
Brain Correlates of the Alcohol Use Disorder Pharmacotherapy Response: A Systematic Review of Neuroimaging Studies

 ,
,  ,
,  , ,
, ,  , ,
, ,  ,
,
Abstract
:1. Introduction
Pharmacotherapies for Alcohol Use Disorder
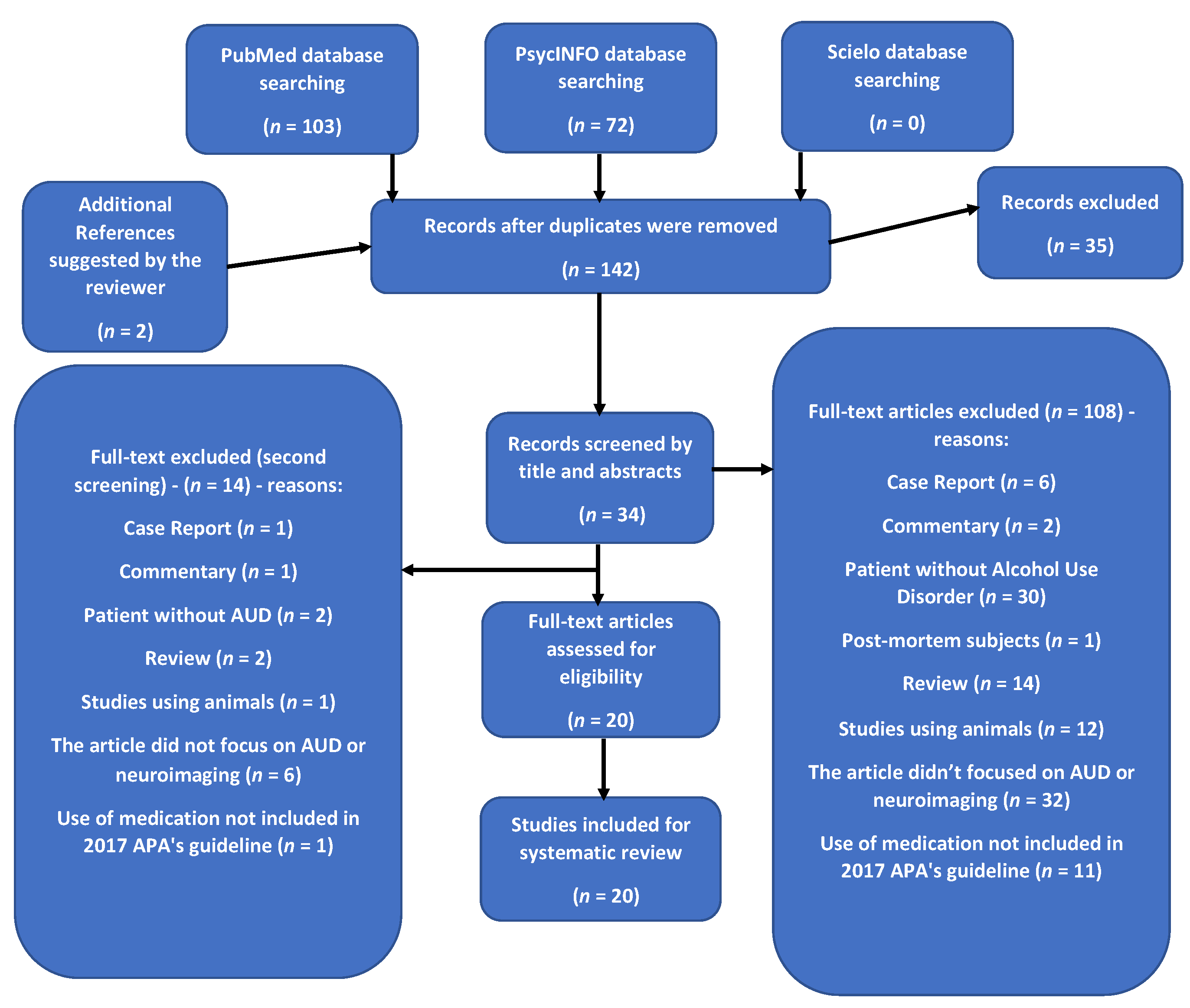
2. Methods
2.1. Eligibility
2.2. Search Strategy
2.3. Study Selection and Data Extraction
2.4. Registration
3. Results
3.1. Sample Characteristics
3.2. Main Findings
3.2.1. Duration of Treatment and Study Design
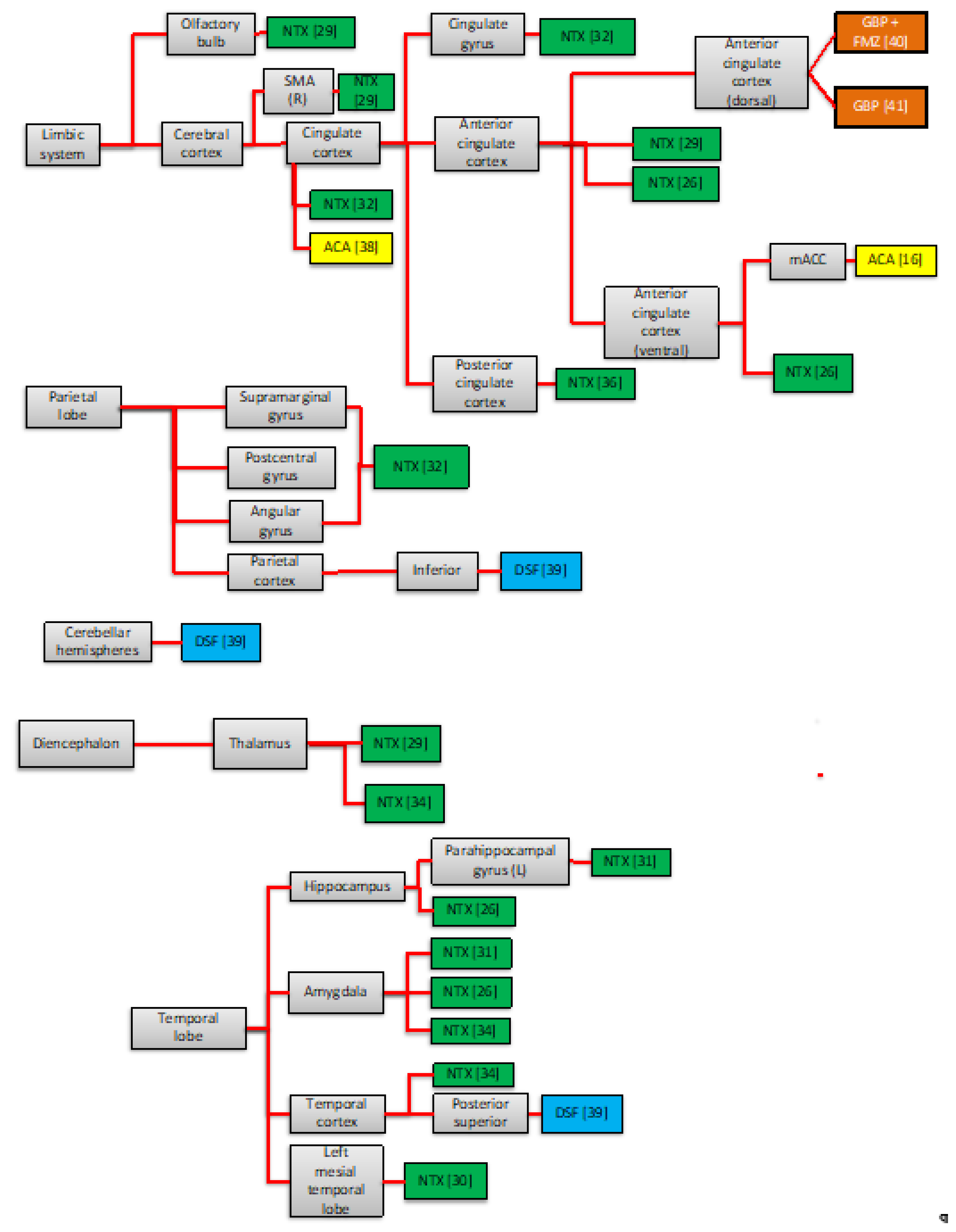
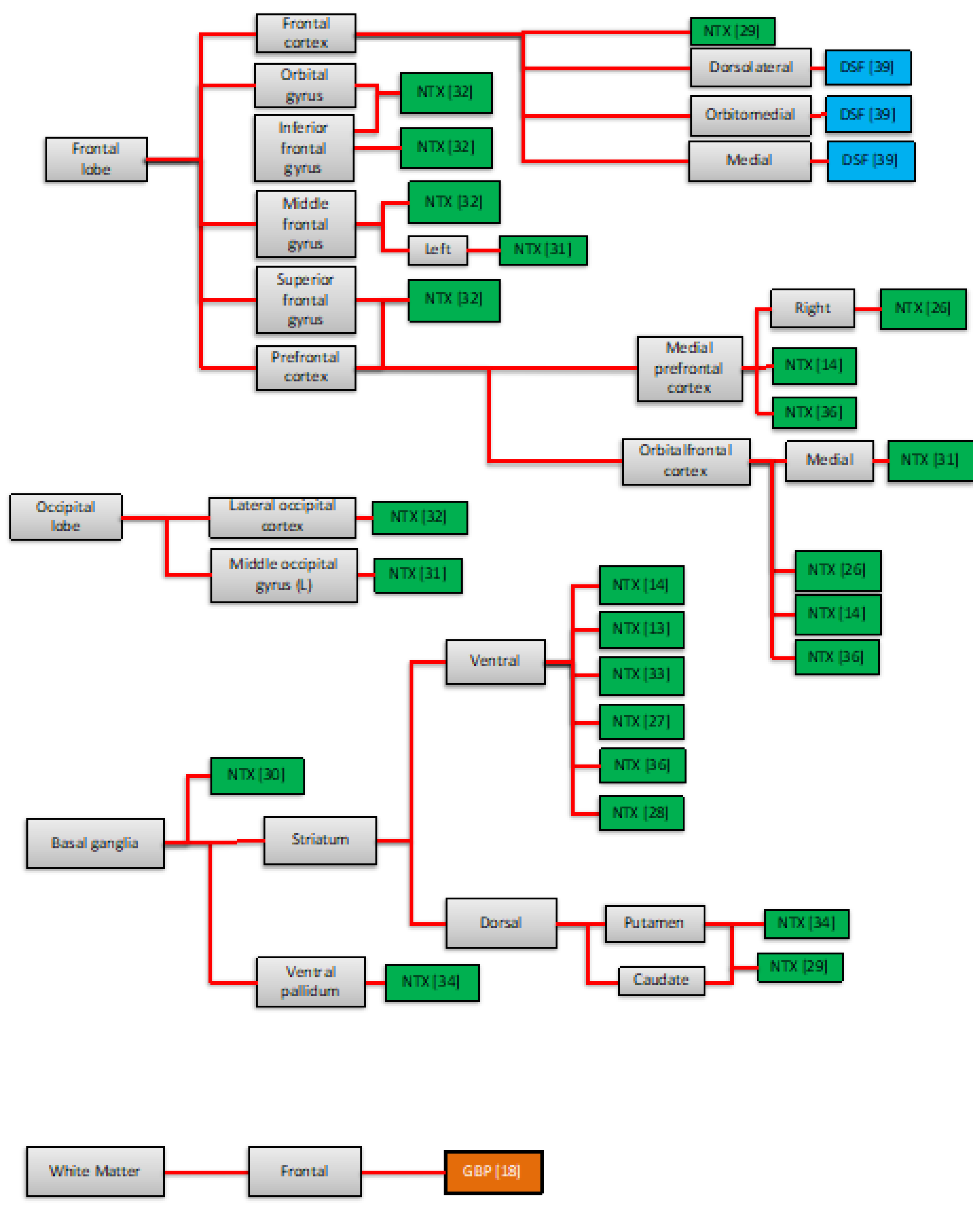
3.2.2. Neuroimaging Findings of the Pharmacotherapy Response
4. Discussion
4.1. Areas Traditionally Involved in the Neurobiology of AUD
4.2. Other Neural Areas
4.3. Implications
4.4. Limitations
4.5. Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Global Burden of Disease Collaborative Network. Global Burden of Disease Study 2017 (GBD 2017) Results; Institute for Health Metrics and Evaluation (IHME): Seattle, DC, USA, 2018. [Google Scholar]
- American Psychiatric Association. DSM-5: Diagnostic and Statistical Manual of Mental Disorder, 5th ed.; American Psychiatric Publishing: Washington, DC, USA, 2013. [Google Scholar]
- World Health Organization. ICD-10: International Statistical Classification of Diseases and Related Health Problems: Tenth Revision, 2nd ed.; World Health Organization: Geneva, Switzerland, 2004. [Google Scholar]
- Degenhardt, L.; Charlson, F.; Ferrari, A.; Santomauro, D.; Erskine, H.; Mantilla-Herrara, A.; Whiteford, H.; Leung, J.; Naghavi, M.; Griswold, M.; et al. The global burden of disease attributable to alcohol and drug use in 195 countries and territories, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Psychiatry 2018, 5, 987–1012. [Google Scholar] [CrossRef] [Green Version]
- Volkow, N.D.; Koob, G.F.; McLellan, A.T. Neurobiologic Advances from the Brain Disease Model of Addiction. N. Engl. J. Med. 2016, 374, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Kranzler, H.R.; Soyka, M. Diagnosis and Pharmacotherapy of Alcohol Use Disorder: A Review. JAMA 2018, 320, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Grodin, E.N.; Ray, L.A. The Use of Functional Magnetic Resonance Imaging to Test Pharmacotherapies for Alcohol Use Disorder: A Systematic Review. Alcohol. Clin. Exp. Res. 2019, 43, 2038–2056. [Google Scholar] [CrossRef] [PubMed]
- Wiers, C.E.; Cabrera, E.; Skarda, E.; Volkow, N.D.; Wang, G.-J. PET imaging for addiction medicine: From neural mechanisms to clinical considerations. Prog. Brain Res. 2015, 224, 175–201. [Google Scholar] [CrossRef] [PubMed]
