
Precision Medicine in Parkinson’s Disease: From Genetic Risk Signals to Personalized Therapy


Abstract
:1. Introduction
2. The Genetic Architecture of PD
3. Autosomal Dominant Genes with High Penetrance
3.1. SNCA (Alpha-Synuclein; OMIM*163890)
α-Synuclein as Therapeutic Target
- (a) α-Synuclein synthesis inhibitors
- (b) Inhibiting α-synuclein aggregation
- (c) Inhibiting α-Synuclein uptake
- (d) Promoting clearance of α-Synuclein: Immunotherapies and Autophagy-enhancing agents
- -
- Immunotherapies
- -
- Autophagy-enhancing agents
4. Autosomal Dominant Genes with Variable Penetrance
4.1. LRRK2 (Leucine-Rich Repeat Kinase 2; OMIM*609007)
Targeting LRRK2 in Clinical Trials
4.2. GBA (Beta-Glucocerebrosidase Acid; OMIM*606463)
4.2.1. Substrate Reduction Therapy
4.2.2. Increasing Glucocerebrosidase Enzymatic Activity
4.2.3. Gene Therapy
4.3. VPS35 (Vacuolar Protein Sorting 35, OMIM*601501)
5. Autosomal Recessive Genes
5.1. Parkin (PRKN or PARK2, OMIM*602544)
5.2. PINK1 (PTEN-Induced Kinase 1, OMIM*608309)
5.3. DJ-1 (Parkinsonism-Associated Deglycase, OMIM*602533)
6. Challenges of Genetic-Driven Disease-Modifying Therapies
7. Role of Genetics in Decision-Making Process on Symptomatic Therapies
8. How Genetic Status Can Help in the Current Clinical Management of PD
8.1. SNCA
8.2. LRRK2
8.3. GBA
8.4. PRKN
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dorsey, E.R.; Bloem, B.R. The Parkinson Pandemic—A Call to Action. JAMA Neurol. 2018, 75, 9–10. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef]
- Hornykiewicz, O. Chemical neuroanatomy of the basal ganglia—Normal and in Parkinson’s disease. J. Chem. Neuroanat. 2001, 22, 3–12. [Google Scholar] [CrossRef]
- Toulorge, D.; Schapira, A.; Hajj, R. Molecular changes in the postmortem parkinsonian brain. J. Neurochem. 2016, 139, 27–58. [Google Scholar] [CrossRef]
- Meissner, W. When does Parkinson’s disease begin? From prodromal disease to motor signs. Rev. Neurol. 2012, 168, 809–814. [Google Scholar] [CrossRef]
- Lang, A.E.; Espay, A.J. Disease Modification in Parkinson’s Disease: Current Approaches, Challenges, and Future Considerations. Mov. Disord. 2018, 33, 660–677. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Fountzilas, E.; Nikanjam, M.; Kurzrock, R. Review of precision cancer medicine: Evolution of the treatment paradigm. Cancer Treat. Rev. 2020, 86, 102019. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Quadri, M.; Mandemakers, W.; Grochowska, M.M.; Masius, R.; Geut, H.; Fabrizio, E.; Breedveld, G.J.; Kuipers, D.; Minneboo, M.; Vergouw, L.J.M.; et al. LRP10 genetic variants in familial Parkinson’s disease and dementia with Lewy bodies: A genome-wide linkage and sequencing study. Lancet Neurol. 2018, 17, 597–608. [Google Scholar] [CrossRef]
- Chen, Y.; Cen, Z.; Zheng, X.; Pan, Q.; Chen, X.; Zhu, L.; Chen, S.; Wu, H.; Xie, F.; Wang, H.; et al. LRP10 in autosomal-dominant Parkinson’s disease. Mov. Disord. 2019, 34, 912–916. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.-X.; Pericak-Vance, M.A.; Siddique, T. Reply to ‘TMEM230 variants in Parkinson’s disease’ and ‘Doubts about TMEM230 as a gene for parkinsonism’. Nat. Genet. 2019, 51, 369–371. [Google Scholar] [CrossRef] [PubMed]
- Farrer, M.J. Doubts about TMEM230 as a gene for parkinsonism. Nat. Genet. 2019, 51, 367–368. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Whelan, E.; Liu, Z.; Liu, C.-F.; Smith, W.W. Controversy of TMEM230 Associated with Parkinson’s Disease. Neuroscience 2020, 453, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Saini, P.; Rudakou, U.; Yu, E.; Ruskey, J.A.; Asayesh, F.; Laurent, S.B.; Spiegelman, D.; Fahn, S.; Waters, C.; Monchi, O.; et al. Association study of DNAJC13, UCHL1, HTRA2, GIGYF2, and EIF4G1 with Parkinson’s disease. Neurobiol. Aging 2020, 100, 119.e7–119.e13. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Kao, A.W.; McKay, A.; Singh, P.P.; Brunet, A.; Huang, E.J. Progranulin, lysosomal regulation and neurodegenerative disease. Nat. Rev. Neurosci. 2017, 18, 325–333. [Google Scholar] [CrossRef]
- Bandres-Ciga, S.; Saez-Atienzar, S.; Bonet-Ponce, L.; Billingsley, K.; Vitale, D.; Blauwendraat, C.; Gibbs, J.R.; Pihlstrøm, L.; Gan-Or, Z.; Cookson, M.R.; et al. The endocytic membrane trafficking pathway plays a major role in the risk of Parkinson’s disease. Mov. Disord. 2019, 34, 460–468. [Google Scholar] [CrossRef]
- Robak, L.A.; Jansen, I.E.; Van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; Heutink, P.; Shulman, J.M. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203. [Google Scholar] [CrossRef]
- Harms, A.S.; Ferreira, S.A.; Romero-Ramos, M. Periphery and brain, innate and adaptive immunity in Parkinson’s disease. Acta Neuropathol. 2021, 141, 527–545. [Google Scholar] [CrossRef]
- Day, J.; Mullin, S. The Genetics of Parkinson’s Disease and Implications for Clinical Practice. Genes 2021, 12, 1006. [Google Scholar] [CrossRef] [PubMed]
- Toffoli, M.; Vieira, S.; Schapira, A. Genetic causes of PD: A pathway to disease modification. Neuropharmacology 2020, 170, 108022. [Google Scholar] [CrossRef] [PubMed]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The Function of α-Synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Edwards, R.H. The physiological role of alpha-synuclein and its relationship to Parkinson’s Disease. J. Neurochem. 2019, 150, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Villar-Piqué, A.; Da Fonseca, T.L.; Outeiro, T.F. Structure, function and toxicity of alpha-synuclein: The Bermuda triangle in synucleinopathies. J. Neurochem. 2015, 139, 240–255. [Google Scholar] [CrossRef]
- Rochet, J.-C.; Hay, B.A.; Guo, M. Molecular Insights into Parkinson’s Disease. Prog. Mol. Biol. Transl. Sci. 2012, 107, 125–188. [Google Scholar] [CrossRef]
- He, S.; Wang, F.; Yung, K.K.L.; Zhang, S.; Qu, S. Effects of α-Synuclein-Associated Post-Translational Modifications in Parkinson’s Disease. ACS Chem. Neurosci. 2021, 12, 1061–1071. [Google Scholar] [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 Is the Dominant Pathological Modification of α-Synuclein in Familial and Sporadic Lewy Body Disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef]
- Lee, K.-W.; Chen, W.; Junn, E.; Im, J.-Y.; Grosso, H.; Sonsalla, P.K.; Feng, X.; Ray, N.; Fernandez, J.R.; Chao, Y.; et al. Enhanced Phosphatase Activity Attenuates -Synucleinopathy in a Mouse Model. J. Neurosci. 2011, 31, 6963–6971. [Google Scholar] [CrossRef]
- Dehay, B.; Bourdenx, M.; Gorry, P.; Przedborski, S.; Vila, M.; Hunot, S.; Singleton, A.; Olanow, C.W.; Merchant, K.M.; Bezard, E.; et al. Targeting α-synuclein for treatment of Parkinson’s disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2015, 14, 855–866. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Yang, C.; Zhang, X.; Li, Y.; Wang, S.; Zheng, L.; Huang, K. C-terminal truncation exacerbates the aggregation and cytotoxicity of α-Synuclein: A vicious cycle in Parkinson’s disease. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2018, 1864, 3714–3725. [Google Scholar] [CrossRef]
- Games, D.; Valera, E.; Spencer, B.; Rockenstein, E.; Mante, M.; Adame, A.; Patrick, C.; Ubhi, K.; Nuber, S.; Sacayon, P.; et al. Reducing C-Terminal-Truncated Alpha-Synuclein by Immunotherapy Attenuates Neurodegeneration and Propagation in Parkinson’s Disease-Like Models. J. Neurosci. 2014, 34, 9441–9454. [Google Scholar] [CrossRef] [PubMed]
- Bassil, F.; Fernagut, P.-O.; Bezard, E.; Pruvost, A.; Leste-Lasserre, T.; Hoang, Q.Q.; Ringe, D.; Petsko, G.A.; Meissner, W.G. Reducing C-terminal truncation mitigates synucleinopathy and neurodegeneration in a transgenic model of multiple system atrophy. Proc. Natl. Acad. Sci. USA 2016, 113, 9593–9598. [Google Scholar] [CrossRef] [PubMed]
- Muntané, G.; Ferrer, I.; Martinez-Vicente, M. α-synuclein phosphorylation and truncation are normal events in the adult human brain. Neuroscience 2012, 200, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front Mol Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.B.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. AlaSOPro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia. Ann. Neurol. 2003, 55, 164–173. [Google Scholar] [CrossRef]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R.; et al. G51D α-synuclein mutation causes a novel Parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Tompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef]
- Proukakis, C.; Dudzik, C.G.; Brier, T.; Mackay, D.S.; Cooper, J.M.; Millhauser, G.L.; Houlden, H.; Schapira, A.H. A novel α-synuclein missense mutation in Parkinson disease. Neurology 2013, 80, 1062–1064. [Google Scholar] [CrossRef] [Green Version]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Pöyhönen, M.; Paetau, A. A novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, e2181–e2185. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein Locus Triplication Causes Parkinson’s Disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.-C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Farrer, M.J.; Bs, J.K.; Forno, L.S.; Lincoln, S.; Wang, D.-S.; Hulihan, M.M.; Maraganore, D.M.; Gwinn, K.; Wszolek, Z.K.; Dickson, D.W.; et al. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann. Neurol. 2004, 55, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.; Gwinn-Hardy, K.; Sharabi, Y.; Li, S.; Holmes, C.; Dendi, R.; Hardy, J.; Singleton, A.; Crawley, A.; Goldstein, D.S. Association between cardiac denervation and parkinsonism caused by α-synuclein gene triplication. Brain 2004, 127, 768–772. [Google Scholar] [CrossRef]
- Tambasco, N.; Nigro, P.; Romoli, M.; Prontera, P.; Simoni, S.; Calabresi, P. A53T in a parkinsonian family: A clinical update of the SNCA phenotypes. J. Neural Transm. 2016, 123, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.Y.; Alcalay, R.N. Genetic Forms of Parkinson’s Disease. Semin Neurol. 2017, 37, 135–146. [Google Scholar] [CrossRef]
- Trinh, J.; Zeldenrust, F.M.; Huang, J.; Kasten, M.; Schaake, S.; Petkovic, S.; Madoev, H.; Grünewald, A.; Almuammar, S.; König, I.R.; et al. Genotype-phenotype relations for the Parkinson’s disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov. Disord. 2018, 33, 1857–1870. [Google Scholar] [CrossRef]
- Kasten, M.; Marras, C.; Klein, C. Nonmotor Signs in Genetic Forms of Parkinson’s Disease. Int. Rev. Neurobiol. 2017, 133, 129–178. [Google Scholar] [CrossRef]
- Chia, R.; Center, T.A.G.; Sabir, M.S.; Bandres-Ciga, S.; Saez-Atienzar, S.; Reynolds, R.H.; Gustavsson, E.; Walton, R.L.; Ahmed, S.; Viollet, C.; et al. Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat. Genet. 2021, 53, 294–303. [Google Scholar] [CrossRef]
