; Wen, A.D.; Li, Y. Resveratrol sensitizes ta-moxifen in antiestrogen-resistant breast cancer cells with epithelial-mesenchymal transition features. Int. J. Mol. Sci. 2013, 14, 15655–15668. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Li, Y. miR-25-3p reverses epithelial-mesenchymal transition via targeting Sema4C in cisplatin-resistance cervical cancer cells. Cancer Sci. 2016, 108, 23–31. [Google Scholar] [CrossRef]
- Gielen, P.R.; Aftab, Q.; Ma, N.; Chen, V.C.; Hong, X.; Lozinsky, S.; Naus, C.C.; Sin, W.C. Connexin43 confers Temozolomide resistance in human glioma cells by modulating the mitochondrial apoptosis pathway. Neuropharmacology 2013, 75, 539–548. [Google Scholar] [CrossRef]
- Bates, D.C.; Sin, W.C.; Aftab, Q.; Naus, C.C. Connexin43 enhances glioma invasion by a mechanism involving the carboxy terminus. Glia 2007, 55, 1554–1564. [Google Scholar] [CrossRef]
- Chen, W.; Wang, N.; Du, X.; He, Y.; Chen, S.; Shao, Q.; Ma, C.; Huang, B.; Chen, A.; Zhao, P.; et al. Glioma cells escaped from cytotoxicity of temozolomide and vincristine by communicating with human astrocytes. Med. Oncol. 2015, 32, 43. [Google Scholar] [CrossRef]
- Tsidulko, A.; Bezier, C.; de la Bourdonnaye, G.; Suhovskih, A.V.; Pankova, T.M.; Kazanskaya, G.M.; Aidagulova, S.; Grigorieva, E. Conventional Anti-glioblastoma Chemotherapy Affects Proteoglycan Composition of Brain Extracellular Matrix in Rat Experimental Model in vivo. Front. Pharmacol. 2018, 9, 1104. [Google Scholar] [CrossRef]
- Tsidulko, A.Y.; Shevelev, O.B.; Khotskina, A.S.; Kolpakova, M.A.; Suhovskih, A.V.; Kazanskaya, G.M.; Volkov, A.M.; Aidagulova, S.V.; Zavyalov, E.L.; Grigorieva, E.V. Chemotherapy-Induced Degradation of Glycosylated Components of the Brain Extracellular Matrix Promotes Glioblastoma Relapse Development in an Animal Model. Front. Oncol. 2021, 11, 713139. [Google Scholar] [CrossRef]
- Vredenburgh, J.J.; Desjardins, A.; Herndon, J.E., 2nd; Dowell, J.M.; Reardon, D.A.; Quinn, J.A.; Rich, J.N.; Sathornsumetee, S.; Gururangan, S.; Wagner, M.; et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin. Cancer Res. 2007, 13, 1253–1259. [Google Scholar] [CrossRef] [Green Version]
- Kreisl, T.N.; Kim, L.; Moore, K.; Duic, P.; Royce, C.; Stroud, I.; Garren, N.; Mackey, M.; Butman, J.A.; Camphausen, K.; et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J. Clin. Oncol. 2009, 27, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy–Temozolomide for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fack, F.; Espedal, H.; Keunen, O.; Golebiewska, A.; Obad, N.; Harter, P.N.; Mittelbronn, M.; Bähr, O.; Weyerbrock, A.; Stuhr, L.; et al. Bevacizumab treatment induces metabolic adaptation toward anaerobic metabolism in glioblastomas. Acta Neuropathol. 2014, 129, 115–131. [Google Scholar] [CrossRef] [Green Version]
- Keunen, O.; Johansson, M.; Oudin, A.; Sanzey, M.; Rahim, S.A.; Fack, F.; Thorsen, F.; Taxt, T.; Bartos, M.; Jirik, R.; et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3749–3754. [Google Scholar] [CrossRef] [Green Version]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity Generated by the Tumor Microenvironment Drives Local Invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim-Hashim, A.; Estrella, V. Acidosis and cancer: From mechanism to neutralization. Cancer Metastasis Rev. 2019, 38, 149–155. [Google Scholar] [CrossRef]