- Hutton, B.F. The origins of SPECT and SPECT/CT. Eur. J. Nucl. Med. Mol. Imaging 2013, 41, 3–16. [Google Scholar] [CrossRef]
- Meyerhoff, D.J. Brain proton magnetic resonance spectroscopy of alcohol use disorders. Handb. Clin. Neurol. 2014, 125, 313–337. [Google Scholar] [CrossRef] [Green Version]
- Reus, V.I.; Fochtmann, L.J.; Bukstein, O.; Eyler, A.E.; Hilty, D.M.; Horvitz-Lennon, M.; Mahoney, J.; Pasic, J.; Weaver, M.; Wills, C.D.; et al. The American Psychiatric Association Practice Guideline for the Pharmacological Treatment of Patients with Alcohol Use Disorder. Am. J. Psychiatry 2018, 175, 86–90. [Google Scholar] [CrossRef]
- Castro, L.A.; Baltieri, D.A. The pharmacologic treatment of the alcohol dependence. Rev. Bras. Psiquiatr. 2004, 26 (Suppl. 1), S43–S46. [Google Scholar] [CrossRef] [Green Version]
- Schacht, J.P.; Randall, P.K.; Latham, P.K.; Voronin, K.; Book, S.W.; Myrick, H.; Anton, R.F. Predictors of Naltrexone Response in a Randomized Trial: Reward-Related Brain Activation, OPRM1 Genotype, and Smoking Status. Neuropsychopharmacology 2017, 42, 2640–2653. [Google Scholar] [CrossRef]
- Schacht, J.P.; Anton, R.F.; Voronin, K.; Randall, P.K.; Li, X.; Henderson, S.; Myrick, H. Interacting Effects of Naltrexone and OPRM1 and DAT1 Variation on the Neural Response to Alcohol Cues. Neuropsychopharmacology 2012, 38, 414–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Center for Substance Abuse Treatment. Incorporating Alcohol Pharmacotherapies Into Medical Practice; Substance Abuse and Mental Health Services Administration (US): Rockville, MD, USA, 2009; Report No.: (SMA) 09-4380. [Google Scholar]
- Frye, M.A.; Hinton, D.J.; Karpyak, V.; Biernacka, J.M.; Gunderson, L.J.; Feeder, S.E.; Choi, D.-S.; Port, J. Anterior Cingulate Glutamate Is Reduced by Acamprosate Treatment in Patients With Alcohol Dependence. J. Clin. Psychopharmacol. 2016, 36, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Naassila, M.; Hammoumi, S.; Legrand, E.; Durbin, P.; Daoust, M. Mechanism of action of acamprosate. Part I. Characterization of spermidine-sensitive acamprosate binding site in rat brain. Alcohol. Clin. Exp. Res. 1998, 22, 802–809. [Google Scholar] [CrossRef]
- Meyerhoff, D.J.; Murray, D.E.; Durazzo, T.C.; Pennington, D.L. Brain GABA and Glutamate Concentrations Following Chronic Gabapentin Administration: A Convenience Sample Studied During Early Abstinence From Alcohol. Front. Psychiatry 2018, 9, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stromberg, M.F.; Mackler, S.A.; Volpicelli, J.R.; O’Brien, C.P. Effect of acamprosate and naltrexone, alone or in combination, on ethanol consumption. Alcohol 2001, 23, 109–116. [Google Scholar] [CrossRef]
