
Shared Genetic Background between Parkinson’s Disease and Schizophrenia: A Two-Sample Mendelian Randomization Study

 , , and
, , and
Abstract
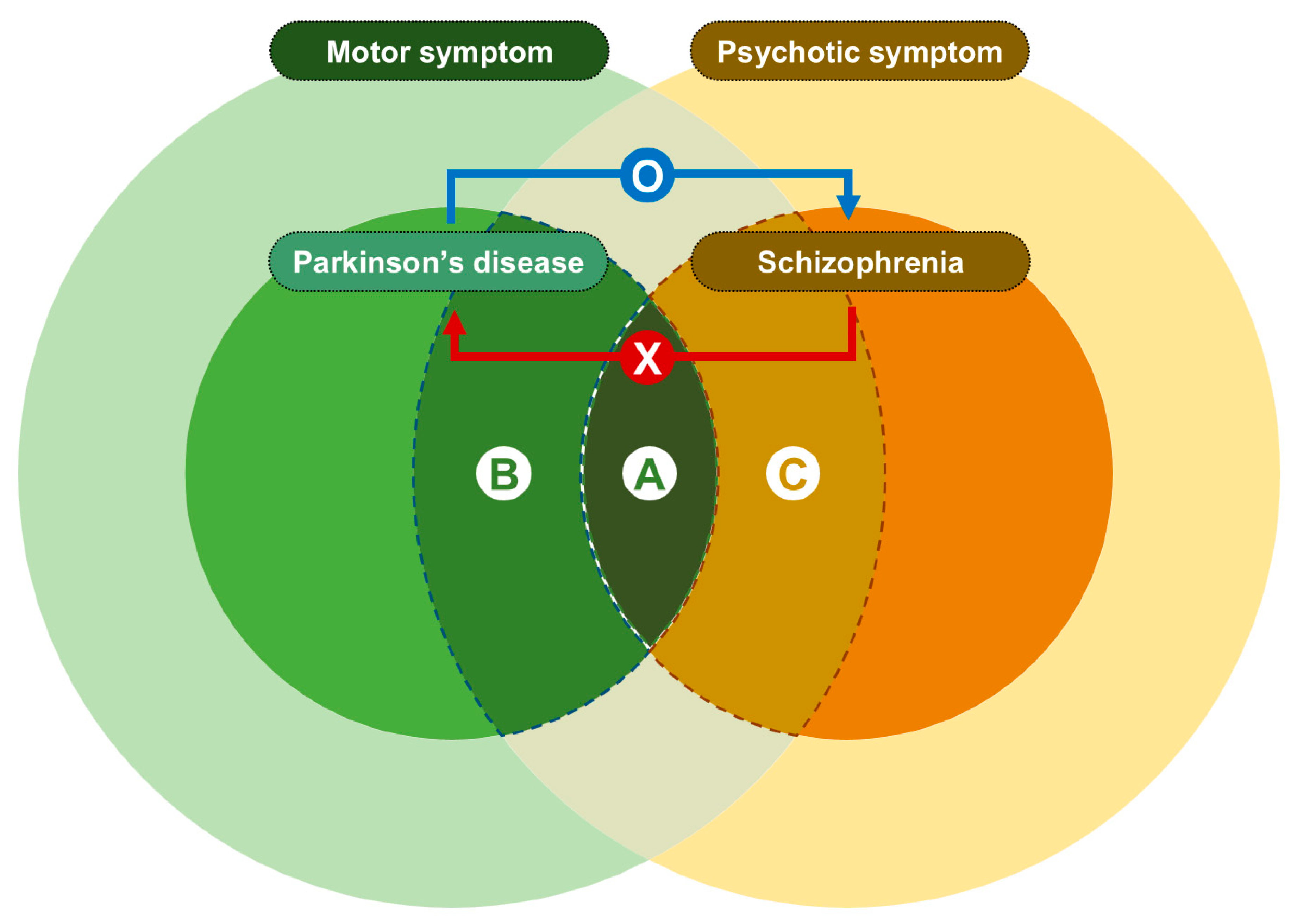
:1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Statistical Analyses
3. Results
3.1. Causality between PD Risk and Schizophrenia Risk
3.2. Shared Genetic Background between PD and Other Psychiatric Disorders or Related Traits
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gelb, D.J.; Oliver, E.; Gilman, S. Diagnostic criteria for Parkinson disease. Arch. Neurol. 1999, 56, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Vescovelli, F.; Sarti, D.; Ruini, C. Well-being and distress of patients with Parkinson’s disease: A comparative investigation. Int. Psychogeriatr. 2019, 31, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Mele, B.; Holroyd-Leduc, J.; Smith, E.E.; Pringsheim, T.; Ismail, Z.; Goodarzi, Z. Detecting anxiety in individuals with Parkinson disease: A systematic review. Neurology 2018, 90, e39–e47. [Google Scholar] [CrossRef]
- Okbay, A.; Baselmans, B.M.; De Neve, J.E.; Turley, P.; Nivard, M.G.; Fontana, M.A.; Meddens, S.F.; Linner, R.K.; Rietveld, C.A.; Derringer, J.; et al. Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nat. Genet. 2016, 48, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Turley, P.; Walters, R.K.; Maghzian, O.; Okbay, A.; Lee, J.J.; Fontana, M.A.; Nguyen-Viet, T.A.; Wedow, R.; Zacher, M.; Furlotte, N.A.; et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat. Genet. 2018, 50, 229–237. [Google Scholar] [CrossRef]
- Ravina, B.; Marder, K.; Fernandez, H.H.; Friedman, J.H.; McDonald, W.; Murphy, D.; Aarsland, D.; Babcock, D.; Cummings, J.; Endicott, J.; et al. Diagnostic criteria for psychosis in Parkinson’s disease: Report of an NINDS, NIMH work group. Mov. Disord. 2007, 22, 1061–1068. [Google Scholar] [CrossRef]
- Inzelberg, R.; Kipervasser, S.; Korczyn, A.D. Auditory hallucinations in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1998, 64, 533–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenelon, G.; Mahieux, F.; Huon, R.; Ziegler, M. Hallucinations in Parkinson’s disease: Prevalence, phenomenology and risk factors. Brain 2000, 123, 733–745. [Google Scholar] [CrossRef]
- Goetz, C.G.; Pappert, E.J.; Blasucci, L.M.; Stebbins, G.T.; Ling, Z.D.; Nora, M.V.; Carvey, P.M. Intravenous levodopa in hallucinating Parkinson’s disease patients: High-dose challenge does not precipitate hallucinations. Neurology 1998, 50, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Gabilondo, A.; Alonso-Moran, E.; Nuno-Solinis, R.; Orueta, J.F.; Iruin, A. Comorbidities with chronic physical conditions and gender profiles of illness in schizophrenia. Results from PREST, a new health dataset. J. Psychosom. Res. 2017, 93, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Walther, S.; Mittal, V.A. Motor System Pathology in Psychosis. Curr. Psychiatry Rep. 2017, 19, 97. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.J.; Lee, H.; Kim, Y.E.; Jeon, B.S. A case of Parkin disease (PARK2) with schizophrenia: Evidence of regional selectivity. Clin. Neurol. Neurosurg. 2014, 126, 35–37. [Google Scholar] [CrossRef]
- Oh, J.; Shen, G.X.; Nan, G.X.; Kim, J.M.; Jung, K.Y.; Jeon, B. Comorbid schizophrenia and Parkinson’s disease: A case series and brief review. Neurol. Asia 2017, 22, 139–142. [Google Scholar]
- Smith, G.D.; Hemani, G. Mendelian randomization: Genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 2014, 23, R89–R98. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.; Nalls, M.A.; Hallgrimsdottir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; International Parkinson’s Disease Genomics Consortium; 23andMe Research Team; Kerchner, G.A.; Ayalon, G.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, F.P.; Borges, M.C.; Horta, B.L.; Bowden, J.; Smith, G.D. Inflammatory Biomarkers and Risk of Schizophrenia: A 2-Sample Mendelian Randomization Study. JAMA Psychiatry 2017, 74, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Sanna, S.; van Zuydam, N.R.; Mahajan, A.; Kurilshikov, A.; Vila, A.V.; Vosa, U.; Mujagic, Z.; Masclee, A.A.M.; Jonkers, D.; Oosting, M.; et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat. Genet. 2019, 51, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Verbanck, M.; Chen, C.Y.; Neale, B.; Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018, 50, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Hemani, G.; Zheng, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 2018, 7, e34408. [Google Scholar] [CrossRef]
- Dastani, Z.; Hivert, M.F.; Timpson, N.; Perry, J.R.; Yuan, X.; Scott, R.A.; Henneman, P.; Heid, I.M.; Kizer, J.R.; Lyytikainen, L.P.; et al. Novel loci for adiponectin levels and their influence on type 2 diabetes and metabolic traits: A multi-ethnic meta-analysis of 45,891 individuals. PLoS Genet. 2012, 8, e1002607. [Google Scholar] [CrossRef] [Green Version]
- International Consortium for Blood Pressure Genome-Wide Association Studies. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 2011, 478, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Burgess, S.; Butterworth, A.; Thompson, S.G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 2013, 37, 658–665. [Google Scholar] [CrossRef] [Green Version]