- Kujawski, M.; Kortylewski, M.; Lee, H.; Herrmann, A.; Kay, H.; Yu, H. Stat3 mediates myeloid cell-dependent tumor angio-genesis in mice. J. Clin. Investig. 2008, 118, 3367–3377. [Google Scholar] [CrossRef]
- De Groot, J.; Liang, J.; Kong, L.-Y.; Wei, J.; Piao, Y.; Fuller, G.; Qiao, W.; Heimberger, A.B. Modulating Antiangiogenic Resistance by Inhibiting the Signal Transducer and Activator of Transcription 3 Pathway in Glioblastoma. Oncotarget 2012, 3, 1036–1048. [Google Scholar] [CrossRef] [Green Version]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Mahase, S.; Rattenni, R.N.; Wesseling, P.; Leenders, W.; Baldotto, C.; Jain, R.; Zagzag, D. Hypoxia-Mediated Mechanisms Associated with Antiangiogenic Treatment Resistance in Glioblastomas. Am. J. Pathol. 2017, 187, 940–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karim, A.B.; Afra, D.; Cornu, P.; Bleehan, N.; Schraub, S.; de Witte, O.; Darcel, F.; Stenning, S.; Pierart, M.; van Glabbeke, M. Randomized trial on the efficacy of radiotherapy for cerebral low-grade glioma in the adult: European Organization for Re-search and Treatment of Cancer Study 22845 with the Medical Research Council study BRO4: An interim analysis. Int. J. Radiat. Oncol. Biol. Phys. 2002, 52, 316–324. [Google Scholar] [CrossRef]
- Dhawan, S.; Patil, C.G.; Chen, C.; Venteicher, A.S. Early versus delayed postoperative radiotherapy for treatment of low-grade gliomas. Cochrane Database Syst. Rev. 2020, 1, CD009229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tom, M.C.; Park, D.Y.J.; Yang, K.; Leyrer, C.M.; Wei, W.; Jia, X.; Varra, V.; Yu, J.S.; Chao, S.T.; Balagamwala, E.H.; et al. Malignant Transformation of Molecularly Classified Adult Low-Grade Glioma. Int. J. Radiat. Oncol. Biol. Phys. 2019, 105, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Wolf, K.J.; Chen, J.; Coombes, J.; Aghi, M.K.; Kumar, S. Dissecting and rebuilding the glioblastoma microenvironment with engineered materials. Nat. Rev. Mater. 2019, 4, 651–668. [Google Scholar] [CrossRef]
- Belka, C.; Budach, W.; Kortmann, R.D.; Bamberg, M. Radiation induced CNS toxicity – molecular and cellular mechanisms. Br. J. Cancer 2001, 85, 1233–1239. [Google Scholar] [CrossRef]
- Kim, J.H.; Brown, S.L.; Jenrow, K.A.; Ryu, S. Mechanisms of radiation-induced brain toxicity and implications for future clinical trials. J. Neuro-Oncol. 2008, 87, 279–286. [Google Scholar] [CrossRef]
- Siu, A.; Wind, J.J.; Iorgulescu, J.B.; Chan, T.A.; Yamada, Y.; Sherman, J.H. Radiation necrosis following treatment of high grade glioma—a review of the literature and current understanding. Acta Neurochir. 2011, 154, 191–201. [Google Scholar] [CrossRef]
- Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol. Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef] [Green Version]
- Mahabir, R.; Tanino, M.; Elmansuri, A.; Wang, L.; Kimura, T.; Itoh, T.; Ohba, Y.; Nishihara, H.; Shirato, H.; Tsuda, M.; et al. Sustained elevation of Snail promotes glial-mesenchymal transition after irradiation in malignant glioma. Neuro-Oncology 2014, 16, 671–685. [Google Scholar] [CrossRef] [Green Version]