- Putzke, J.; Spanagel, R.; Tölle, T.R.; Zieglgänsberger, W. The anti-craving drug acamprosate reduces c-fos expression in rats undergoing ethanol withdrawal. Eur. J. Pharmacol. 1996, 317, 39–48. [Google Scholar] [CrossRef]
- Skinner, M.D.; Lahmek, P.; Pham, H.; Aubin, H.-J. Disulfiram Efficacy in the Treatment of Alcohol Dependence: A Meta-Analysis. PLoS ONE 2014, 9, e87366. [Google Scholar] [CrossRef] [Green Version]
- Fuller, R.K.; Branchey, L.; Brightwell, D.R.; Derman, R.M.; Emrick, C.D.; Iber, F.L.; James, K.E.; Lacoursiere, R.B.; Lee, K.K.; Lowenstam, I. Disulfiram treatment of alcoholism. A Veterans Administration cooperative study. JAMA 1986, 256, 1449–1455. [Google Scholar] [CrossRef]
- Furieri, F.A.; Nakamura-Palacios, E.M. Gabapentin reduces alcohol consumption and craving: A randomized, double-blind, placebo-controlled trial. J. Clin. Psychiatry 2007, 68, 1691–1700. [Google Scholar] [CrossRef]
- Johnson, B.A.; Swift, R.M.; Ait-Daoud, N.; DiClemente, C.C.; Javors, M.A.; Malcolm, R.J., Jr. Development of Novel Pharmacotherapies for the Treatment of Alcohol Dependence: Focus on Antiepileptics. Alcohol. Clin. Exp. Res. 2004, 28, 295–301. [Google Scholar] [CrossRef]
- Johnson, B.A.; Swift, R.M.; Addolorato, G.; Ciraulo, D.A.; Myrick, H. Safety and Efficacy of GABAergic Medications for Treating Alcoholism. Alcohol. Clin. Exp. Res. 2005, 29, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Savulich, G.; Riccelli, R.; Passamonti, L.; Correia, M.; Deakin, J.F.W.; Elliott, R.; Flechais, R.S.A.; Lingford-Hughes, A.R.; Mcgonigle, J.; Murphy, A.; et al. Effects of naltrexone are influenced by childhood adversity during negative emotional processing in addiction recovery. Transl. Psychiatry 2017, 7, e1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, K.; Vollstädt-Klein, S.; Reinhard, I.; Leménager, T.; Fauth-Bühler, M.; Hermann, D.; Hoffmann, S.; Zimmermann, U.S.; Kiefer, F.; Heinz, A.; et al. Predicting naltrexone response in alcohol-dependent patients: The contribution of functional magnetic resonance imaging. Alcohol. Clin. Exp. Res. 2014, 38, 2754–2762. [Google Scholar] [CrossRef] [PubMed]
- Myrick, H.; Anton, R.F.; Li, X.; Henderson, S.; Randall, P.K.; Voronin, K. Effect of Naltrexone and Ondansetron on Alcohol Cue–Induced Activation of the Ventral Striatum in Alcohol-Dependent People. Arch. Gen. Psychiatry 2008, 65, 466–475. [Google Scholar] [CrossRef]