- Stoker, T.B.; Camacho, M.; Winder-Rhodes, S.; Liu, G.; Scherzer, C.R.; Foltynie, T.; Barker, R.A.; Williams-Gray, C.H. A common polymorphism in SNCA is associated with accelerated motor decline in GBA-Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2020, 91, 673–674. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Reed, X.; Krohn, L.; Heilbron, K.; Bandres-Ciga, S.; Tan, M.; Gibbs, J.R.; Hernandez, D.G.; Kumaran, R.; Langston, R.; et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 2020, 143, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Soldner, F.; Stelzer, Y.; Shivalila, C.S.; Abraham, B.J.; Latourelle, J.C.; Barrasa, M.I.; Goldmann, J.; Myers, R.H.; Young, R.A.; Jaenisch, R. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 2016, 533, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Ross, O.A.; Braithwaite, A.T.; Skipper, L.M.; Kachergus, J.; Hulihan, M.M.; Middleton, F.A.; Nishioka, K.; Fuchs, J.; Gasser, T.; Maraganore, D.M.; et al. Genomic investigation of α-synuclein multiplication and parkinsonism. Ann. Neurol. 2008, 63, 743–750. [Google Scholar] [CrossRef]
- Cole, T.A.; Zhao, H.; Collier, T.J.; Sandoval, I.; Sortwell, C.E.; Steece-Collier, K.; Daley, B.F.; Booms, A.; Lipton, J.; Welch, M.; et al. α-Synuclein antisense oligonucleotides as a disease-modifying therapy for Parkinson’s disease. JCI Insight 2021, 6, e135633. [Google Scholar] [CrossRef]
- Bhatt, M.A.; Messer, A.; Kordower, J.H. Can Intrabodies Serve as Neuroprotective Therapies for Parkinson’s Disease? Beginning Thoughts. J. Park. Dis. 2013, 3, 581–591. [Google Scholar] [CrossRef]
- Chatterjee, D.; Bhatt, M.; Butler, D.; De Genst, E.; Dobson, C.M.; Messer, A.; Kordower, J.H. Proteasome-targeted nanobodies alleviate pathology and functional decline in an α-synuclein-based Parkinson’s disease model. npj Park. Dis. 2018, 4, 25. [Google Scholar] [CrossRef]
- Reinle, K.; Mogk, A.; Bukau, B. The Diverse Functions of Small Heat Shock Proteins in the Proteostasis Network. J. Mol. Biol. 2021, 434, 167157. [Google Scholar] [CrossRef]
- Weihofen, A.; Liu, Y.; Arndt, J.W.; Huy, C.; Quan, C.; Smith, B.A.; Baeriswyl, J.-L.; Cavegn, N.; Senn, L.; Su, L.; et al. Development of an aggregate-selective, human-derived α-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson’s disease models. Neurobiol. Dis. 2018, 124, 276–288. [Google Scholar] [CrossRef]
- Brys, M.; Fanning, L.; Hung, S.; Ellenbogen, A.; Penner, N.; Yang, M.; Welch, M.; Koenig, E.; David, E.; Fox, T.; et al. Randomized phase I clinical trial of anti–α-synuclein antibody BIIB054. Mov. Disord. 2019, 34, 1154–1163. [Google Scholar] [CrossRef]
- Kuchimanchi, M.; Monine, M.; Muralidharan, K.K.; Woodward, C.; Penner, N. Phase II Dose Selection for Alpha Synuclein–Targeting Antibody Cinpanemab (BIIB054) Based on Target Protein Binding Levels in the Brain. CPT Pharmacometrics Syst. Pharmacol. 2020, 9, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. α-Synuclein Is Degraded by Both Autophagy and the Proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin Protects against Neuron Death in In Vitro and In Vivo Models of Parkinson’s Disease. J. Neurosci. 2010, 30, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Crews, L.; Spencer, B.; Desplats, P.; Patrick, C.; Paulino, A.; Rockenstein, E.; Hansen, L.; Adame, A.; Galasko, D.; Masliah, E. Selective Molecular Alterations in the Autophagy Pathway in Patients with Lewy Body Disease and in Models of α-Synucleinopathy. PLoS ONE 2010, 5, e9313. [Google Scholar] [CrossRef]
- Uehara, T.; Choong, C.-J.; Nakamori, M.; Hayakawa, H.; Nishiyama, K.; Kasahara, Y.; Baba, K.; Nagata, T.; Yokota, T.; Tsuda, H.; et al. Amido-bridged nucleic acid (AmNA)-modified antisense oligonucleotides targeting α-synuclein as a novel therapy for Parkinson’s disease. Sci. Rep. 2019, 9, 7567. [Google Scholar] [CrossRef]
- Longhena, F.; Spano, P.; Bellucci, A. Targeting of Disordered Proteins by Small Molecules in Neurodegenerative Diseases. Handb. Exp. Pharmacol. 2017, 245, 85–110. [Google Scholar] [CrossRef]
- Li, J.; Zhu, M.; Rajamani, S.; Uversky, V.N.; Fink, A.L. Rifampicin Inhibits α-Synuclein Fibrillation and Disaggregates Fibrils. Chem. Biol. 2004, 11, 1513–1521. [Google Scholar] [CrossRef]
- Tomiyama, T.; Asano, S.; Suwa, Y.; Morita, T.; Kataoka, K.; Mori, H.; Endo, N. Rifampicin Prevents the Aggregation and Neurotoxicity of Amyloid β Protein in Vitro. Biochem. Biophys. Res. Commun. 1994, 204, 76–83. [Google Scholar] [CrossRef]
- Zhu, M.; Rajamani, S.; Kaylor, J.; Han, S.; Zhou, F.; Fink, A.L. The Flavonoid Baicalein Inhibits Fibrillation of α-Synuclein and Disaggregates Existing Fibrils. J. Biol. Chem. 2004, 279, 26846–26857. [Google Scholar] [CrossRef]
- Moree, B.; Yin, G.; Lázaro, D.F.; Munari, F.; Strohäker, T.; Giller, K.; Becker, S.; Outeiro, T.F.; Zweckstetter, M.; Salafsky, J. Small Molecules Detected by Second-Harmonic Generation Modulate the Conformation of Monomeric α-Synuclein and Reduce Its Aggregation in Cells. J. Biol. Chem. 2015, 290, 27582–27593. [Google Scholar] [CrossRef] [Green Version]
- Peña-Díaz, S.; Pujols, J.; Conde-Giménez, M.; Čarija, A.; Dalfo, E.; García, J.; Navarro, S.; Pinheiro, F.; Santos, J.; Salvatella, X.; et al. ZPD-2, a Small Compound That Inhibits α-Synuclein Amyloid Aggregation and Its Seeded Polymerization. Front. Mol. Neurosci. 2019, 12, 306. [Google Scholar] [CrossRef] [PubMed]
- Fonseca-Ornelas, L.; Eisbach, S.E.; Paulat, M.; Giller, K.; Fernandez, C.O.; Outeiro, T.F.; Becker, S.; Zweckstetter, M. Small molecule-mediated stabilization of vesicle-associated helical α-synuclein inhibits pathogenic misfolding and aggregation. Nat. Commun. 2014, 5, 5857. [Google Scholar] [CrossRef] [PubMed]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.-I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.-H.; et al. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016, 353. [Google Scholar] [CrossRef] [PubMed]
- Schenk, D.B.; Koller, M.; Ness, D.K.; Griffith, S.G.; Grundman, M.; Zago, W.; Soto, J.A.; Atiee, G.; Ostrowitzki, S.; Kinney, G.G. First-in-human assessment of PRX002, an anti-α-synuclein monoclonal antibody, in healthy volunteers. Mov. Disord. 2016, 32, 211–218. [Google Scholar] [CrossRef]
- Hebron, M.L.; Lonskaya, I.; Moussa, C.E.-H. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of α-synuclein in Parkinson’s disease models. Hum. Mol. Genet. 2013, 22, 3315–3328. [Google Scholar] [CrossRef]
- Karuppagounder, S.S.; Brahmachari, S.; Lee, Y.; Dawson, V.L.; Dawson, T.M.; Ko, H.S. The c-Abl inhibitor, Nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson’s disease. Sci. Rep. 2014, 4, 4874. [Google Scholar] [CrossRef]
- Wu, J.; Xu, X.; Zheng, L.; Mo, J.; Jin, X.; Bao, Y. Nilotinib inhibits microglia-mediated neuroinflammation to protect against dopaminergic neuronal death in Parkinson’s disease models. Int. Immunopharmacol. 2021, 99, 108025. [Google Scholar] [CrossRef]
- Pagan, F.; Hebron, M.; Valadez, E.H.; Torres-Yaghi, Y.; Huang, X.; Mills, R.R.; Wilmarth, B.M.; Howard, H.; Dunn, C.; Carlson, A.; et al. Nilotinib Effects in Parkinson’s disease and Dementia with Lewy bodies. J. Park. Dis. 2016, 6, 503–517. [Google Scholar] [CrossRef]
- Simuni, T.; Fiske, B.; Merchant, K.; Coffey, C.S.; Klingner, E.; Caspell-Garcia, C.; Lafontant, D.-E.; Matthews, H.; Wyse, R.K.; Brundin, P.; et al. Efficacy of Nilotinib in Patients with Moderately Advanced Parkinson Disease. JAMA Neurol. 2021, 78, 312. [Google Scholar] [CrossRef]
- Pagan, F.L.; Hebron, M.L.; Wilmarth, B.; Torres-Yaghi, Y.; Lawler, A.; Mundel, E.E.; Yusuf, N.; Starr, N.J.; Anjum, M.; Arellano, J.; et al. Nilotinib Effects on Safety, Tolerability, and Potential Biomarkers in Parkinson Disease. JAMA Neurol. 2020, 77, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Pagan, F.L.; Wilmarth, B.; Torres-Yaghi, Y.; Ms, M.L.H.; Mulki, S.; Ms, D.F.; Matar, S.; Ahn, J.; Moussa, C. Long-Term Safety and Clinical Effects of Nilotinib in Parkinson’s Disease. Mov. Disord. 2020, 36, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, S.; Park, Y.J.; Yun, S.P.; Kwon, S.H.; Kim, D.; Kim, D.Y.; Shin, J.S.; Cho, D.J.; Lee, G.Y.; et al. The c-Abl inhibitor, Radotinib HCl, is neuroprotective in a preclinical Parkinson’s disease mouse model. Hum. Mol. Genet. 2018, 27, 2344–2356. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Luo, S.; Zhang, J.; Yu, T.; Fu, Z.; Zheng, Y.; Xu, X.; Liu, C.; Fan, M.; Zhang, Z. Exosome-mediated delivery of antisense oligonucleotides targeting α-synuclein ameliorates the pathology in a mouse model of Parkinson’s disease. Neurobiol. Dis. 2020, 148, 105218. [Google Scholar] [CrossRef]
- Jan, A.; Gonçalves, N.P.; Vaegter, C.B.; Jensen, P.H.; Ferreira, N. The Prion-Like Spreading of Alpha-Synuclein in Parkinson’s Disease: Update on Models and Hypotheses. Int. J. Mol. Sci. 2021, 22, 8338. [Google Scholar] [CrossRef]
- Pagano, G.; Boess, F.G.; Taylor, K.I.; Ricci, B.; Mollenhauer, B.; Poewe, W.; Boulay, A.; Anzures-Cabrera, J.; Vogt, A.; Marchesi, M.; et al. A Phase II Study to Evaluate the Safety and Efficacy of Prasinezumab in Early Parkinson’s Disease (PASADENA): Rationale, Design, and Baseline Data. Front. Neurol. 2021, 12, 705407. [Google Scholar] [CrossRef]
- Werner, M.H.; Olanow, C.W. Parkinson’s Disease Modification Through Abl Kinase Inhibition: An Opportunity. Mov. Disord. 2021, 37, 6–15. [Google Scholar] [CrossRef]
- Zharikov, A.D.; Cannon, J.R.; Tapias, V.; Bai, Q.; Horowitz, M.P.; Shah, V.; El Ayadi, A.; Hastings, T.G.; Greenamyre, J.T.; Burton, E. shRNA targeting alpha-synuclein prevents neurodegeneration in a Parkinson’s disease model. J. Clin. Investig. 2015, 125, 2721–2735. [Google Scholar] [CrossRef]
- Gorbatyuk, O.S.; Li, S.; Nash, K.; Gorbatyuk, M.; Lewin, A.S.; Sullivan, L.F.; Mandel, R.J.; Chen, W.; Meyers, C.; Manfredsson, F.P.; et al. In Vivo RNAi-Mediated alpha-Synuclein Silencing Induces Nigrostriatal Degeneration. Mol. Ther. 2010, 18, 1450–1457. [Google Scholar] [CrossRef]
- Price, D.L.; Koike, M.A.; Khan, A.; Wrasidlo, W.; Rockenstein, E.; Masliah, E.; Bonhaus, D. The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson’s disease. Sci. Rep. 2018, 8, 16165. [Google Scholar] [CrossRef] [Green Version]
- McFarthing, K.; Simuni, T. Clinical Trial Highlights: Targeting Alpha-Synuclein. J. Park. Dis. 2019, 9, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Schofield, D.J.; Irving, L.; Calo, L.; Bogstedt, A.; Rees, G.; Nuccitelli, A.; Narwal, R.; Petrone, M.; Roberts, J.; Brown, L.; et al. Preclinical development of a high affinity α-synuclein antibody, MEDI1341, that can enter the brain, sequester extracellular α-synuclein and attenuate α-synuclein spreading in vivo. Neurobiol. Dis. 2019, 132, 104582. [Google Scholar] [CrossRef] [PubMed]
- Watts, R.J.; Dennis, M.S. Bispecific antibodies for delivery into the brain. Curr. Opin. Chem. Biol. 2013, 17, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Bjørnevik, K.; Im, D.S.; Flierl, A.; Dong, X.; Locascio, J.J.; Abo, K.M.; Long, E.; Jin, M.; Xu, B.; et al. β2-Adrenoreceptor is a regulator of the α-synuclein gene driving risk of Parkinson’s disease. Science 2017, 357, 891–898. [Google Scholar] [CrossRef]