- Lau, J.; Ilkhanizadeh, S.; Wang, S.; Miroshnikova, Y.A.; Salvatierra, N.A.; Wong, R.A.; Schmidt, C.; Weaver, V.M.; Weiss, W.A.; Persson, A.I. STAT3 Blockade Inhibits Radiation-Induced Malignant Progression in Glioma. Cancer Res. 2015, 75, 4302–4311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010, 463, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Iwadate, Y.; Fukuda, K.; Matsutani, T.; Saeki, N. Intrinsic protective mechanisms of the neuron-glia network against glioma invasion. J. Clin. Neurosci. 2016, 26, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT pro-moter (CENTRIC EORTC 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Fink, K.L.; Mikkelsen, T.; Cloughesy, T.F.; O’Neill, A.; Plotkin, S.; Glantz, M.; Ravin, P.; Raizer, J.J.; Rich, K.M.; et al. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glio-blastoma multiforme. J. Clin. Oncol. 2008, 26, 5610–5617. [Google Scholar] [CrossRef] [PubMed]
- Gerstner, E.R.; Ye, X.; Duda, D.G.; Levine, M.A.; Mikkelsen, T.; Kaley, T.J.; Olson, J.J.; Nabors, B.L.; Ahluwalia, M.S.; Wen, P.Y.; et al. A phase I study of cediranib in combination with cilengitide in patients with recurrent glioblastoma. Neuro-Oncology 2015, 17, 1386–1392. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Furuta, T.; Sabit, H.; Dong, Y.; Miyashita, K.; Kinoshita, M.; Uchiyama, N.; Hayashi, Y.; Hayashi, Y.; Minamoto, T.; Nakada, M. Biological basis and clinical study of glycogen synthase kinase- 3β-targeted therapy by drug repositioning for glioblastoma. Oncotarget 2017, 8, 22811–22824. [Google Scholar] [CrossRef] [Green Version]
- Carcelén, M.; Velásquez, C.; Vidal, V.; Gutierrez, O.; Fernandez-Luna, J.L. HIF2α Upregulates the Migration Factor ODZ1 under Hypoxia in Glioblastoma Stem Cells. Int. J. Mol. Sci. 2022, 23, 741. [Google Scholar] [CrossRef]
- Kessler, J.; Hahnel, A.; Wichmann, H.; Rot, S.; Kappler, M.; Bache, M.; Vordermark, D. HIF-1α inhibition by siRNA or chetomin in human malignant glioma cells: Effects on hypoxic radioresistance and monitoring via CA9 expression. BMC Cancer 2010, 10, 605. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Li, Y.; Shu, M.; Tang, J.; Huang, Y.; Zhou, Y.; Liang, Y.; Yan, G. Hypoxia-inducible factor-1α blocks differentiation of malignant gliomas. FEBS J. 2009, 276, 7291–7304. [Google Scholar] [CrossRef] [PubMed]
- Kurozumi, K.; Hardcastle, J.; Thakur, R.; Yang, M.; Christoforidis, G.; Fulci, G.; Hochberg, F.H.; Weissleder, R.; Carson, W.; Chiocca, E.A.; et al. Effect of Tumor Microenvironment Modulation on the Efficacy of Oncolytic Virus Therapy. JNCI J. Natl. Cancer Inst. 2007, 99, 1768–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Wong, E.T.; Kanner, A.A.; Steinberg, D.; Engelhard, H.; Heidecke, V.; Kirson, E.D.; Taillibert, S.; Liebermann, F.; Dbalý, V.; et al. NovoTTF-100A versus physician’s choice chemotherapy in recurrent glioblastoma: A randomised phase III trial of a novel treatment modality. Eur. J. Cancer 2012, 48, 2192–2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.; Zhu, J.-J. Tumor treating fields: A novel and effective therapy for glioblastoma: Mechanism, efficacy, safety and future perspectives. Chin. Clin. Oncol. 2017, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamai, S.; Ichinose, T.; Tsutsui, T.; Tanaka, S.; Garaeva, F.; Sabit, H.; Nakada, M. Tumor Microenvironment in Glioma Invasion. Brain Sci. 2022, 12, 505. https://doi.org/10.3390/brainsci12040505
Tamai S, Ichinose T, Tsutsui T, Tanaka S, Garaeva F, Sabit H, Nakada M. Tumor Microenvironment in Glioma Invasion. Brain Sciences. 2022; 12(4):505. https://doi.org/10.3390/brainsci12040505
Chicago/Turabian StyleTamai, Sho, Toshiya Ichinose, Taishi Tsutsui, Shingo Tanaka, Farida Garaeva, Hemragul Sabit, and Mitsutoshi Nakada. 2022. "Tumor Microenvironment in Glioma Invasion" Brain Sciences 12, no. 4: 505. https://doi.org/10.3390/brainsci12040505