- Weerts, E.M.; Kim, Y.K.; Wand, G.S.; Dannals, R.F.; Lee, J.S.; Frost, J.J.; McCaul, M.E. Differences in δ- and μ-Opioid Receptor Blockade Measured by Positron Emission Tomography in Naltrexone-Treated Recently Abstinent Alcohol-Dependent Subjects. Neuropsychopharmacology 2007, 33, 653–665. [Google Scholar] [CrossRef] [Green Version]
- Catafau, A.M.; Etcheberrigaray, A.; Cobos, J.P.D.L.; Estorch, M.; Guardia, J.; Flotats, A.; Bernà, L.; Marí, C.; Casas, M.; Carrió, I. Regional cerebral blood flow changes in chronic alcoholic patients induced by naltrexone challenge during detoxification. J. Nucl. Med. 1999, 40, 19–24. [Google Scholar]
- Morris, L.S.; Baek, K.; Tait, R.; Elliott, R.; Ersche, K.D.; Flechais, R.; Mcgonigle, J.; Murphy, A.; Nestor, L.J.; Orban, C.; et al. Naltrexone ameliorates functional network abnormalities in alcohol-dependent individuals. Addict. Biol. 2017, 23, 425–436. [Google Scholar] [CrossRef]
- Lukas, S.E.; Lowen, S.B.; Lindsey, K.P.; Conn, N.; Tartarini, W.; Rodolico, J.; Mallya, G.; Palmer, C.; Penetar, D.M. Extended-release naltrexone (XR-NTX) attenuates brain responses to alcohol cues in alcohol-dependent volunteers: A bold FMRI study. NeuroImage 2013, 78, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Spagnolo, P.A.; Ramchandani, V.A.; Schwandt, M.L.; Zhang, L.; Blaine, S.K.; Usala, J.M.; Diamond, K.A.; Phillips, M.J.; George, D.T.; Momenan, R.; et al. Effects of Naltrexone on Neural and Subjective Response to Alcohol in Treatment-Seeking Alcohol-Dependent Patients. Alcohol. Clin. Exp. Res. 2014, 38, 3024–3032. [Google Scholar] [CrossRef] [Green Version]
- Bach, P.; Weil, G.; Pompili, E.; Hoffmann, S.; Hermann, D.; Vollstädt-Klein, S.; Mann, K.; Perez-Ramirez, U.; Moratal, D.; Canals, S.; et al. Incubation of neural alcohol cue reactivity after withdrawal and its blockade by naltrexone. Addict. Biol. 2019, 25, e12717. [Google Scholar] [CrossRef]
- Lim, A.C.; Ghahremani, D.G.; Grodin, E.N.; Green, R.; Bujarski, S.; Hartwell, E.E.; Courtney, K.E.; Hutchison, K.; Miotto, K.; Ray, L.A. Neuroimaging findings from an experimental pharmacology trial of naltrexone in heavy drinkers of East Asian descent. Drug Alcohol Depend. 2019, 200, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Nestor, L.J.; Paterson, L.; Murphy, A.; Mcgonigle, J.; Orban, C.; Reed, L.; Taylor, E.; Flechais, R.; Smith, D.; Bullmore, E.T.; et al. Naltrexone differentially modulates the neural correlates of motor impulse control in abstinent alcohol-dependent and polysubstance-dependent individuals. Eur. J. Neurosci. 2018, 50, 2311–2321. [Google Scholar] [CrossRef] [PubMed]