- Gronich, N.; Abernethy, D.R.; Auriel, E.; Lavi, I.; Rennert, G.; Saliba, W. β2-adrenoceptor agonists and antagonists and risk of Parkinson’s disease. Mov. Disord. 2018, 33, 1465–1471. [Google Scholar] [CrossRef]
- Hopfner, F.; Höglinger, G.U.; Kuhlenbäumer, G.; Pottegård, A.; Wod, M.; Christensen, K.; Tanner, C.M.; Deuschl, G. β-adrenoreceptors and the risk of Parkinson’s disease. Lancet Neurol. 2020, 19, 247–254. [Google Scholar] [CrossRef]
- Krishnan, R.; Tsubery, H.; Proschitsky, M.Y.; Asp, E.; Lulu, M.; Gilead, S.; Gartner, M.; Waltho, J.P.; Davis, P.J.; Hounslow, A.M.; et al. A Bacteriophage Capsid Protein Provides a General Amyloid Interaction Motif (GAIM) That Binds and Remodels Misfolded Protein Assemblies. J. Mol. Biol. 2014, 426, 2500–2519. [Google Scholar] [CrossRef]
- Nordström, E.; Eriksson, F.; Sigvardson, J.; Johannesson, M.; Kasrayan, A.; Jones-Kostalla, M.; Appelkvist, P.; Söderberg, L.; Nygren, P.; Blom, M.; et al. ABBV-0805, a novel antibody selective for soluble aggregated α-synuclein, prolongs lifespan and prevents buildup of α-synuclein pathology in mouse models of Parkinson’s disease. Neurobiol. Dis. 2021, 161, 105543. [Google Scholar] [CrossRef]
- Wagner, J.; Ryazanov, S.; Leonov, A.; Levin, J.; Shi, S.; Schmidt, F.; Prix, C.; Pan-Montojo, F.; Bertsch, U.; Mitteregger-Kretzschmar, G.; et al. Anle138b: A novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol. 2013, 125, 795–813. [Google Scholar] [CrossRef]
- Ghosh, A.; Tyson, T.; George, S.; Hildebrandt, E.N.; Steiner, J.A.; Madaj, Z.; Schulz, E.; Machiela, E.; McDonald, W.G.; Galvis, M.L.E.; et al. Mitochondrial pyruvate carrier regulates autophagy, inflammation, and neurodegeneration in experimental models of Parkinson’s disease. Sci. Transl. Med. 2016, 8, 368ra174. [Google Scholar] [CrossRef]
- O’Hara, D.; Davis, G.M.; Adlesic, N.A.; Hayes, J.M.; Davey, G.P. Dichloroacetate Stabilizes Mitochondrial Fusion Dynamics in Models of Neurodegeneration. Front. Mol. Neurosci. 2019, 12, 219. [Google Scholar] [CrossRef] [PubMed]
- Divakaruni, A.S.; Wallace, M.; Buren, C.; Martyniuk, K.; Andreyev, A.Y.; Li, E.; Fields, J.A.; Cordes, T.; Reynolds, I.J.; Bloodgood, B.; et al. Inhibition of the mitochondrial pyruvate carrier protects from excitotoxic neuronal death. J. Cell Biol. 2017, 216, 1091–1105. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. Targeting the Mitochondrial Pyruvate Carrier for Neuroprotection. Brain Sci. 2019, 9, 238. [Google Scholar] [CrossRef]
- Moors, T.E.; Hoozemans, J.J.M.; Ingrassia, A.; Beccari, T.; Parnetti, L.; Chartier-Harlin, M.-C.; Van De Berg, W.D.J. Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol. Neurodegener. 2017, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, F.; Tampi, R.R. Valproic Acid–Induced Parkinsonism in the Elderly: A Comprehensive Review of the Literature. Am. J. Geriatr. Pharmacother. 2011, 9, 405–412. [Google Scholar] [CrossRef]
- Schwab, K.; Frahm, S.; Horsley, D.; Rickard, J.E.; Melis, V.; Goatman, E.A.; Magbagbeolu, M.; Douglas, M.; Leith, M.G.; Baddeley, T.C.; et al. A Protein Aggregation Inhibitor, Leuco-Methylthioninium Bis(Hydromethanesulfonate), Decreases α-Synuclein Inclusions in a Transgenic Mouse Model of Synucleinopathy. Front. Mol. Neurosci. 2018, 10, 447. [Google Scholar] [CrossRef]
- Volc, D.; Poewe, W.; Kutzelnigg, A.; Lührs, P.; Thun-Hohenstein, C.; Schneeberger, A.; Galabova, G.; Majbour, N.; Vaikath, N.; El-Agnaf, O.; et al. Safety and immunogenicity of the α-synuclein active immunotherapeutic PD01A in patients with Parkinson’s disease: A randomised, single-blinded, phase 1 trial. Lancet Neurol. 2020, 19, 591–600. [Google Scholar] [CrossRef]
- Fjord-Larsen, L.; Thougaard, A.; Wegener, K.M.; Christiansen, J.; Larsen, F.; Schrøder-Hansen, L.M.; Kaarde, M.; Ditlevsen, D.K. Nonclinical safety evaluation, pharmacokinetics, and target engagement of Lu AF82422, a monoclonal IgG1 antibody against alpha-synuclein in development for treatment of synucleinopathies. mAbs 2021, 13, 1994690. [Google Scholar] [CrossRef]
- Zhang, P.; Park, H.-J.; Zhang, J.; Junn, E.; Andrews, R.J.; Velagapudi, S.P.; Abegg, D.; Vishnu, K.; Costales, M.G.; Childs-Disney, J.L.; et al. Translation of the intrinsically disordered protein α-synuclein is inhibited by a small molecule targeting its structured mRNA. Proc. Natl. Acad. Sci. USA 2020, 117, 1457–1467. [Google Scholar] [CrossRef]
- Bengoa-Vergniory, N.; Faggiani, E.; Ramos-Gonzalez, P.; Kirkiz, E.; Connor-Robson, N.; Brown, L.V.; Siddique, I.; Li, Z.; Vingill, S.; Cioroch, M.; et al. CLR01 protects dopaminergic neurons in vitro and in mouse models of Parkinson’s disease. Nat. Commun. 2020, 11, 4885. [Google Scholar] [CrossRef]
- Mandler, M.; Valera, E.; Rockenstein, E.; Weninger, H.; Patrick, C.; Adame, A.; Santic, R.; Meindl, S.; Vigl, B.; Smrzka, O.; et al. Next-generation active immunization approach for synucleinopathies: Implications for Parkinson’s disease clinical trials. Acta Neuropathol. 2014, 127, 861–879. [Google Scholar] [CrossRef] [PubMed]
- Myöhänen, T.; Hannula, M.; Van Elzen, R.; Gerard, M.; Van Der Veken, P.; García-Horsman, J.; Baekelandt, V.; Männistö, P.; Lambeir, A. A prolyl oligopeptidase inhibitor, KYP-2047, reduces α-synuclein protein levels and aggregates in cellular and animal models of Parkinson’s disease. J. Cereb. Blood Flow Metab. 2012, 166, 1097–1113. [Google Scholar] [CrossRef] [PubMed]
- Savolainen, M.H.; Richie, C.T.; Harvey, B.K.; Männistö, P.T.; Maguire-Zeiss, K.A.; Myöhänen, T.T. The beneficial effect of a prolyl oligopeptidase inhibitor, KYP-2047, on alpha-synuclein clearance and autophagy in A30P transgenic mouse. Neurobiol. Dis. 2014, 68, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Svarcbahs, R.; Julku, U.H.; Myöhänen, T.T. Inhibition of Prolyl Oligopeptidase Restores Spontaneous Motor Behavior in the α-Synuclein Virus Vector–Based Parkinson’s Disease Mouse Model by Decreasing α-Synuclein Oligomeric Species in Mouse Brain. J. Neurosci. 2016, 36, 12485–12497. [Google Scholar] [CrossRef]
- Rostami, J.; Jäntti, M.; Cui, H.; Rinne, M.K.; Kukkonen, J.P.; Falk, A.; Erlandsson, A.; Myöhänen, T. Prolyl oligopeptidase inhibition by KYP-2407 increases alpha-synuclein fibril degradation in neuron-like cells. Biomed. Pharmacother. 2020, 131, 110788. [Google Scholar] [CrossRef]
- Poewe, W.; Volc, D.; Seppi, K.; Medori, R.; Lührs, P.; Kutzelnigg, A.; Djamshidian, A.; Thun-Hohenstein, C.; Meissner, W.G.; Rascol, O.; et al. Safety and Tolerability of Active Immunotherapy Targeting α-Synuclein with PD03A in Patients with Early Parkinson’s Disease: A Randomized, Placebo-Controlled, Phase 1 Study. J. Park. Dis. 2021, 11, 1079–1089. [Google Scholar] [CrossRef]
- Wrasidlo, W.; Tsigelny, I.F.; Price, D.L.; Dutta, G.; Rockenstein, E.; Schwarz, T.C.; Ledolter, K.; Bonhaus, D.; Paulino, A.; Eleuteri, S.; et al. A de novocompound targeting α-synuclein improves deficits in models of Parkinson’s disease. Brain 2016, 139, 3217–3236. [Google Scholar] [CrossRef]
- Daniels, M.J.; Nourse, J.B., Jr.; Kim, H.; Sainati, V.; Schiavina, M.; Murrali, M.G.; Pan, B.; Ferrie, J.J.; Haney, C.M.; Moons, R.; et al. Cyclized NDGA modifies dynamic α-synuclein monomers preventing aggregation and toxicity. Sci. Rep. 2019, 9, 2937. [Google Scholar] [CrossRef]
- Tatenhorst, L.; Eckermann, K.; Dambeck, V.; Fonseca-Ornelas, L.; Walle, H.; Da Fonseca, T.L.; Koch, J.C.; Becker, S.; Tönges, L.; Bähr, M.; et al. Fasudil attenuates aggregation of α-synuclein in models of Parkinson’s disease. Acta Neuropathol. Commun. 2016, 4, 39. [Google Scholar] [CrossRef]
- Perni, M.; Galvagnion, C.; Maltsev, A.; Meisl, G.; Müller, M.B.D.; Challa, P.K.; Kirkegaard, J.B.; Flagmeier, P.; Cohen, S.I.A.; Cascella, R.; et al. A natural product inhibits the initiation of α-synuclein aggregation and suppresses its toxicity. Proc. Natl. Acad. Sci. USA 2017, 114, E1009–E1017. [Google Scholar] [CrossRef] [Green Version]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Wild, E.J.; Tabrizi, S.J. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol. 2017, 16, 837–847. [Google Scholar] [CrossRef]
- Chen, C.-L.; Wang, S.-Y.; Chen, T.-C.; Chuang, C.-S. Association between β2-Adrenoreceptor Medications and Risk of Parkinson’s Disease: A Meta-Analysis. Medicina 2021, 57, 1006. [Google Scholar] [CrossRef] [PubMed]
- Alexander, G.M.; Schwartzman, R.J.; Nukes, T.A.; Grothusen, J.R.; Hooker, M.D. β2-Adrenergic agonist as adjunct therapy to levodopa in Parkinson’s disease. Neurology 1994, 44, 1511. [Google Scholar] [CrossRef]
- Hishida, R.; Kurahashi, K.; Narita, S.; Baba, T.; Matsunaga, M. “Wearing-off” and β2-adrenoceptor agonist in Parkinson’s disease. Lancet 1992, 339, 870. [Google Scholar] [CrossRef]
- Uc, E.Y.; Lambert, C.P.; Harik, S.I.; Rodnitzky, R.L.; Evans, W.J. Albuterol Improves Response to Levodopa and Increases Skeletal Muscle Mass in Patients with Fluctuating Parkinson Disease. Clin. Neuropharmacol. 2003, 26, 207–212. [Google Scholar] [CrossRef]
- Magistrelli, L.; Comi, C. Beta2-Adrenoceptor Agonists in Parkinson’s Disease and Other Synucleinopathies. J. Neuroimmune Pharmacol. 2019, 15, 74–81. [Google Scholar] [CrossRef]
- Meissner, W.G.; Traon, A.P.; Foubert-Samier, A.; Galabova, G.; Galitzky, M.; Kutzelnigg, A.; Laurens, B.; Lührs, P.; Medori, R.; Péran, P.; et al. A Phase 1 Randomized Trial of Specific Active α-Synuclein Immunotherapies PD01A and PD03A in Multiple System Atrophy. Mov. Disord. 2020, 35, 1957–1965. [Google Scholar] [CrossRef]
- Wagner, J.; Krauss, S.; Shi, S.; Ryazanov, S.; Steffen, J.; Miklitz, C.; Leonov, A.; Kleinknecht, A.; Göricke, B.; Weishaupt, J.H.; et al. Reducing tau aggregates with anle138b delays disease progression in a mouse model of tauopathies. Acta Neuropathol. 2015, 130, 619–631. [Google Scholar] [CrossRef]
- Levin, J.; Schmidt, F.; Boehm, C.; Prix, C.; Bötzel, K.; Ryazanov, S.; Leonov, A.; Griesinger, C.; Giese, A. The oligomer modulator anle138b inhibits disease progression in a Parkinson mouse model even with treatment started after disease onset. Acta Neuropathol. 2014, 127, 779–780. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, A.M.; Urbanke, H.; Gillman, A.L.; Lee, J.; Ryazanov, S.; Agbemenyah, H.Y.; Benito, E.; Jain, G.; Kaurani, L.; Grigorian, G.; et al. The diphenylpyrazole compound anle138b blocks Aβ channels and rescues disease phenotypes in a mouse model for amyloid pathology. EMBO Mol. Med. 2017, 10, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Heras-Garvin, A.; Weckbecker, D.; Ryazanov, S.; Leonov, A.; Griesinger, C.; Giese, A.; Wenning, G.K.; Stefanova, N. Anle138b modulates α-synuclein oligomerization and prevents motor decline and neurodegeneration in a mouse model of multiple system atrophy. Mov. Disord. 2018, 34, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Sinnige, T.; Yu, A.; Morimoto, R.I. Challenging Proteostasis: Role of the Chaperone Network to Control Aggregation-Prone Proteins in Human Disease. J. Pathol. 2020, 1243, 53–68. [Google Scholar] [CrossRef]