- Bowden, J.; Smith, G.D.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, D.C.; Lawlor, D.A.; Thompson, J.R. Re: Estimation of bias in nongenetic observational studies using “Mendelian triangulation” by Bautista et al. Ann. Epidemiol. 2007, 17, 511–513. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.; Smith, G.D.; Haycock, P.C.; Burgess, S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar] [CrossRef] [Green Version]
- Simon-Sanchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Gage, S.H.; Jones, H.J.; Taylor, A.E.; Burgess, S.; Zammit, S.; Munafo, M.R. Investigating causality in associations between smoking initiation and schizophrenia using Mendelian randomization. Sci. Rep. 2017, 7, 40653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gage, S.H.; Jones, H.J.; Burgess, S.; Bowden, J.; Smith, G.D.; Zammit, S.; Munafo, M.R. Assessing causality in associations between cannabis use and schizophrenia risk: A two-sample Mendelian randomization study. Psychol. Med. 2017, 47, 971–980. [Google Scholar] [CrossRef] [Green Version]
- Ffytche, D.H.; Creese, B.; Politis, M.; Chaudhuri, K.R.; Weintraub, D.; Ballard, C.; Aarsland, D. The psychosis spectrum in Parkinson disease. Nat. Rev. Neurol. 2017, 13, 81–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5®): American Psychiatric Pub. 2013. Available online: https://dsm.psychiatryonline.org/doi/book/10.1176/appi.books.9780890425596 (accessed on 3 August 2021).
- Hacksell, U.; Burstein, E.S.; McFarland, K.; Mills, R.G.; Williams, H. On the discovery and development of pimavanserin: A novel drug candidate for Parkinson’s psychosis. Neurochem. Res. 2014, 39, 2008–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballanger, B.; Strafella, A.P.; van Eimeren, T.; Zurowski, M.; Rusjan, P.M.; Houle, S.; Fox, S.H. Serotonin 2A receptors and visual hallucinations in Parkinson disease. Arch. Neurol. 2010, 67, 416–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huot, P.; Johnston, T.H.; Darr, T.; Hazrati, L.N.; Visanji, N.P.; Pires, D.; Brotchie, J.M.; Fox, S.H. Increased 5-HT2A receptors in the temporal cortex of parkinsonian patients with visual hallucinations. Mov. Disord. 2010, 25, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Purkayastha, S.; Ford, J.; Kanjilal, B.; Diallo, S.; Del Rosario Inigo, J.; Neuwirth, L.; El Idrissi, A.; Ahmed, Z.; Wieraszko, A.; Azmitia, E.C.; et al. Clozapine functions through the prefrontal cortex serotonin 1A receptor to heighten neuronal activity via calmodulin kinase II-NMDA receptor interactions. J. Neurochem. 2012, 120, 396–407. [Google Scholar] [CrossRef]
- Cummings, J.; Isaacson, S.; Mills, R.; Williams, H.; Chi-Burris, K.; Corbett, A.; Dhall, R.; Ballard, C. Pimavanserin for patients with Parkinson’s disease psychosis: A randomised, placebo-controlled phase 3 trial. Lancet 2014, 383, 533–540. [Google Scholar] [CrossRef]