- Langosch, J.M.; Spiegelhalder, K.; Jahnke, K.; Feige, B.; Regen, W.; Kiemen, A.; Hennig, J.; Olbrich, H.M. The impact of acamprosate on cue reactivity in alcohol dependent individuals: A functional magnetic resonance imaging study. J. Clin. Psychopharmacol. 2012, 32, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Umhau, J.C.; Momenan, R.; Schwandt, M.L.; Singley, E.; Lifshitz, M.; Doty, L.; Adams, L.J.; Vengeliene, V.; Spanagel, R.; Zhang, Y.; et al. Effect of acamprosate on magnetic resonance spectroscopy measures of central glutamate in detoxified alcohol-dependent individuals a randomized controlled experimental medicine study. Arch. Gen. Psychiatry 2010, 67, 1069–1077. [Google Scholar] [CrossRef]
- Gilman, S.; Adams, K.M.; Johnson-Greene, D.; Koeppe, R.A.; Junck, L.; Kluin, K.J.; Martorello, S.; Heumann, M.; Hill, E. Effects of Disulfiram on Positron Emission Tomography and Neuropsychological Studies in Severe Chronic Alcoholism. Alcohol. Clin. Exp. Res. 1996, 20, 1456–1461. [Google Scholar] [CrossRef] [Green Version]
- Schacht, J.P.; Anton, R.F.; Randall, P.K.; Li, X.; Henderson, S.; Myrick, H. Effects of a GABA-ergic medication combination and initial alcohol withdrawal severity on cue-elicited brain activation among treatment-seeking alcoholics. Psychopharmacology 2013, 227, 627–637. [Google Scholar] [CrossRef] [Green Version]
- Prisciandaro, J.J.; Hoffman, M.; Brown, T.R.; Voronin, K.; Book, S.; Bristol, E.; Anton, R.F. Effects of Gabapentin on Dorsal Anterior Cingulate Cortex GABA and Glutamate Levels and Their Associations With Abstinence in Alcohol Use Disorder: A Randomized Clinical Trial. Am. J. Psychiatry 2021, 178, 829–837. [Google Scholar] [CrossRef]
- de Laat, B.; Goldberg, A.; Shi, J.; Tetrault, J.M.; Nabulsi, N.; Zheng, M.-Q.; Najafzadeh, S.; Gao, H.; Kapinos, M.; Ropchan, J.; et al. The Kappa Opioid Receptor Is Associated With Naltrexone-Induced Reduction of Drinking and Craving. Biol. Psychiatry 2019, 86, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Pierri, J.N.; Lewis, D.A. Functional neuroanatomy. In Kaplan and Sadock’s Comprehensive Textbook of Psychiatry, 8th ed.; Sadock, B.J., Sadock, V.A., Eds.; Lippincott William and Wilkins: Phildelphia, PA, USA, 2012; pp. 3–33. [Google Scholar]