- Horonchik, L.; Tzaban, S.; Ben-Zaken, O.; Yedidia, Y.; Rouvinski, A.; Papy-Garcia, D.; Barritault, D.; Vlodavsky, I.; Taraboulos, A. Heparan Sulfate Is a Cellular Receptor for Purified Infectious Prions. J. Biol. Chem. 2005, 280, 17062–17067. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; Devos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; MacLeod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Autophagy and Alpha-Synuclein: Relevance to Parkinson’s Disease and Related Synucleopathies. Mov. Disord. 2016, 31, 178–192. [Google Scholar] [CrossRef]
- Zondler, L.; Kostka, M.; Garidel, P.; Heinzelmann, U.; Hengerer, B.; Mayer, B.; Weishaupt, J.H.; Gillardon, F.; Danzer, K.M. Proteasome impairment by α-synuclein. PLoS ONE 2017, 12, e0184040. [Google Scholar] [CrossRef]
- Lee, B.-H.; Lee, M.J.; Park, S.; Oh, D.-C.; Elsasser, S.; Chen, P.-C.; Gartner, C.; Dimova, N.; Hanna, J.; Gygi, S.P.; et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010, 467, 179–184. [Google Scholar] [CrossRef]
- Banerjee, C.; Roy, M.; Mondal, R.; Chakraborty, J. USP14 as a Therapeutic Target Against Neurodegeneration: A Rat Brain Perspective. Front. Cell Dev. Biol. 2020, 8, 727. [Google Scholar] [CrossRef]
- Nguyen, A.P.T.; Tsika, E.; Kelly, K.; Levine, N.; Chen, X.; West, A.B.; Boularand, S.; Barneoud, P.; Moore, D.J. Dopaminergic neurodegeneration induced by Parkinson’s disease-linked G2019S LRRK2 is dependent on kinase and GTPase activity. Proc. Natl. Acad. Sci. USA 2020, 117, 17296–17307. [Google Scholar] [CrossRef] [PubMed]
- Rubio, J.P.; Topp, S.; Warren, L.; Jean, P.L.S.; Wegmann, D.; Kessner, D.; Novembre, J.; Shen, J.; Fraser, D.; Aponte, J.; et al. Deep sequencing of the LRRK2gene in 14,002 individuals reveals evidence of purifying selection and independent origin of the p.Arg1628Pro mutation in Europe. Hum. Mutat. 2012, 33, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef]
- Vinagre-Aragón, A.; Campo-Caballero, D.; Mondragón-Rezola, E.; Pardina-Vilella, L.; Eguiazu, H.H.; Gorostidi, A.; Croitoru, I.; Bergareche, A.; Ruiz-Martinez, J. A More Homogeneous Phenotype in Parkinson’s Disease Related to R1441G Mutation in the LRRK2 Gene. Front. Neurol. 2021, 12, 635396. [Google Scholar] [CrossRef]
- Hentati, F.; Trinh, J.; Thompson, C.; Nosova, E.; Farrer, M.J.; Aasly, J.O. LRRK2 parkinsonism in Tunisia and Norway: A comparative analysis of disease penetrance. Neurology 2014, 83, 568–569. [Google Scholar] [CrossRef]
- Ms, A.J.L.; Wang, Y.; Alcalay, M.R.N.; Ms, H.M.; Saunders-Pullman, M.R.; Bressman, S.; Corvol, J.; Brice, A.; Lesage, S.; Mangone, G.; et al. Penetrance estimate of LRRK2 p.G2019S mutation in individuals of non-Ashkenazi Jewish ancestry. Mov. Disord. 2017, 32, 1432–1438. [Google Scholar] [CrossRef]
- Marder, K.; Wang, Y.; Alcalay, R.N.; Mejia-Santana, H.; Tang, M.-X.; Lee, A.; Raymond, D.; Mirelman, A.; Saunders-Pullman, R.; Clark, L.; et al. Age-specific penetrance of LRRK2G2019S in the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium. Neurology 2015, 85, 89–95. [Google Scholar] [CrossRef]
- Ross, O.A.; Soto-Ortolaza, A.I.; Heckman, M.G.; Aasly, J.O.; Abahuni, N.; Annesi, G.; Bacon, J.A.; Bardien, S.; Bozi, M.; Brice, A.; et al. Association of LRRK2 exonic variants with susceptibility to Parkinson’s disease: A case–control study. Lancet Neurol. 2011, 10, 898–908. [Google Scholar] [CrossRef]
- Kestenbaum, M.; Alcalay, R.N. Clinical Features of LRRK2 Carriers with Parkinson’s Disease. LRRK2 2017, 14, 31–48. [Google Scholar] [CrossRef]
- Marras, C.; Alcalay, R.N.; Caspell-Garcia, C.; Coffey, C.; Chan, P.; Duda, J.E.; Facheris, M.F.; Fernández-Santiago, R.; Ruíz-Martínez, J.; Mestre, T.; et al. Motor and nonmotor heterogeneity of LRRK2 -related and idiopathic Parkinson’s disease. Mov. Disord. 2016, 31, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Rui, Q.; Ni, H.; Li, D.; Gao, R.; Chen, G. The Role of LRRK2 in Neurodegeneration of Parkinson Disease. Curr. Neuropharmacol. 2018, 16, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Howlett, E.H.; Jensen, N.; Belmonte, F.; Zafar, F.; Hu, X.; Kluss, J.; Schüle, B.; Kaufman, B.A.; Greenamyre, J.T.; Sanders, L.H. LRRK2 G2019S-induced mitochondrial DNA damage is LRRK2 kinase dependent and inhibition restores mtDNA integrity in Parkinson’s disease. Hum. Mol. Genet. 2017, 26, 4340–4351. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.D.; Shin, J.H.; VanKampen, J.; Petrucelli, L.; West, A.; Ko, H.S.; Lee, Y.I.; Maguire-Zeiss, K.A.; Bowers, W.J.; Federoff, H.J.; et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat. Med. 2010, 16, 998–1000. [Google Scholar] [CrossRef] [PubMed]
- Tsika, E.; Nguyen, A.P.T.; Dusonchet, J.; Colin, P.; Schneider, B.L.; Moore, D.J. Adenoviral-mediated expression of G2019S LRRK2 induces striatal pathology in a kinase-dependent manner in a rat model of Parkinson’s disease. Neurobiol. Dis. 2015, 77, 49–61. [Google Scholar] [CrossRef]
- Yakhine-Diop, S.M.S.; Rodríguez-Arribas, M.; Canales-Cortés, S.; Martínez-Chacón, G.; Uribe-Carretero, E.; Blanco-Benítez, M.; Duque-González, G.; Paredes-Barquero, M.; Alegre-Cortés, E.; Climent, V.; et al. The parkinsonian LRRK2 R1441G mutation shows macroautophagy-mitophagy dysregulation concomitant with endoplasmic reticulum stress. Cell Biol. Toxicol. 2021, 1–23. [Google Scholar] [CrossRef]
- Di Maio, R.; Hoffman, E.K.; Rocha, E.M.; Keeney, M.T.; Sanders, L.H.; De Miranda, B.R.; Zharikov, A.; Van Laar, A.; Stepan, A.F.; Lanz, T.A.; et al. LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med. 2018, 10, eaar5429. [Google Scholar] [CrossRef]
- Mir, R.; Tonelli, F.; Lis, P.; Macartney, T.; Polinski, N.K.; Martinez, T.N.; Chou, M.-Y.; Howden, A.J.; König, T.; Hotzy, C.; et al. The Parkinson’s disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human. Biochem. J. 2018, 475, 1861–1883. [Google Scholar] [CrossRef]
- Bonello, F.; Hassoun, S.-M.; Mouton-Liger, F.; Shin, Y.S.; Muscat, A.; Tesson, C.; Lesage, S.; Beart, P.M.; Brice, A.; Krupp, J.; et al. LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: Pathologic insights into Parkinson’s disease. Hum. Mol. Genet. 2019, 28, 1645–1660. [Google Scholar] [CrossRef]
- Bae, E.J.; Kim, D.K.; Kim, C.; Mante, M.; Adame, A.; Rockenstein, E.; Ulusoy, A.; Klinkenberg, M.; Jeong, G.R.; Bae, J.R.; et al. The physiological role of α-synuclein and its relationship to Parkinson’s Disease. J. Neurochem. 2019, 150, 475–486. [Google Scholar]
- Wojewska, D.; Kortholt, A. LRRK2 Targeting Strategies as Potential Treatment of Parkinson’s Disease. Biomolecules 2021, 11, 1101. [Google Scholar] [CrossRef] [PubMed]
- Estrada, A.A.; Liu, X.; Baker-Glenn, C.; Beresford, A.; Burdick, D.J.; Chambers, M.; Chan, B.K.; Chen, H.; Ding, X.; DiPasquale, A.G.; et al. Discovery of Highly Potent, Selective, and Brain-Penetrable Leucine-Rich Repeat Kinase 2 (LRRK2) Small Molecule Inhibitors. J. Med. Chem. 2012, 55, 9416–9433. [Google Scholar] [CrossRef] [PubMed]
- Estrada, A.A.; Chan, B.K.; Baker-Glenn, C.; Beresford, A.; Burdick, D.J.; Chambers, M.; Chen, H.; Dominguez, S.L.; Dotson, J.; Drummond, J.; et al. Discovery of Highly Potent, Selective, and Brain-Penetrant Aminopyrazole Leucine-Rich Repeat Kinase 2 (LRRK2) Small Molecule Inhibitors. J. Med. Chem. 2014, 57, 921–936. [Google Scholar] [CrossRef]
- Hatcher, J.M.; Zhang, J.; Choi, H.G.; Ito, G.; Alessi, D.R.; Gray, N.S. Discovery of a Pyrrolopyrimidine (JH-II-127), a Highly Potent, Selective, and Brain Penetrant LRRK2 Inhibitor. ACS Med. Chem. Lett. 2015, 6, 584–589. [Google Scholar] [CrossRef]
- Henderson, J.L.; Kormos, B.L.; Hayward, M.M.; Coffman, K.J.; Jasti, J.; Kurumbail, R.G.; Wager, T.T.; Verhoest, P.R.; Noell, G.S.; Chen, Y.; et al. Discovery and Preclinical Profiling of 3-[4-(Morpholin-4-yl)-7 H-pyrrolo[2,3-d]pyrimidin-5-yl]benzonitrile (PF-06447475), a Highly Potent, Selective, Brain Penetrant, and in Vivo Active LRRK2 Kinase Inhibitor. J. Med. Chem. 2014, 58, 419–432. [Google Scholar] [CrossRef]
- Zhang, J.; Deng, X.; Choi, H.G.; Alessi, D.R.; Gray, N.S. Characterization of TAE684 as a potent LRRK2 kinase inhibitor. Bioorganic Med. Chem. Lett. 2012, 22, 1864–1869. [Google Scholar] [CrossRef]
- Choi, H.G.; Zhang, J.; Deng, X.; Hatcher, J.M.; Patricelli, M.P.; Zhao, Z.; Alessi, D.R.; Gray, N.S. Brain Penetrant LRRK2 Inhibitor. ACS Med. Chem. Lett. 2012, 3, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Fell, M.J.; Mirescu, C.; Basu, K.; Cheewatrakoolpong, B.; DeMong, D.E.; Ellis, J.M.; Hyde, L.A.; Lin, Y.; Markgraf, C.G.; Mei, H.; et al. MLi-2, a Potent, Selective, and Centrally Active Compound for Exploring the Therapeutic Potential and Safety of LRRK2 Kinase Inhibition. J. Pharmacol. Exp. Ther. 2015, 355, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.A.; Wegener, K.M.; Larsen, S.; Badolo, L.; Smith, G.P.; Jeggo, R.; Jensen, P.H.; Sotty, F.; Christensen, K.V.; Thougaard, A. PFE-360-induced LRRK2 inhibition induces reversible, non-adverse renal changes in rats. Toxicology 2018, 395, 15–22. [Google Scholar] [CrossRef]
- Andersen, M.A.; Christensen, K.V.; Badolo, L.; Smith, G.P.; Jeggo, R.; Jensen, P.H.; Andersen, K.J.; Sotty, F. Parkinson’s disease-like burst firing activity in subthalamic nucleus induced by AAV-α-synuclein is normalized by LRRK2 modulation. Neurobiol. Dis. 2018, 116, 13–27. [Google Scholar] [CrossRef]
- Andersen, M.A.; Sotty, F.; Jensen, P.H.; Badolo, L.; Jeggo, R.; Smith, G.P.; Christensen, K.V. Long-Term Exposure to PFE-360 in the AAV-α-Synuclein Rat Model: Findings and Implications. Eneuro 2019, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennings, D.; LeWitt, P.; Kern, D.; Goodman, I.; Siderowf, A.; Omidvar, O.; Ellenbogen, A.; Aldred, J.; Macuica, R.; Huntwork-Rodriguez, S.; et al. Safety, Tolerability, and Pharmacodynamic Profile of DNL201 at dose levels demonstrating LRRK2 inhibition in Parkinson’s Disease Patients with and without LRRK2 mutations. Mov. Disord. 2020, 35 (Suppl. S1). [Google Scholar]
- Baptista, M.A.S.; Merchant, K.; Barrett, T.; Bhargava, S.; Bryce, D.K.; Ellis, J.M.; Estrada, A.A.; Fell, M.J.; Fiske, B.K.; Fuji, R.N.; et al. LRRK2 inhibitors induce reversible changes in nonhuman primate lungs without measurable pulmonary deficits. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Singh, R.K.; Soliman, A.; Guaitoli, G.; Störmer, E.; von Zweydorf, F.; Maso, T.D.; Oun, A.; Van Rillaer, L.; Schmidt, S.H.; Chatterjee, D.; et al. Nanobodies as allosteric modulators of Parkinson’s disease-associated LRRK2. Proc. Natl. Acad. Sci. USA 2022, 119, e2112712119. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.T.; John, N.; Delic, V.; Ikeda-Lee, K.; Kim, A.; Weihofen, A.; Swayze, E.E.; Kordasiewicz, H.B.; West, A.B.; Volpicelli-Daley, L.A. LRRK2 Antisense Oligonucleotides Ameliorate α-Synuclein Inclusion Formation in a Parkinson’s Disease Mouse Model. Mol. Ther. -Nucleic Acids 2017, 8, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.; West, A.B. Pharmacodynamic Biomarkers for Emerging LRRK2 Therapeutics. Front. Neurosci. 2020, 14, 807. [Google Scholar] [CrossRef]