- Psychosis Endophenotypes International Consortium; Wellcome Trust Case-Control Consortium; Bramon, E.; Pirinen, M.; Strange, A.; Lin, K.; Freeman, C.; Bellenguez, C.; Su, Z.; Band, G.; et al. A genome-wide association analysis of a broad psychosis phenotype identifies three loci for further investigation. Biol. Psychiatry 2014, 75, 386–397. [Google Scholar] [CrossRef] [Green Version]
- Caldani, S.; Amado, I.; Bendjemaa, N.; Vialatte, F.; Mam-Lam-Fook, C.; Gaillard, R.; Krebs, M.O.; Bucci, M.P. Oculomotricity and Neurological Soft Signs: Can we refine the endophenotype? A study in subjects belonging to the spectrum of schizophrenia. Psychiatry Res. 2017, 256, 490–497. [Google Scholar] [CrossRef]
- Morrens, M.; Docx, L.; Walther, S. Beyond boundaries: In search of an integrative view on motor symptoms in schizophrenia. Front. Psychiatry 2014, 5, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateos, J.J.; Lomena, F.; Parellada, E.; Mireia, F.; Fernandez-Egea, E.; Pavia, J.; Prats, A.; Pons, F.; Bernardo, M. Lower striatal dopamine transporter binding in neuroleptic-naive schizophrenic patients is not related to antipsychotic treatment but it suggests an illness trait. Psychopharmacology 2007, 191, 805–811. [Google Scholar] [CrossRef]
- Schmitt, G.J.; Meisenzahl, E.M.; Frodl, T.; La Fougere, C.; Hahn, K.; Moller, H.J.; Dresel, S. Increase of striatal dopamine transmission in first episode drug-naive schizophrenic patients as demonstrated by [(123)I]IBZM SPECT. Psychiatry Res. 2009, 173, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Plaschke, R.N.; Cieslik, E.C.; Muller, V.I.; Hoffstaedter, F.; Plachti, A.; Varikuti, D.P.; Goosses, M.; Latz, A.; Caspers, S.; Jockwitz, C.; et al. On the integrity of functional brain networks in schizophrenia, Parkinson’s disease, and advanced age: Evidence from connectivity-based single-subject classification. Hum. Brain Mapp. 2017, 38, 5845–5858. [Google Scholar] [CrossRef] [Green Version]
- Gaig, C.; Tolosa, E. When does Parkinson’s disease begin? Mov. Disord. 2009, 24, S656–S664. [Google Scholar] [CrossRef]
- Vingerhoets, F.J.; Snow, B.J.; Lee, C.S.; Schulzer, M.; Mak, E.; Calne, D.B. Longitudinal fluorodopa positron emission tomographic studies of the evolution of idiopathic parkinsonism. Ann. Neurol. 1994, 36, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Scherman, D.; Desnos, C.; Darchen, F.; Pollak, P.; Javoy-Agid, F.; Agid, Y. Striatal dopamine deficiency in Parkinson’s disease: Role of aging. Ann. Neurol. 1989, 26, 551–557. [Google Scholar] [CrossRef]
- Cohen, B.M. Embracing Complexity in Psychiatric Diagnosis, Treatment, and Research. JAMA Psychiatry 2016, 73, 1211–1212. [Google Scholar] [CrossRef] [PubMed]