- Kakko, J.; Alho, H.; Baldacchino, A.; Molina, R.; Nava, F.A.; Shaya, G. Craving in Opioid Use Disorder: From Neurobiology to Clinical Practice. Front. Psychiatry 2019, 10, 592. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, R.Z.; Volkow, N.D. Drug Addiction and Its Underlying Neurobiological Basis: Neuroimaging Evidence for the Involvement of the Frontal Cortex. Am. J. Psychiatry 2002, 159, 1642–1652. [Google Scholar] [CrossRef]
- Latt, N.C.; Jurd, S.; Houseman, J.; Wutzke, S.E. Naltrexone in alcohol dependence: A randomised controlled trial of effectiveness in a standard clinical setting. Med. J. Aust. 2002, 176, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Sugrue, L.P.; Corrado, G.S.; Newsome, W.T. Matching Behavior and the Representation of Value in the Parietal Cortex. Science 2004, 304, 1782–1787. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Rius, J.; Miquel, M. The cerebellum in drug craving. Drug Alcohol Depend. 2017, 173, 151–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koob, G.F.; Volkow, N.D. Neurocircuitry of addiction. Neuropsychopharmacology 2010, 35, 217–238. [Google Scholar] [CrossRef] [Green Version]
- Claus, E.D.; Ewing, S.W.F.; Filbey, F.M.; Sabbineni, A.; E Hutchison, K. Identifying Neurobiological Phenotypes Associated with Alcohol Use Disorder Severity. Neuropsychopharmacology 2011, 36, 2086–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Tian, F.; Zhang, H.; Zeng, J.; Chen, T.; Wang, S.; Jia, Z.; Gong, Q. Cortical and subcortical gray matter shrinkage in alcohol-use disorders: A voxel-based meta-analysis. Neurosci. Biobehav. Rev. 2016, 66, 92–103. [Google Scholar] [CrossRef]
- Postuma, R.B.; Dagher, A. Basal Ganglia Functional Connectivity Based on a Meta-Analysis of 126 Positron Emission Tomography and Functional Magnetic Resonance Imaging Publications. Cereb. Cortex 2005, 16, 1508–1521. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, N.H.; Bechara, A. The hidden island of addiction: The insula. Trends Neurosci. 2009, 32, 56–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Reference | NTX | XR-NTX | NTX + ODT | ODT | ACA | DSF | GBP | GBP + FMZ | PLA | IWT | HCS |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bach et al. (2019) [34] | 22 | 13 | 35 | ||||||||
| Catafau et al. (1999) [30] | 29 | ||||||||||
| Frye et al. (2016) [16] | 9 | 16 | |||||||||
| Gilman et al. (1996) [39] | 11 | ||||||||||
| Langosch et al. (2012) [37] | 15 | 14 | |||||||||
| Lim et al. (2019) * [35] | 41 | 41 | |||||||||
| Lukas et al. (2013) [32] | 15 | 13 | |||||||||