- Blandini, F.; Cilia, R.; Cerri, S.; Pezzoli, G.; Schapira, A.; Mullin, S.; Lanciego, J.L. Glucocerebrosidase mutations and synucleinopathies: Toward a model of precision medicine. Mov. Disord. 2018, 34, 9–21. [Google Scholar] [CrossRef]
- Mignot, C.; Gelot, A.; De Villemeur, T.B. Gaucher disease. Handb. Clin. Neurol. 2013, 113, 1709–1715. [Google Scholar] [CrossRef]
- Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher disease: Mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef]
- Beutler, E.; Gelbart, T.; Scott, C.R. Hematologically important mutations: Gaucher disease. Blood Cells Mol. Dis. 2005, 35, 355–364. [Google Scholar] [CrossRef]
- Petrucci, S.; Ginevrino, M.; Trezzi, I.; Monfrini, E.; Ricciardi, L.; Albanese, A.; Avenali, M.; Barone, P.; Bentivoglio, A.R.; Bonifati, V.; et al. GBA -Related Parkinson’s Disease: Dissection of Genotype–Phenotype Correlates in a Large Italian Cohort. Mov. Disord. 2020, 35, 2106–2111. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Nalls, M.; Aasly, J.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migdalska-Richards, A.; Schapira, A.H.V. The relationship between glucocerebrosidase mutations and Parkinson disease. J. Neurochem. 2016, 139, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Straniero, L.; Asselta, R.; Bonvegna, S.; Rimoldi, V.; Melistaccio, G.; Soldà, G.; Aureli, M.; Della Porta, M.; Lucca, U.; Di Fonzo, A.; et al. The SPID-GBA study. Neurol. Genet. 2020, 6, e523. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, J.; Xu, C.; Feng, J.; Li, J. Effects of glucocerebrosidase gene polymorphisms and mutations on the risk of Parkinson’s disease dementia: A meta-analysis. Neurosci. Lett. 2019, 714, 134544. [Google Scholar] [CrossRef]
- Shiner, T.; Mirelman, A.; Weisz, M.G.; Bar-Shira, A.; Ash, E.; Cialic, R.; Nevler, N.; Gurevich, T.; Bregman, N.; Orr-Urtreger, A.; et al. High Frequency of GBA Gene Mutations in Dementia with Lewy Bodies Among Ashkenazi Jews. JAMA Neurol. 2016, 73, 1448–1453. [Google Scholar] [CrossRef]
- Setó-Salvia, N.; Pagonabarraga, J.; Houlden, H.; Sedano, B.M.P.; Icardo, O.D.; Tucci, A.; Paisán-Ruiz, C.; Campolongo, A.; Antón-Aguirre, S.; Martín, I.; et al. Glucocerebrosidase mutations confer a greater risk of dementia during Parkinson’s disease course. Mov. Disord. 2011, 27, 393–399. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Giladi, N.; Rozovski, U.; Shifrin, C.; Rosner, S.; Gurevich, T.; Bar-Shira, A.; Orr-Urtreger, A. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 2008, 70, 2277–2283. [Google Scholar] [CrossRef]
- Malek, N.; Weil, R.S.; Bresner, C.; Lawton, M.A.; A Grosset, K.; Tan, M.; Bajaj, N.; Barker, R.A.; Burn, D.J.; Foltynie, T.; et al. Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study. J. Neurol. Neurosurg. Psychiatry 2018, 89, 702–709. [Google Scholar] [CrossRef]
- Cilia, R.; Tunesi, S.; Marotta, G.; Cereda, E.; Siri, C.; Tesei, S.; Zecchinelli, A.L.; Canesi, M.; Mariani, C.B.; Meucci, N.; et al. Survival and dementia in GBA -associated Parkinson’s disease: The mutation matters. Ann. Neurol. 2016, 80, 662–673. [Google Scholar] [CrossRef]
- Lerche, S.; Wurster, I.; Roeben, B.; Zimmermann, M.; Riebenbauer, B.; Deuschle, C.; Hauser, A.; Schulte, C.; Berg, D.; Maetzler, W.; et al. Parkinson’s Disease: Glucocerebrosidase 1 Mutation Severity Is Associated with CSF Alpha-Synuclein Profiles. Mov. Disord. 2019, 35, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Sardi, S.P.; Cheng, S.H.; Shihabuddin, L.S. Gaucher-related synucleinopathies: The examination of sporadic neurodegeneration from a rare (disease) angle. Prog. Neurobiol. 2015, 125, 47–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gegg, M.E.; Burke, D.; Heales, S.J.R.; Cooper, J.M.; Hardy, J.; Wood, N.W.; Schapira, A.H.V. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol. 2012, 72, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Parnetti, L.; Paciotti, S.; Eusebi, P.; Dardis, A.; Zampieri, S.; Chiasserini, D.; Tasegian, A.; Tambasco, N.; Bembi, B.; Calabresi, P.; et al. Cerebrospinal fluid β-glucocerebrosidase activity is reduced in parkinson’s disease patients. Mov. Disord. 2017, 32, 1423–1431. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Levy, O.A.; Waters, C.H.; Fahn, S.; Ford, B.; Kuo, S.-H.; Mazzoni, P.; Pauciulo, M.W.; Nichols, W.C.; Gan-Or, Z.; et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBAmutations. Brain 2015, 138, 2648–2658. [Google Scholar] [CrossRef]
- Pang, S.Y.-Y.; Lo, R.C.N.; Ho, P.W.-L.; Liu, H.-F.; Chang, E.E.S.; Leung, C.-T.; Malki, Y.; Choi, Z.Y.-K.; Wong, W.Y.; Kung, M.H.-W.; et al. LRRK2, GBA and their interaction in the regulation of autophagy: Implications on therapeutics in Parkinson’s disease. Transl. Neurodegener. 2022, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, E.; Deroma, L.; Bembi, B.; Deegan, P.; Hollak, C.; Weinreb, N.J.; Cox, T.M. Enzyme replacement and substrate reduction therapy for Gaucher disease. Cochrane Database Syst. Rev. 2015, 2015, CD010324. [Google Scholar] [CrossRef]
- Cabrera-Salazar, M.A.; Deriso, M.; Bercury, S.D.; Li, L.; Lydon, J.T.; Weber, W.; Pande, N.; Cromwell, M.A.; Copeland, D.; Leonard, J.; et al. Systemic Delivery of a Glucosylceramide Synthase Inhibitor Reduces CNS Substrates and Increases Lifespan in a Mouse Model of Type 2 Gaucher Disease. PLoS ONE 2012, 7, e43310. [Google Scholar] [CrossRef]
- Sardi, S.P.; Clarke, J.; Kinnecom, C.; Tamsett, T.J.; Li, L.; Stanek, L.M.; Passini, M.A.; Grabowski, G.A.; Schlossmacher, M.G.; Sidman, R.L.; et al. CNS expression of glucocerebrosidase corrects α-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc. Natl. Acad. Sci. USA 2011, 108, 12101–12106. [Google Scholar] [CrossRef]
- Viel, C.; Clarke, J.; Kayatekin, C.; Richards, A.M.; Chiang, M.S.R.; Park, H.; Wang, B.; Shihabuddin, L.S.; Sardi, S.P. Preclinical pharmacology of glucosylceramide synthase inhibitor venglustat in a GBA-related synucleinopathy model. Sci. Rep. 2021, 11, 20945. [Google Scholar] [CrossRef]
- Peterschmitt, M.J.; Saiki, H.; Hatano, T.; Gasser, T.; Isaacson, S.H.; Gaemers, S.J.; Minini, P.; Saubadu, S.; Sharma, J.; Walbillic, S.; et al. Safety, Pharmacokinetics, and Pharmacodynamics of Oral Venglustat in Patients with Parkinson’s Disease and a GBA Mutation: Results from Part 1 of the Randomized, Double-Blinded, Placebo-Controlled MOVES-PD Trial. J. Park. Dis. 2022, 12, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Sardi, S.P.; Clarke, J.; Viel, C.; Chan, M.; Tamsett, T.J.; Treleaven, C.M.; Bu, J.; Sweet, L.; Passini, M.A.; Dodge, J.C.; et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc. Natl. Acad. Sci. USA 2013, 110, 3537–3542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawkar, A.R.; Cheng, W.-C.; Beutler, E.; Wong, C.-H.; Balch, W.E.; Kelly, J.W. Chemical chaperones increase the cellular activity of N370S β-glucosidase: A therapeutic strategy for Gaucher disease. Proc. Natl. Acad. Sci. USA 2002, 99, 15428–15433. [Google Scholar] [CrossRef] [PubMed]
- de la Mata, M.; Cotán, D.; Oropesa-Ávila, M.; Garrido-Maraver, J.; Cordero, M.D.; Paz, M.V.; Pavón, A.D.; Alcocer-Gómez, E.; de Lavera, I.; Ybot-González, P.; et al. Pharmacological Chaperones and Coenzyme Q10 Treatment Improves Mutant β-Glucocerebrosidase Activity and Mitochondrial Function in Neuronopathic Forms of Gaucher Disease. Sci. Rep. 2015, 5, 10903. [Google Scholar] [CrossRef]
- Kato, A.; Nakagome, I.; Sato, K.; Yamamoto, A.; Adachi, I.; Nash, R.J.; Fleet, G.W.J.; Natori, Y.; Watanabe, Y.; Imahori, T.; et al. Docking study and biological evaluation of pyrrolidine-based iminosugars as pharmacological chaperones for Gaucher disease. Org. Biomol. Chem. 2015, 14, 1039–1048. [Google Scholar] [CrossRef]
- Steet, R.A.; Chung, S.; Wustman, B.; Powe, A.; Do, H.; Kornfeld, S.A. The iminosugar isofagomine increases the activity of N370S mutant acid β-glucosidase in Gaucher fibroblasts by several mechanisms. Proc. Natl. Acad. Sci. USA 2006, 103, 13813–13818. [Google Scholar] [CrossRef]
- Khanna, R.; Benjamin, E.R.; Pellegrino, L.; Schilling, A.; Rigat, B.A.; Soska, R.; Nafar, H.; Ranes, B.E.; Feng, J.; Lun, Y.; et al. The pharmacological chaperone isofagomine increases the activity of the Gaucher disease L444P mutant form of β-glucosidase. FEBS J. 2010, 277, 1618–1638. [Google Scholar] [CrossRef]
- Richter, F.; Fleming, S.M.; Watson, M.; Lemesre, V.; Pellegrino, L.; Ranes, B.; Zhu, C.; Mortazavi, F.; Mulligan, C.; Sioshansi, P.C.; et al. A GCase Chaperone Improves Motor Function in a Mouse Model of Synucleinopathy. Neurotherapeutics 2014, 11, 840–856. [Google Scholar] [CrossRef]
- Boyd, R.E.; Lee, G.; Rybczynski, P.; Benjamin, E.R.; Khanna, R.; Wustman, B.A.; Valenzano, K.J. Pharmacological Chaperones as Therapeutics for Lysosomal Storage Diseases. J. Med. Chem. 2013, 56, 2705–2725. [Google Scholar] [CrossRef]
- Maegawa, G.H.B.; Tropak, M.B.; Buttner, J.D.; Rigat, B.A.; Fuller, M.; Pandit, D.; Tang, L.; Kornhaber, G.J.; Hamuro, Y.; Clarke, J.T.R.; et al. Identification and Characterization of Ambroxol as an Enzyme Enhancement Agent for Gaucher Disease. J. Biol. Chem. 2009, 284, 23502–23516. [Google Scholar] [CrossRef]
- Bendikov-Bar, I.; Maor, G.; Filocamo, M.; Horowitz, M. Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol. Dis. 2013, 50, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Luan, Z.; Li, L.; Higaki, K.; Nanba, E.; Suzuki, Y.; Ohno, K. The chaperone activity and toxicity of ambroxol on Gaucher cells and normal mice. Brain Dev. 2013, 35, 317–322. [Google Scholar] [CrossRef] [PubMed]
- McNeill, A.; Magalhaes, J.; Shen, C.; Chau, K.-Y.; Hughes, D.; Mehta, A.; Foltynie, T.; Cooper, J.M.; Abramov, A.Y.; Gegg, M.; et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 2014, 137, 1481–1495. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, G.; Ghezzi, C.; Zangaglia, R.; Levandis, G.; Pacchetti, C.; Blandini, F. Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson’s disease cells. Neurobiol. Dis. 2015, 82, 235–242. [Google Scholar] [CrossRef]
- Migdalska-Richards, A.; Daly, L.; Bezard, E.; Schapira, A.H.V. Ambroxol effects in glucocerebrosidase and α-synuclein transgenic mice. Ann. Neurol. 2016, 80, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Zimran, A.; Altarescu, G.; Elstein, D. Pilot study using ambroxol as a pharmacological chaperone in type 1 Gaucher disease. Blood Cells Mol. Dis. 2013, 50, 134–137. [Google Scholar] [CrossRef]