- Gadit, A. Schizophrenia and Parkinson’s disease: Challenges in management. BMJ Case Rep. 2011, 2011. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Lawlor, D.A. Commentary: Two-sample Mendelian randomization: Opportunities and challenges. Int. J. Epidemiol. 2016, 45, 908–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| SNP | Chromosome | Position (hg19) | Gene Region | Effective Allele | PD | SCZ | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Beta | SE of Beta | p Value | Beta | SE of Beta | p Value | |||||
| rs4889730 | 17 | 21717727 | None | G | −0.32 | 0.05 | 2.83 × 10−11 | −0.02 | 0.01 | 0.04 |
| rs2736990 | 4 | 90678541 | SNCA | G | 0.24 | 0.04 | 5.69 × 10−9 | 0.03 | 0.01 | 1.60 × 10−3 |
| rs3784847 | 16 | 61977449 | CDH8 | G | 0.46 | 0.08 | 1.66 × 10−9 | 0.02 | 0.02 | 0.22 |
| rs415430 | 17 | 44859144 | WNT3 | C | −0.29 | 0.05 | 4.50 × 10−8 | −0.04 | 0.01 | 1.65 × 10−3 |
| Subthreshold for PD GWAS p-Value | 5 × 10−8 | 5 × 10−7 | 5 × 10−6 | |||
|---|---|---|---|---|---|---|
| p-Value | OR (95% CI) | p-Value | OR (95% CI) | p-Value | OR (95% CI) | |
| SCZ | 3.49 × 10−5 | 1.10 (1.05–1.15) | 7.00 × 10−7 | 1.10 (1.06–1.14) | 6.64 × 10−5 | 1.06 (1.03–1.09) |
| ADHD | 0.78 | 0.99 (0.91–1.07) | 1.00 | 1.00 (0.93–1.07) | 0.31 | 1.02 (0.98–1.05) |
| ASD | 0.07 | 0.92 (0.83–1.00) | 0.10 | 0.93 (0.85–1.01) | 0.10 | 0.93 (0.85–1.01) |
| BP | 0.22 | 1.10 (0.93–1.26) | 0.11 | 1.10 (0.97–1.23) | 0.16 | 1.05 (0.98–1.12) |
| MDD | 0.92 | 1.00 (0.89–1.10) | 0.67 | 0.98 (0.90–1.07) | 0.22 | 0.97 (0.91–1.02) |
| AD | 0.67 | 1.03 (0.91–1.15) | 0.86 | 0.99 (0.86–1.11) | 0.39 | 0.98 (0.92–1.03) |
| Alcohol dependence | 0.91 | 0.98 (0.68–1.28) | 0.80 | 0.97 (0.73–1.21) | 0.58 | 0.96 (0.81–1.11) |
| Smoking | 0.99 | 1.00 (0.64–1.36) | 0.46 | 1.21 (0.60–1.82) | 0.25 | 1.15 (0.87–1.43) |
| Cannabis use | 0.005 | 1.02 (1.01–1.03) | 0.15 | 1.01 (0.99–1.03) | 0.19 | 1.01 (0.99–1.02) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.; Kim, S.; Myung, W.; Shim, I.; Lee, H.; Kim, B.; Cho, S.K.; Yoon, J.; Kim, D.K.; Won, H.-H. Shared Genetic Background between Parkinson’s Disease and Schizophrenia: A Two-Sample Mendelian Randomization Study. Brain Sci. 2021, 11, 1042. https://doi.org/10.3390/brainsci11081042
Kim K, Kim S, Myung W, Shim I, Lee H, Kim B, Cho SK, Yoon J, Kim DK, Won H-H. Shared Genetic Background between Parkinson’s Disease and Schizophrenia: A Two-Sample Mendelian Randomization Study. Brain Sciences. 2021; 11(8):1042. https://doi.org/10.3390/brainsci11081042
Chicago/Turabian StyleKim, Kiwon, Soyeon Kim, Woojae Myung, Injeong Shim, Hyewon Lee, Beomsu Kim, Sung Kweon Cho, Joohyun Yoon, Doh Kwan Kim, and Hong-Hee Won. 2021. "Shared Genetic Background between Parkinson’s Disease and Schizophrenia: A Two-Sample Mendelian Randomization Study" Brain Sciences 11, no. 8: 1042. https://doi.org/10.3390/brainsci11081042