| Mann et al. (2014) [27] | 36 | 28 | |||||||||
| Meyerhoff et al. (2018) [18] | 13 | ||||||||||
| Morris et al. (2017) [31] | 45 | 48 | |||||||||
| Myrick et al. (2008) [28] | 23 | 20 | 23 | 24 | 17 | ||||||
| Nestor et al. (2019) [36] | NA | NA | 35 | ||||||||
| Prisciandaro et al. (2021) [41] | 31 | 37 | |||||||||
| Savulich et al. (2017) * [26] | 18 | 18 | 21 | ||||||||
| Schacht et al. (2012) [14] | 33 | 39 | |||||||||
| Schacht et al. (2013) [40] | 28 | 20 | |||||||||
| Schacht et al. (2017) [13] | 76 | 56 | |||||||||
| Spagnolo et al. (2014) [33] | 31 | 32 | |||||||||
| Umhau et al. (2010) [38] | 15 | 18 | |||||||||
| Weerts et al. (2008) [29] | 36 | ||||||||||
| TOTAL | 390 | 15 | 20 | 23 | 67 | 11 | 44 | 28 | 360 | 124 |
| Reference | Treatment | Neuroimage Study | Follow-Up | Alcohol Use Scales |
|---|---|---|---|---|
| Bach et al. (2019) [34] | NTX 21 days | fMRI at baseline (after 2–4 weeks of controlled abstinence) and after 2 weeks of treatment | 3 month follow-up | BDI, ADS Score, OCDS Score, CIWA-Ar, TLFB |
| Catafau et al. (1999) [30] | NTX 1 day | SPECT on the tenth day of abstinence and on day 12 after 150 mg NTX (oral) | NA | MAIPY, MTAA |
| Frye et al. (2016) [16] | ACA 4 weeks | H-MRS shortly after admission and after 4 weeks of NTX treatment | NA | BDI-II, PHQ-9, TLFB, DSLD, PACS, AUQ, CIWA |
| Gilman et al. (1996) [39] | DSF 30 days | PET Scan was conducted after at least 30 days of sobriety, except for one patient | NA | LTAC, YHD |
| Langosch et al. (2012) [37] | ACA 2 weeks | fMRI before treatment initiation and after 2 weeks of treatment | NA | PSS, BDI-II, CIWA |
| Lim et al. (2019) [35] | NTX 8 days | One fMRI session after 4 days of NTX and another after 4 days on placebo | NA | AUDIT, TLFB, DD, DPDD |
| Lukas et al. (2013) [32] | XR-NTX 4 weeks | fMRI immediatly before and two weeks after injection | 4 visits | ADH, NDW, DSB |
| Mann et al. (2014) [27] | NTX 6 months | first fMRI was after withdrawal symptoms had subsided and the other 2 weeks after treatment beginning | 1 year | ADS Score, OCDS Score, AUDIT, AUQ |
| Meyerhoff et al. (2018) [18] | GBP 1 week | MR spectroscopy after at least 1 week taking GBP | NA | SCID 2.0, LDH, CIWA |
| Morris et al. (2017) [31] | NTX 1 day | fMRI—2 h after NTX or placebo intake | NA | BDI-II, STAI |
| Myrick et al. (2008) [28] | NTX 8 days | fMRI on day 7 after at least 24 h of abstinence | NA | ADS Score, OCDS Score, TLFB, DD |
| Nestor et al. (2019) [36] | NTX 1 day | fMRI—2 h after NTX or placebo intake | The study has mentioned a follow-up, but not its length | ASSIST, TLFB |