- Narita, A.; Shirai, K.; Itamura, S.; Matsuda, A.; Ishihara, A.; Matsushita, K.; Fukuda, C.; Kubota, N.; Takayama, R.; Shigematsu, H.; et al. Ambroxol chaperone therapy for neuronopathic Gaucher disease: A pilot study. Ann. Clin. Transl. Neurol. 2016, 3, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Pawlinski, L.; Malecki, M.T.; Kiec-Wilk, B. The additive effect on the antiepileptic treatment of ambroxol in type 3 Gaucher patient. The early observation. Blood Cells Mol. Dis. 2018, 68, 192–193. [Google Scholar] [CrossRef]
- Kim, Y.M.; Yum, M.S.; Heo, S.H.; Kim, T.; Jin, H.K.; Bae, J.S.; Seo, G.H.; Oh, A.; Yoon, H.M.; Lim, H.T.; et al. Pharmacologic properties of high-dose ambroxol in four patients with Gaucher disease and myoclonic epilepsy. J. Med. Genet. 2019, 57, 124–131. [Google Scholar] [CrossRef]
- Mullin, S.; Smith, L.; Lee, K.; D’Souza, G.; Woodgate, P.; Elflein, J.; Hällqvist, J.; Toffoli, M.; Streeter, A.; Hosking, J.; et al. Ambroxol for the Treatment of Patients with Parkinson Disease with and without Glucocerebrosidase Gene Mutations: A Nonrandomized, Noncontrolled Trial. JAMA Neurol. 2020, 77, 427–434. [Google Scholar] [CrossRef]
- Silveira, C.R.A.; MacKinley, J.; Coleman, K.; Li, Z.; Finger, E.; Bartha, R.; Morrow, S.A.; Wells, J.; Borrie, M.; Tirona, R.G.; et al. Ambroxol as a novel disease-modifying treatment for Parkinson’s disease dementia: Protocol for a single-centre, randomized, double-blind, placebo-controlled trial. BMC Neurol. 2019, 19, 20. [Google Scholar] [CrossRef] [PubMed]
- Aflaki, E.; Borger, D.; Moaven, N.; Stubblefield, B.K.; Rogers, S.A.; Patnaik, S.; Schoenen, F.; Westbroek, W.; Zheng, W.; Sullivan, P.; et al. A New Glucocerebrosidase Chaperone Reduces -Synuclein and Glycolipid Levels in iPSC-Derived Dopaminergic Neurons from Patients with Gaucher Disease and Parkinsonism. J. Neurosci. 2016, 36, 7441–7452. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Zunke, F.; Tsunemi, T.; Toker, N.J.; Jeon, S.; Burbulla, L.F.; Patnaik, S.; Sidransky, E.; Marugan, J.J.; Sue, C.M.; et al. Activation of -Glucocerebrosidase Reduces Pathological -Synuclein and Restores Lysosomal Function in Parkinson’s Patient Midbrain Neurons. J. Neurosci. 2016, 36, 7693–7706. [Google Scholar] [CrossRef]
- Heijer, J.M.D.; Kruithof, A.C.; van Amerongen, G.; de Kam, M.L.; Thijssen, E.; Grievink, H.W.; Moerland, M.; Walker, M.; Been, K.; Skerlj, R.; et al. A randomized single and multiple ascending dose study in healthy volunteers of LTI-291, a centrally penetrant glucocerebrosidase activator. Br. J. Clin. Pharmacol. 2021, 87, 3561–3573. [Google Scholar] [CrossRef]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hayes, M.A.; Beagan, J.; McLean, J.R.; Izen, S.C.; Perez-Torres, E.; et al. Glucocerebrosidase gene therapy prevents α-synucleinopathy of midbrain dopamine neurons. Neurobiol. Dis. 2015, 82, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Morabito, G.; Giannelli, S.G.; Ordazzo, G.; Bido, S.; Castoldi, V.; Indrigo, M.; Cabassi, T.; Cattaneo, S.; Luoni, M.; Cancellieri, C.; et al. AAV-PHP.B-Mediated Global-Scale Expression in the Mouse Nervous System Enables GBA1 Gene Therapy for Wide Protection from Synucleinopathy. Mol. Ther. 2017, 25, 2727–2742. [Google Scholar] [CrossRef] [PubMed]
- Enquist, I.B.; Nilsson, E.; Ooka, A.; Månsson, J.-E.; Olsson, K.; Ehinger, M.; Brady, R.O.; Richter, J.; Karlsson, S. Effective cell and gene therapy in a murine model of Gaucher disease. Proc. Natl. Acad. Sci. USA 2006, 103, 13819–13824. [Google Scholar] [CrossRef]
- Dahl, M.; Doyle, A.; Olsson, K.; Månsson, J.-E.; Marques, A.R.A.; Mirzaian, M.; Aerts, J.M.; Ehinger, M.; Rothe, M.; Modlich, U.; et al. Lentiviral Gene Therapy Using Cellular Promoters Cures Type 1 Gaucher Disease in Mice. Mol. Ther. 2015, 23, 835–844. [Google Scholar] [CrossRef]
- Vilariño-Güell, C.; Wider, C.; Ross, O.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 Mutations in Parkinson Disease. Am. J. Hum. Genet. 2011, 89, 162–167. [Google Scholar] [CrossRef]
- Zimprich, A.; Benet-Pagès, A.; Struhal, W.; Graf, E.; Eck, S.H.; Offman, M.N.; Haubenberger, D.; Spielberger, S.; Schulte, E.C.; Lichtner, P.; et al. A Mutation in VPS35, Encoding a Subunit of the Retromer Complex, Causes Late-Onset Parkinson Disease. Am. J. Hum. Genet. 2011, 89, 168–175. [Google Scholar] [CrossRef]
- Eleuteri, S.; Albanese, A. VPS35-Based Approach: A Potential Innovative Treatment in Parkinson’s Disease. Front. Neurol. 2019, 10, 1272. [Google Scholar] [CrossRef] [PubMed]
- Mecozzi, V.J.; Berman, D.E.; Simoes, S.; Vetanovetz, C.; Awal, M.R.; Patel, V.M.; Schneider, R.T.; Petsko, G.A.; Ringe, D.; Small, S.A. Pharmacological chaperones stabilize retromer to limit APP processing. Nat. Chem. Biol. 2014, 10, 443–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merola, A.; Van Laar, A.; Lonser, R.; Bankiewicz, K. Gene therapy for Parkinson’s disease: Contemporary practice and emerging concepts. Expert Rev. Neurother. 2020, 20, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Kamienieva, I.; Duszyński, J.; Szczepanowska, J. Multitasking guardian of mitochondrial quality: Parkin function and Parkinson’s disease. Transl. Neurodegener. 2021, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Sassone, J.; Serratto, G.M.; Valtorta, F.; Silani, V.; Passafaro, M.; Ciammola, A. The synaptic function of parkin. Brain 2017, 140, 2265–2272. [Google Scholar] [CrossRef]
- Mortiboys, H.; Aasly, J.; Bandmann, O. Ursocholanic acid rescues mitochondrial function in common forms of familial Parkinson’s disease. Brain 2013, 136, 3038–3050. [Google Scholar] [CrossRef]
- Abdelkader, N.F.; Safar, M.M.; Salem, H.A. Ursodeoxycholic Acid Ameliorates Apoptotic Cascade in the Rotenone Model of Parkinson’s Disease: Modulation of Mitochondrial Perturbations. Mol. Neurobiol. 2014, 53, 810–817. [Google Scholar] [CrossRef]
- Ms, A.G.S.; Tuite, P.; Chen, C.; Ma, Y.; Chen, W.; Cloyd, J.; Low, W.C.; Steer, C.J.; Lee, B.; Zhu, X.; et al. Pharmacokinetics, Safety, and Tolerability of Orally Administered Ursodeoxycholic Acid in Patients with Parkinson’s Disease—A Pilot Study. J. Clin. Pharmacol. 2020, 60, 744–750. [Google Scholar] [CrossRef]
- Payne, T.; Sassani, M.; Buckley, E.; Moll, S.; Anton, A.; Appleby, M.; Maru, S.; Taylor, R.; McNeill, A.; Hoggard, N.; et al. Ursodeoxycholic acid as a novel disease-modifying treatment for Parkinson’s disease: Protocol for a two-centre, randomised, double-blind, placebo-controlled trial, The ’UP’ study. BMJ Open 2020, 10, e038911. [Google Scholar] [CrossRef]
- Park, D.; Lee, M.N.; Jeong, H.; Koh, A.; Yang, Y.R.; Suh, P.-G.; Ryu, S.H. Parkin ubiquitinates mTOR to regulate mTORC1 activity under mitochondrial stress. Cell. Signal. 2014, 26, 2122–2130. [Google Scholar] [CrossRef]
- Tain, L.; Mortiboys, H.; Tao, R.N.; Ziviani, E.; Bandmann, O.; Whitworth, A.J. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat. Neurosci. 2009, 12, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vizziello, M.; Borellini, L.; Franco, G.; Ardolino, G. Disruption of Mitochondrial Homeostasis: The Role of PINK1 in Parkinson’s Disease. Cells 2021, 10, 3022. [Google Scholar] [CrossRef] [PubMed]
- Hijioka, M.; Inden, M.; Yanagisawa, D.; Kitamura, Y. DJ-1/PARK7: A New Therapeutic Target for Neurodegenerative Disorders. Biol. Pharm. Bull. 2017, 40, 548–552. [Google Scholar] [CrossRef]
- Sun, S.-Y.; An, C.-N.; Pu, X.-P. DJ-1 protein protects dopaminergic neurons against 6-OHDA/MG-132-induced neurotoxicity in rats. Brain Res. Bull. 2012, 88, 609–616. [Google Scholar] [CrossRef]
- Gao, H.; Yang, W.; Qi, Z.; Lu, L.; Duan, C.; Zhao, C.; Yang, H. DJ-1 Protects Dopaminergic Neurons against Rotenone-Induced Apoptosis by Enhancing ERK-Dependent Mitophagy. J. Mol. Biol. 2012, 423, 232–248. [Google Scholar] [CrossRef] [PubMed]
- Lev, N.; Barhum, Y.; Ben-Zur, T.; Aharony, I.; Trifonov, L.; Regev, N.; Melamed, E.; Gruzman, A.; Offen, D. A DJ-1 Based Peptide Attenuates Dopaminergic Degeneration in Mice Models of Parkinson’s Disease via Enhancing Nrf2. PLoS ONE 2015, 10, e0127549. [Google Scholar] [CrossRef]
- Miyazaki, S.; Yanagida, T.; Nunome, K.; Ishikawa, S.; Inden, M.; Kitamura, Y.; Nakagawa, S.; Taira, T.; Hirota, K.; Niwa, M.; et al. DJ-1-binding compounds prevent oxidative stress-induced cell death and movement defect in Parkinson’s disease model rats. J. Neurochem. 2008, 105, 2418–2434. [Google Scholar] [CrossRef]
- Inden, M.; Kitamura, Y.; Takahashi, K.; Takata, K.; Ito, N.; Niwa, R.; Funayama, R.; Nishimura, K.; Taniguchi, T.; Honda, T.; et al. Protection Against Dopaminergic Neurodegeneration in Parkinson’s Disease–Model Animals by a Modulator of the Oxidized Form of DJ-1, a Wild-type of Familial Parkinson’s Disease–Linked PARK7. J. Pharmacol. Sci. 2011, 117, 189–203. [Google Scholar] [CrossRef]
- Kitamura, Y.; Watanabe, S.; Taguchi, M.; Takagi, K.; Kawata, T.; Takahashi-Niki, K.; Yasui, H.; Maita, H.; Iguchi-Ariga, S.M.; Ariga, H. Neuroprotective effect of a new DJ-1-binding compound against neurodegeneration in Parkinson’s disease and stroke model rats. Mol. Neurodegener. 2011, 6, 48. [Google Scholar] [CrossRef]
- Takahashi-Niki, K.; Inafune, A.; Michitani, N.; Hatakeyama, Y.; Suzuki, K.; Sasaki, M.; Kitamura, Y.; Niki, T.; Iguchi-Ariga, S.M.; Ariga, H. DJ-1-dependent protective activity of DJ-1-binding compound no. 23 against neuronal cell death in MPTP-treated mouse model of Parkinson’s disease. J. Pharmacol. Sci. 2015, 127, 305–310. [Google Scholar] [CrossRef]
- Feng, C.-W.; Hung, H.-C.; Huang, S.-Y.; Chen, C.-H.; Chen, Y.-R.; Yang, S.-N.; Wang, H.-M.D.; Sung, P.-J.; Sheu, J.-H.; Tsui, K.-H.; et al. Neuroprotective Effect of the Marine-Derived Compound 11-Dehydrosinulariolide through DJ-1-Related Pathway in In Vitro and In Vivo Models of Parkinson’s Disease. Mar. Drugs 2016, 14, 187. [Google Scholar] [CrossRef] [PubMed]
- Ablat, N.; Lv, D.; Ren, R.; Xiaokaiti, Y.; Ma, X.; Zhao, X.; Sun, Y.; Lei, H.; Xu, J.; Ma, Y.; et al. Neuroprotective Effects of a Standardized Flavonoid Extract from Safflower against a Rotenone-Induced Rat Model of Parkinson’s Disease. Molecules 2016, 21, 1107. [Google Scholar] [CrossRef] [PubMed]
- Koprich, J.B.; Kalia, L.V.; Brotchie, J.M. Animal models of α-synucleinopathy for Parkinson disease drug development. Nat. Rev. Neurosci. 2017, 18, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Espay, A.J.; Vizcarra, J.A.; Marsili, L.; Lang, A.E.; Simon, D.K.; Merola, A.; Josephs, K.A.; Fasano, A.; Morgante, F.; Savica, R.; et al. Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases. Neurology 2019, 92, 329–337. [Google Scholar] [CrossRef]
- Markesbery, W.R.; Jicha, G.A.; Liu, H.; Schmitt, F.A. Lewy Body Pathology in Normal Elderly Subjects. J. Neuropathol. Exp. Neurol. 2009, 68, 816–822. [Google Scholar] [CrossRef]
- Doherty, K.M.; Hardy, J. Parkin disease and the Lewy body conundrum. Mov. Disord. 2013, 28, 702–704. [Google Scholar] [CrossRef]
- Johansen, K.K.; Torp, S.H.; Farrer, M.J.; Gustavsson, E.K.; Aasly, J.O. A Case of Parkinson’s Disease with No Lewy Body Pathology due to a Homozygous Exon Deletion in Parkin. Case Rep. Neurol. Med. 2018, 2018, 6838965. [Google Scholar] [CrossRef]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef]
- Ingelsson, M. Alpha-Synuclein Oligomers—Neurotoxic Molecules in Parkinson’s Disease and Other Lewy Body Disorders. Front. Neurosci. 2016, 10, 408. [Google Scholar] [CrossRef]
- Cascella, R.; Bigi, A.; Cremades, N.; Cecchi, C. Effects of oligomer toxicity, fibril toxicity and fibril spreading in synucleinopathies. Experientia 2022, 79, 174. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, H.; Tsuji, A.; Hashimoto, Y.; Takata, M.; Koga, S.; Nishida, K.; Futamura, N.; Kawamoto, M.; Kohara, N.; Dickson, D.W.; et al. Discrepancy between distribution of alpha-synuclein oligomers and Lewy-related pathology in Parkinson’s disease. Acta Neuropathol. Commun. 2022, 10, 133. [Google Scholar] [CrossRef] [PubMed]
- Collier, T.J.; Redmond, D.E., Jr.; Esteece-Collier, K.; Lipton, J.W.; Manfredsson, F.P. Is Alpha-Synuclein Loss-of-Function a Contributor to Parkinsonian Pathology? Evidence from Non-human Primates. Front. Neurosci. 2016, 10, 12. [Google Scholar] [CrossRef]
- Espay, A.J. Movement disorders research in 2021: Cracking the paradigm. Lancet Neurol. 2022, 21, 10–11. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Corvol, J.; Poewe, W. Pharmacogenetics of Parkinson’s Disease in Clinical Practice. Mov. Disord. Clin. Pr. 2016, 4, 173–180. [Google Scholar] [CrossRef]
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; García-Martín, E.; Agúndez, J.A.G. Advances in understanding genomic markers and pharmacogenetics of Parkinson’s disease. Expert Opin. Drug Metab. Toxicol. 2016, 12, 433–448. [Google Scholar] [CrossRef]
- Kalinderi, K.; Papaliagkas, V.; Fidani, L. Pharmacogenetics and levodopa induced motor complications. Int. J. Neurosci. 2018, 129, 384–392. [Google Scholar] [CrossRef]
- De Oliveira, L.M.; Barbosa, E.R.; Aquino, C.C.; Munhoz, R.P.; Fasano, A.; Cury, R.G. Deep Brain Stimulation in Patients with Mutations in Parkinson’s Disease–Related Genes: A Systematic Review. Mov. Disord. Clin. Pr. 2019, 6, 359–368. [Google Scholar] [CrossRef]
- Kuusimäki, T.; Korpela, J.; Pekkonen, E.; Martikainen, M.H.; Antonini, A.; Kaasinen, V. Deep brain stimulation for monogenic Parkinson’s disease: A systematic review. J. Neurol. 2019, 267, 883–897. [Google Scholar] [CrossRef]
- Kielb, S.; Kisanuki, Y.Y.; Dawson, E. Neuropsychological profile associated with an alpha-synuclein gene (SNCA) duplication. Clin. Neuropsychol. 2021, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Over, L.; Brüggemann, N.; Lohmann, K. Therapies for Genetic Forms of Parkinson’s Disease: Systematic Literature Review. J. Neuromuscul. Dis. 2021, 8, 341–356. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.; Oyama, G.; Hattori, N.; Shimo, Y.; Kuusimäki, T.; Kaasinen, V.; Antonini, A.; Kim, D.; Lee, J.; Cho, K.R.; et al. Subthalamic deep brain stimulation in Parkinson’s disease with SNCA mutations: Based on the follow-up to 10 years. Brain Behav. 2022, 12, e2503. [Google Scholar] [CrossRef]
- Salles, P.A.; Mata, I.F.; Fernandez, H.H. Should we start integrating genetic data in decision-making on device-aided therapies in Parkinson disease? A point of view. Park. Relat. Disord. 2021, 88, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Artusi, C.A.; Dwivedi, A.K.; Romagnolo, A.; Pal, G.; Kauffman, M.; Mata, I.F.; Patel, D.; Vizcarra, J.A.; Duker, A.; Marsili, L.; et al. Association of Subthalamic Deep Brain Stimulation with Motor, Functional, and Pharmacologic Outcomes in Patients with Monogenic Parkinson Disease. JAMA Netw. Open 2019, 2, e187800. [Google Scholar] [CrossRef] [PubMed]
- Martikainen, M.H.; Päivärinta, M.; Hietala, M.; Kaasinen, V. Clinical and imaging findings in Parkinson disease associated with the A53E SNCAmutation. Neurol. Genet. 2015, 1, e27. [Google Scholar] [CrossRef]
- Perandones, C.; Olivos, N.A.; Raina, G.B.; Pellene, L.A.; Giugni, J.C.; Calvo, D.S.; Radrizzani, M.; Piedimonte, F.; Micheli, F.E. Successful GPi stimulation in genetic Parkinson’s disease caused by mosaicism of alpha-synuclein gene duplication: First description. J. Neurol. 2014, 262, 222–223. [Google Scholar] [CrossRef]
- Ahn, T.B.; Kim, S.Y.; Kim, J.Y.; Park, S.S.; Lee, D.S.; Min, H.J.; Kim, Y.K.; Kim, H.J.; Cho, J.; Jeon, B.S. Synuclein gene duplication is present in sporadic Parkinson disease. Neurology 2007, 70, 43–49. [Google Scholar] [CrossRef]
- Ligaard, J.; Sannæs, J.; Pihlstrøm, L. Deep brain stimulation and genetic variability in Parkinson’s disease: A review of the literature. npj Park. Dis. 2019, 5, 18. [Google Scholar] [CrossRef]
- Elia, A.E.; Petrucci, S.; Fasano, A.; Guidi, M.; Valbonesi, S.; Bernardini, L.; Consoli, F.; Ferraris, A.; Albanese, A.; Valente, E.M. Alpha-synuclein gene duplication: Marked intrafamilial variability in two novel pedigrees. Mov. Disord. 2013, 28, 813–817. [Google Scholar] [CrossRef]
- Antonini, A.; Pilleri, M.; Padoan, A.; Landi, A.; Ferla, S.; Biundo, R.; D’Avella, D. Successful subthalamic stimulation in genetic Parkinson’s disease caused by duplication of the α-synuclein gene. J. Neurol. 2011, 259, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Shimo, Y.; Natori, S.; Oyama, G.; Nakajima, M.; Ishii, H.; Arai, H.; Hattori, N. Subthalamic Deep Brain Stimulation for a Parkinson’s Disease Patient with Duplication of SNCA. Neuromodulation Technol. Neural Interface 2013, 17, 102–103. [Google Scholar] [CrossRef] [PubMed]
- Salles, P.A.; Liao, J.; Shuaib, U.; Mata, I.F.; Fernandez, H.H. A Review on Response to Device-Aided Therapies Used in Monogenic Parkinsonism and GBA Variants Carriers: A Need for Guidelines and Comparative Studies. J. Park. Dis. 2022, 12, 1703–1725. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Esteban, J.C.; Lezcano, E.; Zarranz, J.J.; González, C.; Bilbao, G.; Lambarri, I.; Rodríguez, O.; Garibi, J. Outcome of bilateral deep brain subthalamic stimulation in patients carrying the R1441G mutation in the LRRK2 dardarin gene. Neurosurgery 2008, 62, 857–863. [Google Scholar] [CrossRef]
- Sayad, M.; Zouambia, M.; Chaouch, M.; Ferrat, F.; Nebbal, M.; Bendini, M.; Lesage, S.; Brice, A.; Errahmani, M.B.; Asselah, B. Greater improvement in LRRK2 G2019S patients undergoing Subthalamic Nucleus Deep Brain Stimulation compared to non-mutation carriers. BMC Neurosci. 2016, 17, 6. [Google Scholar] [CrossRef]
- Schüpbach, M.; Lohmann, E.; Anheim, M.; Lesage, S.; Ma, V.C.; Yaici, S.; Worbe, Y.; Charles, P.; Welter, M.; Pollak, P.; et al. Subthalamic nucleus stimulation is efficacious in patients with Parkinsonism and LRRK2 mutations. Mov. Disord. 2007, 22, 119–122. [Google Scholar] [CrossRef]
- Foltynie, T.; Magee, C.; James, C.; Webster, G.J.M.; Lees, A.J.; Limousin, P. Impact of Duodopa on Quality of Life in Advanced Parkinson’s Disease: A UK Case Series. Park. Dis. 2013, 2013, 362908. [Google Scholar] [CrossRef]
- Thaler, A.; Hillel, A.; Shabtai, H.; Giladi, N.; Gurevich, T. Assessing the response to L-dopa/carbidopa intestinal gel infusion (Deudopa) based on genetic status. Mov. Disord. 2017, 32, 1015. [Google Scholar]
- Thaler, A.; Livneh, V.; Hillel, A.; Strauss, H.; Yahalom, H.; Shabtai, H.; Migirov Senderovich, A.; Israel-Korn, S.; Fay-Carmon, T.; Giladi, N.; et al. Assessing the response to L-dopa/carbidopa intestinal gel infusion based on genetic status. Mov. Disord. 2019, 34, 434. [Google Scholar]
- Khan, N.L.; Jain, S.; Lynch, J.M.; Pavese, N.; Abou-Sleiman, P.; Holton, J.L.; Healy, D.G.; Gilks, W.; Sweeney, M.G.; Ganguly, M.; et al. Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson’s disease: Clinical, pathological, olfactory and functional imaging and genetic data. Brain 2005, 128, 2786–2796. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Amshalom, I.; Kilarski, L.L.; Bar-Shira, A.; Gana-Weisz, M.; Mirelman, A.; Marder, K.; Bressman, S.; Giladi, N.; Orr-Urtreger, A. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 2015, 84, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Aharon-Peretz, J.; Rosenbaum, H.; Gershoni-Baruch, R. Mutations in the Glucocerebrosidase Gene and Parkinson’s Disease in Ashkenazi Jews. N. Engl. J. Med. 2004, 351, 1972–1977. [Google Scholar] [CrossRef] [PubMed]
- Oeda, T.; Umemura, A.; Mori, Y.; Tomita, S.; Kohsaka, M.; Park, K.; Inoue, K.; Fujimura, H.; Hasegawa, H.; Sugiyama, H.; et al. Impact of glucocerebrosidase mutations on motor and nonmotor complications in Parkinson’s disease. Neurobiol. Aging 2015, 36, 3306–3313. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Castillo-Torres, S.A.; Merello, M. Early motor response to dopamine replacement therapy in Parkinson’s disease patients carrying GBA variants. J. Neurol. Sci. 2022, 440, 120354. [Google Scholar] [CrossRef] [PubMed]
- Winder-Rhodes, S.E.; Evans, J.R.; Ban, M.; Mason, S.L.; Williams-Gray, C.; Foltynie, T.; Duran, R.; Mencacci, N.E.; Sawcer, S.J.; Barker, R.A. Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain 2013, 136, 392–399. [Google Scholar] [CrossRef]
- Brockmann, K.; Srulijes, K.; Pflederer, S.; Hauser, A.; Schulte, C.; Maetzler, W.; Gasser, T.; Berg, D. GBA-associated Parkinson’s disease: Reduced survival and more rapid progression in a prospective longitudinal study. Mov. Disord. 2014, 30, 407–411. [Google Scholar] [CrossRef]
- Davis, M.Y.; Johnson, C.O.; Leverenz, J.B.; Weintraub, D.; Trojanowski, J.Q.; Chen-Plotkin, A.; Van Deerlin, V.M.; Quinn, J.F.; Chung, K.A.; Peterson-Hiller, A.L.; et al. Association of GBA Mutations and the E326K Polymorphism with Motor and Cognitive Progression in Parkinson Disease. JAMA Neurol. 2016, 73, 1217–1224. [Google Scholar] [CrossRef]
- Beavan, M.; McNeill, A.; Proukakis, C.; Hughes, D.A.; Mehta, A.; Schapira, A.H.V. Evolution of Prodromal Clinical Markers of Parkinson Disease in a GBAMutation–Positive Cohort. JAMA Neurol. 2015, 72, 201–208. [Google Scholar] [CrossRef]
- Gatto, E.M.; Etcheverry, J.L.; Sanguinetti, A.; Cesarini, M.; Escobar, N.F.; Drelichman, G. Prodromal Clinical Markers of Parkinson disease in Gaucher Disease Individuals. Eur. Neurol. 2016, 76, 19–21. [Google Scholar] [CrossRef]
- Wilke, M.V.M.B.; Dornelles, A.D.; Schuh, A.S.; Vairo, F.; Basgalupp, S.P.; Siebert, M.; Nalin, T.; Piltcher, O.B.; Schwartz, I.V.D. Evaluation of the frequency of non-motor symptoms of Parkinson’s disease in adult patients with Gaucher disease type 1. Orphanet J. Rare Dis. 2019, 14, 103. [Google Scholar] [CrossRef]