| Priscindaro et al. (2021) [41] | GBP 16 weeks | MR spectroscopy were acquired before start of treatment and again approximately 14 days after randomization. | NA | CIWA |
| Savulich et al. (2017) [26] | NTX 4 weeks | 4 fMRIs—2 h prior MRI NTX or PLA (4 days/times) | NA | WTAR, CTQ, PSS, AUDIT, BDI-II, STAI |
| Schacht et al. (2012) [14] | NTX 7 days | fMRI conducted on the sixth day of treatment | On the second visit | ADS Score, OCDS Score, DPD, HDD, DPDD, AASE, AI |
| Schacht et al. (2013) [40] | GBP + FMZ 6 weeks | fMRI was performed between the second and third week of treatment (mean scan day = 15; SD 2.5 days) | NA | ADS Score, OCDS Score, HDD, CIWA |
| Schacht et al. (2017) [13] | NTX 16 weeks | fMRI conducted at baseline and week 2 | 9 visits | ADS Score, OCDS Score, DPD, HDD, DPDD |
| Spagnolo et al. (2014) [30] | NTX 9 days | the fMRI was conducted on day 9 | 3 weeks | ADS Score, TLFB, ANDD, DD, HDD, DAPA |
| Umhau et al. (2010) [38] | ACA 4 weeks | H-MRS measures were obtained on the 4th and 25th day of the study | NA | ADS Score, TLFB, CIWA |
| Weerts et al. (2008) [29] | NTX 5 days | PET Scan before day 5 (no NTX) and on eighteenth day | Further evaluated in follow-up visits and continued naltrexone treatment | ADS Socre, ANDPDD, ANDDW |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Florence, L.; Lassi, D.L.S.; Kortas, G.T.; Lima, D.R.; de Azevedo-Marques Périco, C.; Andrade, A.G.; Torales, J.; Ventriglio, A.; De Berardis, D.; De Aquino, J.P.; et al. Brain Correlates of the Alcohol Use Disorder Pharmacotherapy Response: A Systematic Review of Neuroimaging Studies. Brain Sci. 2022, 12, 386. https://doi.org/10.3390/brainsci12030386
Florence L, Lassi DLS, Kortas GT, Lima DR, de Azevedo-Marques Périco C, Andrade AG, Torales J, Ventriglio A, De Berardis D, De Aquino JP, et al. Brain Correlates of the Alcohol Use Disorder Pharmacotherapy Response: A Systematic Review of Neuroimaging Studies. Brain Sciences. 2022; 12(3):386. https://doi.org/10.3390/brainsci12030386
Chicago/Turabian StyleFlorence, Luiza, Dângela Layne Silva Lassi, Guilherme T. Kortas, Danielle R. Lima, Cintia de Azevedo-Marques Périco, Arthur G. Andrade, Julio Torales, Antonio Ventriglio, Domenico De Berardis, João P. De Aquino, and et al. 2022. "Brain Correlates of the Alcohol Use Disorder Pharmacotherapy Response: A Systematic Review of Neuroimaging Studies" Brain Sciences 12, no. 3: 386. https://doi.org/10.3390/brainsci12030386