- Jesús, S.; Huertas, I.; Bernal, I.; Bonilla-Toribio, M.; Cáceres-Redondo, M.T.; Vargas-González, L.; Gómez-Llamas, M.; Carrillo, F.; Calderón, E.; Carballo, M.; et al. GBA Variants Influence Motor and Non-Motor Features of Parkinson’s Disease. PLoS ONE 2016, 11, e0167749. [Google Scholar] [CrossRef] [PubMed]
- Saunders-Pullman, R.; Hagenah, J.; Dhawan, V.; Stanley, K.; Pastores, G.; Sathe, S.; Tagliati, M.; Condefer, K.; Palmese, C.; Brüggemann, N.; et al. Gaucher disease ascertained through a Parkinson’s center: Imaging and clinical characterization. Mov. Disord. 2010, 25, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Mullin, S.; Beavan, M.; Bestwick, J.; McNeill, A.; Proukakis, C.; Cox, T.; Hughes, D.; Mehta, A.; Zetterberg, H.; Schapira, A.H. Evolution and clustering of prodromal parkinsonian features in GBA1 carriers. Mov. Disord. 2019, 34, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Thaler, A.; Bregman, N.; Gurevich, T.; Shiner, T.; Dror, Y.; Zmira, O.; Gan-Or, Z.; Bar-Shira, A.; Gana-Weisz, M.; Orr-Urtreger, A.; et al. Parkinson’s disease phenotype is influenced by the severity of the mutations in the GBA gene. Park. Relat. Disord. 2018, 55, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Boot, B.; Locascio, J.J.; Ms, I.E.J.; Winder-Rhodes, S.; Eberly, S.; Elbaz, A.; Brice, A.; Ravina, B.; Van Hilten, J.J.; et al. Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann. Neurol. 2016, 80, 674–685. [Google Scholar] [CrossRef]
- Creese, B.; Bell, E.; Johar, I.; Francis, P.T.; Ballard, C.; Aarsland, D. Glucocerebrosidase mutations and neuropsychiatric phenotypes in Parkinson’s disease and Lewy body dementias: Review and meta-analyses. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2017, 177, 232–241. [Google Scholar] [CrossRef]
- Weiss, D.; Brockmann, K.; Srulijes, K.; Meisner, C.; Klotz, R.; Reinbold, S.; Hauser, A.-K.; Schulte, C.; Berg, D.; Gasser, T.; et al. Long-term follow-up of subthalamic nucleus stimulation in glucocerebrosidase-associated Parkinson’s disease. J. Neurol. 2012, 259, 1970–1972. [Google Scholar] [CrossRef]
- Pal, G.D.; Hall, D.; Ouyang, B.; Phelps, J.; Alcalay, R.; Pauciulo, M.W.; Nichols, W.C.; Clark, L.; Mejia-Santana, H.; Blasucci, L.; et al. Genetic and Clinical Predictors of Deep Brain Stimulation in Young-Onset Parkinson’s Disease. Mov. Disord. Clin. Pr. 2016, 3, 465–471. [Google Scholar] [CrossRef]
- Mangone, G.; Bekadar, S.; Cormier-Dequaire, F.; Tahiri, K.; Welaratne, A.; Czernecki, V.; Pineau, F.; Karachi, C.; Castrioto, A.; Durif, F.; et al. Early cognitive decline after bilateral subthalamic deep brain stimulation in Parkinson’s disease patients with GBA mutations. Park. Relat. Disord. 2020, 76, 56–62. [Google Scholar] [CrossRef]
- Angeli, A.; Mencacci, N.E.; Duran, R.; Aviles-Olmos, I.; Kefalopoulou, Z.; Candelario, J.; Rusbridge, S.; Foley, J.; Pradhan, P.; Jahanshahi, M.; et al. Genotype and phenotype in Parkinson’s disease: Lessons in heterogeneity from deep brain stimulation. Mov. Disord. 2013, 28, 1370–1375. [Google Scholar] [CrossRef]
- Lythe, V.; Athauda, D.; Foley, J.; Mencacci, N.E.; Jahanshahi, M.; Cipolotti, L.; Hyam, J.; Zrinzo, L.; Hariz, M.; Hardy, J.; et al. GBA-Associated Parkinson’s Disease: Progression in a Deep Brain Stimulation Cohort. J. Park. Dis. 2017, 7, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Anheim, M.; Condroyer, C.; Pollak, P.; Durif, F.; Dupuits, C.; Viallet, F.; Lohmann, E.; Corvol, J.-C.; Honoré, A.; et al. Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum. Mol. Genet. 2010, 20, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Kurtis, M.M.; Rajah, T.; Delgado, L.F.; Dafsari, H.S. The effect of deep brain stimulation on the non-motor symptoms of Parkinson’s disease: A critical review of the current evidence. npj Park. Dis. 2017, 3, 16024. [Google Scholar] [CrossRef]
- Pal, G.; Mangone, G.; Hill, E.J.; Ouyang, B.; Liu, Y.; Lythe, V.; Ehrlich, D.; Saunders-Pullman, R.; Shanker, V.; Bressman, S.; et al. Parkinson Disease and Subthalamic Nucleus Deep Brain Stimulation: Cognitive Effects in GBA Mutation Carriers. Ann. Neurol. 2022, 91, 424–435. [Google Scholar] [CrossRef]
- Kasten, M.; Hartmann, C.; Hampf, J.; Schaake, S.; Westenberger, A.; Vollstedt, E.-J.; Balck, A.; Domingo, A.; Vulinovic, F.; Dulovic, M.; et al. Genotype-Phenotype Relations for the Parkinson’s Disease Genes Parkin, PINK1, DJ1: MDSGene Systematic Review. Mov. Disord. 2018, 33, 730–741. [Google Scholar] [CrossRef]
- Kim, H.J.; Yun, J.Y.; Kim, Y.-E.; Lee, J.-Y.; Kim, H.-J.; Kim, J.-Y.; Park, S.S.; Paek, S.H.; Jeon, B.S. Parkin mutation and deep brain stimulation outcome. J. Clin. Neurosci. 2014, 21, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Rieu, I.; Degos, B.; Castelnovo, G.; Vial, C.; Durand, E.; Pereira, B.; Simonetta-Moreau, M.; Sangla, S.; Fluchère, F.; Guehl, D.; et al. Incobotulinum toxin A in Parkinson’s disease with foot dystonia: A double blind randomized trial. Park. Relat. Disord. 2018, 46, 9–15. [Google Scholar] [CrossRef]
- Inzelberg, R.; Hattori, N.; Nisipeanu, P.; Mouch, S.A.; Blumen, S.; Carasso, R.; Mizuno, Y. Camptocormia, axial dystonia, and parkinsonism: Phenotypic heterogeneity of a parkin mutation. Neurology 2003, 60, 1393–1394. [Google Scholar] [CrossRef]
- Hubsher, G.; Haider, M.; Okun, M. Amantadine: The journey from fighting flu to treating Parkinson disease. Neurology 2012, 78, 1096–1099. [Google Scholar] [CrossRef]
- Espay, A.J.; Lang, A. Common Myths in the Use of Levodopa in Parkinson Disease. JAMA Neurol. 2017, 74, 633–634. [Google Scholar] [CrossRef]
- Yoritaka, A.; Shimo, Y.; Shimo, Y.; Inoue, Y.; Yoshino, H.; Hattori, N. Nonmotor Symptoms in Patients withPARK2Mutations. Park. Dis. 2011, 2011, 473640. [Google Scholar] [CrossRef]
- Tijero, B.; Gabilondo, I.; Lezcano, E.; Teran-Villagrá, N.; Llorens, V.; Ruiz-Martinez, J.; Marti-Masso, J.F.; Carmona, M.; Luquin, M.R.; Berganzo, K.; et al. Autonomic involvement in Parkinsonian carriers of PARK2 gene mutations. Park. Relat. Disord. 2015, 21, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Shen, B.; Yang, Y.-J.; Liu, F.-T.; Zhao, J.; Tang, Y.-L.; Chen, C.; Ding, Z.-T.; An, Y.; Wu, J.-J.; et al. Non-motor Symptoms in Parkinson’s Disease Patients with Parkin Mutations: More Depression and Less Executive Dysfunction. J. Mol. Neurosci. 2020, 70, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, R.N.; Caccappolo, E.; Mejia-Santana, H.; Tang, M.X.; Rosado, L.; Reilly, M.O.; Ruiz, D.; Louis, E.D.; Comella, C.L.; Nance, M.A.; et al. Cognitive and Motor Function in Long-Duration PARKIN-Associated Parkinson Disease. JAMA Neurol. 2014, 71, 62–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Le, W. Profiling Non-motor Symptoms in Monogenic Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 591183. [Google Scholar] [CrossRef]
- Morgante, F.; Fasano, A.; Ginevrino, M.; Petrucci, S.; Ricciardi, L.; Bove, F.; Criscuolo, C.; Moccia, M.; De Rosa, A.; Sorbera, C.; et al. Impulsive-compulsive behaviors in parkin-associated Parkinson disease. Neurology 2016, 87, 1436–1441. [Google Scholar] [CrossRef]
- Faouzi, J.; Corvol, J.-C.; Mariani, L.-L. Impulse control disorders and related behaviors in Parkinson’s disease: Risk factors, clinical and genetic aspects, and management. Curr. Opin. Neurol. 2021, 34, 547–555. [Google Scholar] [CrossRef]
- Stahl, S.M. Drugs for psychosis and mood: Unique actions at D3, D2, and D1 dopamine receptor subtypes. CNS Spectr. 2017, 22, 375–384. [Google Scholar] [CrossRef]
- Paul, D.A.; Qureshi, A.R.M.; Rana, A.Q. Peripheral neuropathy in Parkinson’s disease. Neurol. Sci. 2020, 41, 2691–2701. [Google Scholar] [CrossRef]
- Lohmann, E.; Welter, M.; Fraix, V.; Krack, P.; Lesage, S.; Laine, S.; Tanguy, M.; Houeto, J.; Mesnage, V.; Pollak, P.; et al. Are parkin patients particularly suited for deep-brain stimulation? Mov. Disord. 2008, 23, 740–743. [Google Scholar] [CrossRef]
- Lefaucheur, R.; Derrey, S.; Guyant-Maréchal, L.; Chastan, N.; Maltête, D. Whatever the disease duration, stimulation of the subthalamic nucleus improves Parkin disease. Park. Relat. Disord. 2010, 16, 482–483. [Google Scholar] [CrossRef] [PubMed]
- Capecci, M.; Passamonti, L.; Annesi, F.; Annesi, G.; Bellesi, M.; Ricciuti, R.; Iacoangeli, M.; Scerrati, M.; Zappia, M.; Tarantino, P.; et al. Chronic bilateral subthalamic deep brain stimulation in a patient with homozygous deletion in the Parkin gene. Mov. Disord. 2004, 19, 1450–1452. [Google Scholar] [CrossRef] [PubMed]
- Romito, L.M.A.; Contarino, M.F.; Ghezzi, D.; Franzini, A.; Garavaglia, B.; Albanese, A. High frequency stimulation of the subthalamic nucleus is efficacious in Parkin disease. J. Neurol. 2005, 252, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Moro, E.; Volkmann, J.; König, I.; Winkler, S.; Hiller, A.; Hassin-Baer, S.; Herzog, J.; Schnitzler, A.; Lohmann, K.; Pinsker, M.O.; et al. Bilateral subthalamic stimulation in Parkin and PINK1 parkinsonism. Neurology 2008, 70, 1186–1191. [Google Scholar] [CrossRef]
- Johansen, K.K.; Jørgensen, J.V.; White, L.R.; Farrer, M.; Aasly, J.O. Parkinson-related genetics in patients treated with deep brain stimulation. Acta Neurol. Scand. 2011, 123, 201–206. [Google Scholar] [CrossRef]
- Bohlega, S.; Al-Shaar, H.A.; Alkhairallah, T.; Al-Ajlan, F.; Hasan, N.; Alkahtani, K. Levodopa-Carbidopa Intestinal Gel Infusion Therapy in Advanced Parkinson’s Disease: Single Middle Eastern Center Experience. Eur. Neurol. 2015, 74, 227–236. [Google Scholar] [CrossRef]
- Khan, N.L.; Graham, E.; Critchley, P.; Schrag, A.E.; Wood, N.; Lees, A.J.; Bhatia, K.P.; Quinn, N. Parkin disease: A phenotypic study of a large case series. Brain 2003, 126, 1279–1292. [Google Scholar] [CrossRef]



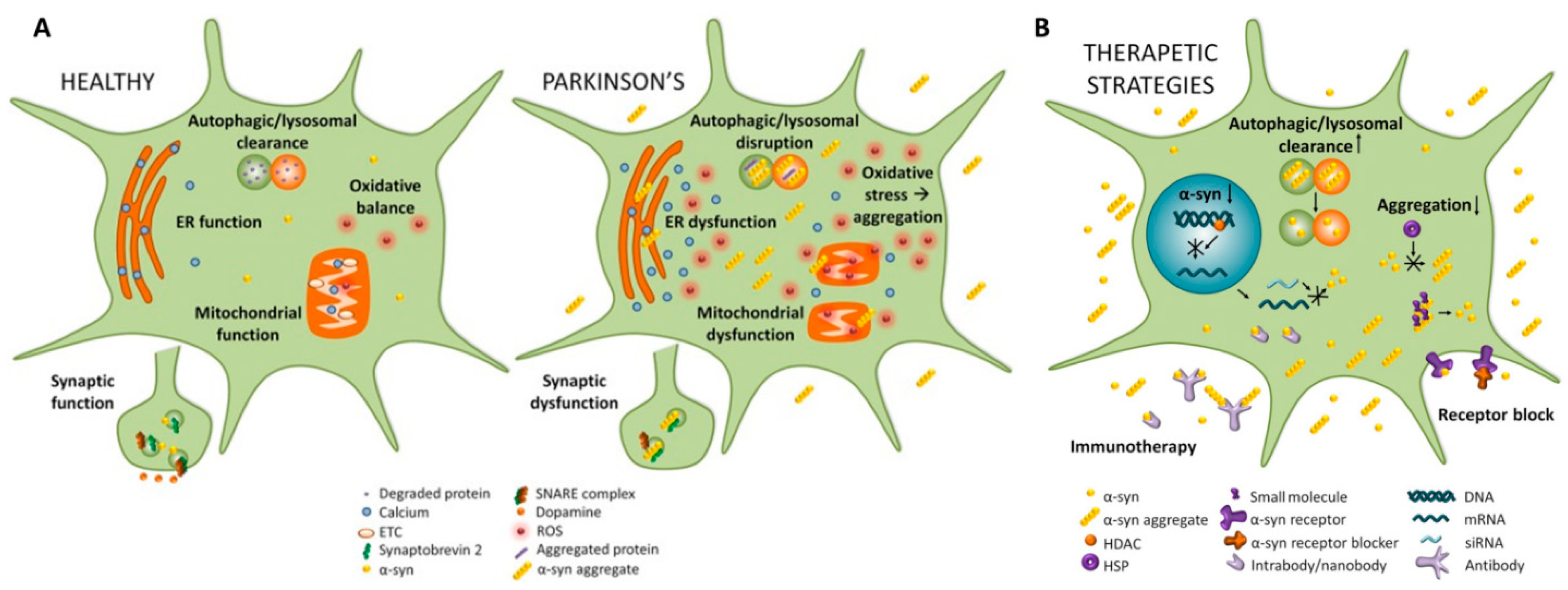{kind=link}
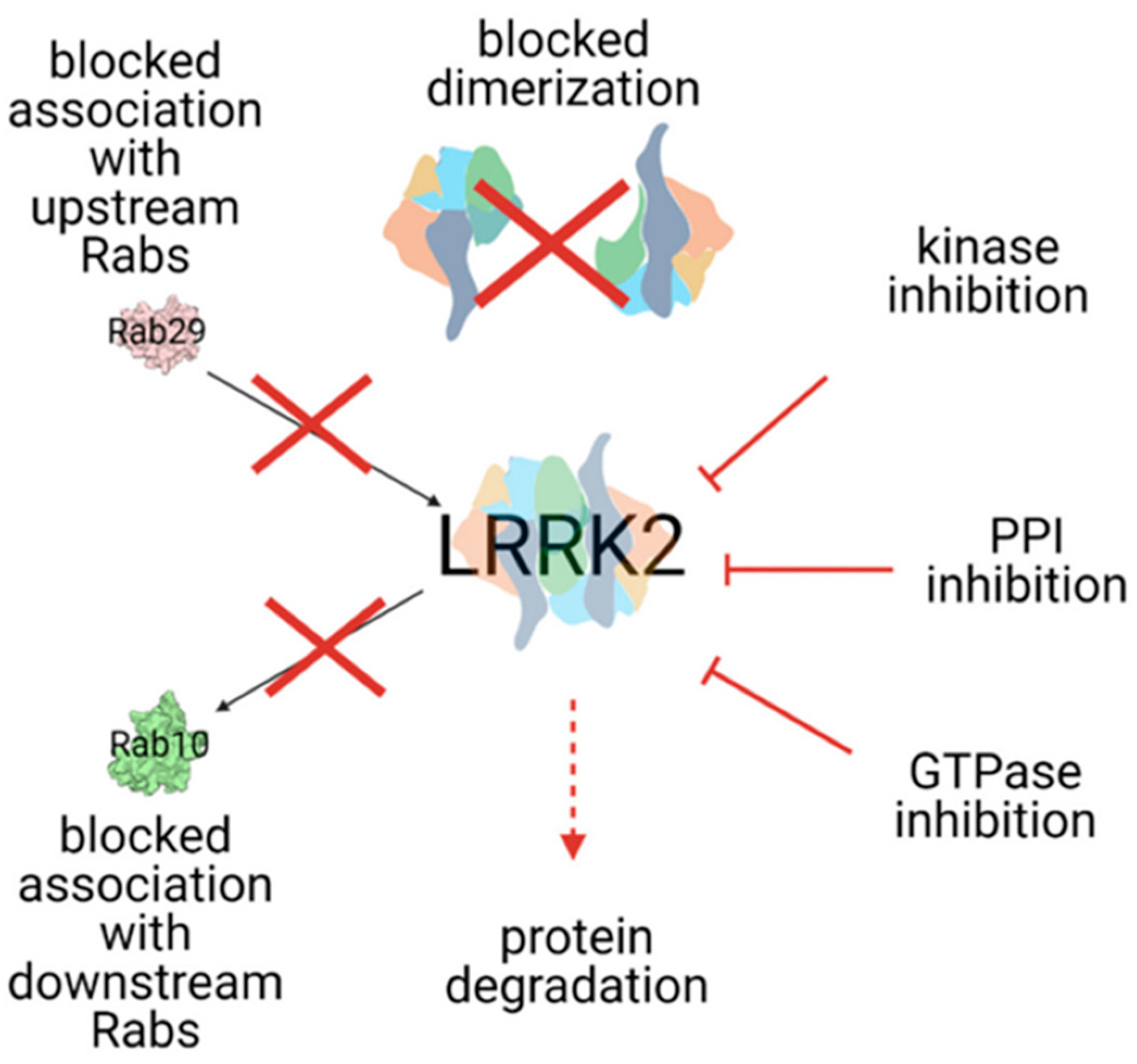{kind=link}
| α-Syn Synthesis Inhibitors | Inhibiting α-Syn Aggregation | Inhibiting α-Syn Uptake | Immunotherapy | Autophagy-Enhancers |
|---|---|---|---|---|
| Passive | ||||
|
| |||
|
|
| ||
|
|
|
| |
|
| |||
|
|
| ||
|
| Active | ||
| (a) Histone acetylase modulators [94,105] |
| |||
| (b) IRE-block (Synucleozid) [109] |
|
| ||
| ||||
|
| |||
| ||||
| ||||
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Straccia, G.; Colucci, F.; Eleopra, R.; Cilia, R. Precision Medicine in Parkinson’s Disease: From Genetic Risk Signals to Personalized Therapy. Brain Sci. 2022, 12, 1308. https://doi.org/10.3390/brainsci12101308
Straccia G, Colucci F, Eleopra R, Cilia R. Precision Medicine in Parkinson’s Disease: From Genetic Risk Signals to Personalized Therapy. Brain Sciences. 2022; 12(10):1308. https://doi.org/10.3390/brainsci12101308
Chicago/Turabian StyleStraccia, Giulia, Fabiana Colucci, Roberto Eleopra, and Roberto Cilia. 2022. "Precision Medicine in Parkinson’s Disease: From Genetic Risk Signals to Personalized Therapy" Brain Sciences 12, no. 10: 1308. https://doi.org/10.3390/brainsci12101308